
Abstract
In adult articular cartilage, the extracellular matrix is maintained by a balance between the degradation and the synthesis of matrix components. Chondrocytes that sparsely reside in the matrix and rarely proliferate are the key cellular mediators for cartilage homeostasis. There are indications for the involvement of the WNT signaling pathway in maintaining articular cartilage. Various WNTs are involved in the subsequent stages of chondrocyte differentiation during development, and deregulation of WNT signaling was observed in cartilage degeneration. Even though gene expression and protein synthesis can be activated upon injury, articular cartilage has a limited ability of self-repair and efforts to regenerate articular cartilage have so far not been successful. Because WNT signaling was found to be involved in the development and maintenance of cartilage as well as in the degeneration of cartilage, interfering with this pathway might contribute to improving cartilage regeneration. However, most of the studies on elucidating the role of WNT signaling in these processes were conducted using in vitro or in vivo animal models. Discrepancies have been found in the role of WNT signaling between chondrocytes of mouse and human origin, and extrapolation of results from mouse models to the human situation remains a challenge. Elucidation of detailed WNT signaling functions will provide knowledge to improve cartilage regeneration.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Adult articular cartilage is an avascular tissue composed of a dense extracellular matrix (ECM) predominantly composed of collagens, proteoglycans and glycosaminoglycans, which provide compressive and tensile strength to the cartilage tissue. The chondrocytes residing in this ECM maintain it by remodeling. Because the metabolic activity of chondrocytes is relatively low and cartilage is scarcely populated with cells, cartilage remodeling is a slow process. Articular cartilage is composed of four distinct regions, the superficial zone which faces the synovial cavity, the middle zone, the deep zone and the calcified cartilage zone adjacent to the subchondral bone plate. These zones differ in cell density and matrix composition. Also the chondrocyte morphology differs between zones, as chondrocytes in the superficial zone are small and flattened, middle zone chondrocytes are more rounded and in the deep zone chondrocytes are grouped in columns or clusters. Because chondrocytes rarely divide and are separated from each other by the matrix, it is unclear how they are regulated to maintain the ECM under homeostatic conditions. It is known, however, that gene expression and protein synthesis can be activated upon injury.
Aging and joint disease disrupt the equilibrium within the cartilage tissue and the synthesis of new matrix components is exceeded by the loss of collagens and proteoglycans [1]. This disbalance between anabolic and catabolic processes results in progressive cartilage degeneration. Even though the exact etiology and pathophysiology of cartilage degenerative diseases is largely unknown and there are probably multiple conditions that may trigger its onset, cartilage destruction is considered to be the result of increased expression and activity of proteolytic enzymes, for example as a response to abnormal mechanical loading, genetic predisposition, trauma or inflammation. Matrix metalloproteinases (MMPs) and aggrecanases are the major proteinases that degrade collagens and proteoglycans in joint disease.
Articular cartilage has a limited ability for self-repair. In spite of extensive research efforts to develop treatment options for cartilage degeneration by regeneration of articular cartilage, so far no curative treatment is available to regenerate or restore native articular cartilage.
In order to reverse degeneration and induce regeneration of articular cartilage, it is crucial to have a detailed understanding of chondrogenic differentiation during skeletal development and the maintenance of articular cartilage in adulthood. Using this knowledge, it may be possible to induce chondrogenic differentiation and shifting the balance from catabolism to anabolism. In this review, we describe the processes and molecular components that are involved in skeletal development with emphasis on articular cartilage formation and homeostasis. Because the WNT pathway was found to play an important role in embryonic development and indications of deregulated WNT signaling have been found in degenerative cartilage disease, we focus on the role of the WNT signaling pathway and its components in skeletal development and the maintenance of cartilage homeostasis. We also touch upon the role of WNT signaling in cartilage degeneration. The different components of the WNT signaling pathway that are involved in cartilage development and disease are listed in Table 1.
WNT Signaling
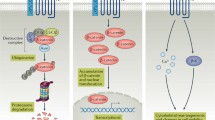
The WNT family of secreted glycoproteins, which are characterized by several conserved cysteine residues, consists of 19 members, of which several encode distinct isoforms arising from differential splicing [2]. The WNT signaling pathway is composed of several components and plays a fundamental role in controlling cell proliferation, cell fate determination, and differentiation by inducing changes in gene expression during embryonic development and in adult cartilage [2, 3]. At least three distinct intracellular signaling pathways are known that are activated by distinct sets of WNTs and Frizzled (FZD) receptors and that lead to unique cellular responses [4]. The canonical WNT/β-catenin pathway is the best described pathway for WNT signal transduction (Fig. 1). In an inactive state, in the absence of a WNT ligand, β-catenin (CTNNB1) is phosphorylated at the NH2 terminus by glycogen synthase kinase 3β (GSK3β) and casein kinase I (CKI) in a destruction complex, which is brought together by the two scaffolding proteins axin and adenomatous polyposis coli (APC). This phosphorylation targets β-catenin for subsequent ubiquitylation and proteasomal degradation. When WNTs bind to the seven transmembrane frizzled receptor in combination with a coreceptor of the LDL related proteins (LRP) 5 or 6, disheveled (DSH) is activated, resulting in suppression of GSK3β activity. As a result, β-catenin will not undergo phosphorylation and is stabilized in the cytoplasm. Upon reaching a certain level, β-catenin translocates to the nucleus, where it interacts with transcription factors of the T-cell-specific transcription factor/lymphoid enhancer-binding factor (TCF/LEF) family to initiate the transcription of target genes [5, 6].

WNT/β-catenin signaling pathway In the inactive state, β-catenin is phosphorylated by a degradation complex consisting of GSK3β, APC, AXIN and CKI, targeting β-catenin for ubiquitylation and subsequent proteasomal degradation. When a WNT ligand binds to the FZD receptor and coreceptors LRP5/6, the degradation complex is disrupted, resulting in stabilization of β-catenin in the cytoplasm. Subsequently β-catenin is translocated to the nucleus and binds to transcription factors TCF/LEF to initiate gene transcription
Another intracellular WNT pathway was first identified in Drosophila. This pathway is involved in regulating planar cell polarity by inducing cytoskeletal organization relative to the plane of the tissue in which the cells reside [4, 7]. However, although FZD and DSH were shown to play a role in this pathway, no involvement of LRP, β-catenin or TCF was found and current evidence suggests that no WNT ligands are involved in the regulation of planar cell polarity [4]. FZD is also involved in the WNT/Ca2+ pathway, which is another pathway activated by subsets of WNT ligands [4, 6, 8]. In this pathway, binding of a WNT ligand to FZD induces activation of a heterotrimeric G protein, resulting in an increase in intracellular levels of Ca2+. This activates Ca2+-dependent effector molecules such as the transcription factor Nuclear Factor Associated with T cells (NFAT) [4].
The specificity of activation downstream of WNT is determined by selective receptor activation, receptor-mediated endocytosis and the presence of cofactors such as heparin and sulfate proteoglycans [9]. Accumulating evidence has suggested that WNT may activate multiple pathways depending on the engagement of distinct receptors [4]. For example, WNT-5A, which is most often associated with noncanonical pathway, also activates the canonical β-catenin signaling [10]; the canonical WNT-3A also activates the noncanonical Ca2+ pathway [11].
WNT signals can be modulated extracellularly by secreted proteins, including members of the secreted Frizzled-related protein (sFRP) family, WNT-inhibitory factor (WIF) 1, Cerberus, SOST and Dickkopf 1 (DKK1). sFRPs, WIF-1 and Cerberus can bind to WNTs directly, thereby preventing their interaction with FZD receptors, whereas sFRPs can also bind FZD receptors to form nonfunctional complexes [2, 12]. Both SOST and DKK-1 antagonize WNT signaling by binding to LRP5/6 coreceptors, albeit to distinct regions, and thereby preventing complex formation with the FZD/WNTs [13]. Intracellularly, WNT/β-catenin signaling can be modulated by Inhibitor of β-catenin and T-cell factor (ICAT), which disturbs the interaction between β-catenin and transcription factors TCF/LEF, thereby inhibiting β-catenin-mediated transcription [14, 15].
Skeletal Development
The formation of most of the vertebral skeleton occurs via endochondral bone formation, a process starting with the aggregation, proliferation and condensation of mesenchymal stromal cells (MSCs) at specific locations within the embryo. These MSCs originate from the neural crest (forming craniofacial bones), the sclerotome of the paraxial mesoderm (forming the axial skeleton), or the lateral plate mesoderm (forming the appendicular skeleton) [16]. MSCs commit to the skeletal lineage once they have differentiated into skeletal precursor cells (SPCs), from which chondrocytes and osteoblasts can be derived. Cellular condensations form as the result of altered mitotic activity, and failure of cells to move away from a center or aggregation of cells toward a center, as occurs in limb formation. This process leads to increased mesenchymal cell density, without an increase in cell proliferation. Consequently, cellular condensation is associated with an increase in cell–cell contacts through cell–cell adhesion molecules and gap junctions that facilitate intercellular communication [17]. Before condensation, MSCs secrete an ECM rich in hyaluronan and collagen type I, preventing intimate cell–cell interaction. As condensation begins, an increase in hyaluronidase activity is observed, so the ECM hyaluronan content decreases, allowing cell migration. The increased cellular interaction as observed during condensation is thought to be involved in triggering signal transduction pathways that initiate chondrogenic differentiation. Adhesion molecules like neural cadherin (N-cadherin) and neural cell adhesion molecule (N-CAM) are expressed in condensing mesenchyme, while they disappear in differentiating chondrocytes. Furthermore, cell–matrix interactions involving fibronectin play an important role in mesenchymal condensation. Fibronectin expression was found to be up-regulated in cellular condensation, and its expression decreases when chondrogenic differentiation continues. At the periphery of these condensations, SPCs form a perichondrial layer, while in the core they differentiate into chondrocytes that produce cartilage specific extracellular matrix (ECM) proteins and continue to proliferate [17].
The differentiation of chondroprogenitor cells is characterized by the deposition of a cartilaginous extracellular matrix containing collagen types II, IX and XI and aggrecan. One of the earliest markers expressed in cells undergoing condensation is SOX9, which is required for the expression of the collagen type II α1 (COL2A1) gene and other cartilage specific extracellular matrix proteins [18, 19]. Continuous proliferation of chondrocytes and secretion of ECM contribute to the elongation of the cartilage template, which prefigures the shape of the future bone.
After formation of the cartilaginous template, the core chondrocytes mature and become hypertrophic, secreting a progressively calcified ECM. Simultaneously, perichondrial SPCs differentiate into osteoblasts, forming the future periosteum, which modulates the final shape and size of the cartilage template. After mineralization of the cartilage ECM, vascular invasion and apoptosis of terminal hypertrophic chondrocytes initiate the formation of the primary ossification center. This complex differentiation program radiates centrifugally, leading to the development of trabecular bone [16, 20–24].
Involvement of WNT Signaling in Skeletal Development
In skeletal development, the fate of MSCs to differentiate into either chondrocytes or osteoblasts depends on the expression of the transcription factors SOX9 or RUNX2, respectively. Skeletal precursor cells express both transcription factors, and the intracellular expression levels of β-catenin determine the fate of these cells. High levels of β-catenin inhibit SOX9 expression and activity, while potentiating RUNX2, which results in osteoblast differentiation. In contrast, low β-catenin levels induce SOX9 expression and thereby chondrocyte differentiation [25, 26].
An increasing amount of evidence indicates the important role of WNT/β-catenin signaling in essentially all aspects of skeletal development and maintenance. The role of canonical WNT/β-catenin signaling at subsequent stages of skeletogenesis has been suggested on the basis of the expression patterns of many WNT pathway members, as well as WNT-reporter expression in mice [25–31]. Involvement of WNT signaling in chondrogenic differentiation is depicted in Fig. 2.

Involvement of WNT signaling in consecutive stages of chondrogenic differentiation WNT-3A induces proliferation and self-renewal of MSCs, which form mesenchymal condensations at the initial stage of differentiation. Subsequently, chondrocyte differentiation is induced by low levels of β-catenin, WNT-5A, WNT-5B and WIF-1, whereas WNT-1, WNT-3A, WNT-7A and WNT-14 block chondrocyte differentiation. In adult articular cartilage, a fine balance of β-catenin levels is involved in maintaining the chondrocyte phenotype. Hypertrophic differentiation of chondrocytes is induced by WNT-4 and high levels of β-catenin and blocked by WNT antagonists DKK-1 and FRZB
Because β-catenin is a key molecule in the canonical WNT signaling pathway, it is the most studied molecule involved in this pathway. In vitro and in vivo data suggest that β-catenin plays an essential role in cell fate determination in skeletal development, as it acts as a molecular switch between chondrocyte and osteoblast differentiation in SPCs. Up-regulation of β-catenin in mesenchymal condensations was found before expression of the osteoblast-specific transcription factors RUNX2 and OSX was detected, indicating that high levels of β-catenin precede osteoblast differentiation. In contrast, β-catenin expression was down-regulated in chondrogenic mesenchymal condensations [25]. Because conditional deletion of β-catenin is prenatally lethal, an essential role for β-catenin in early skeletal development was indicated [25]. Deletion of β-catenin in early embryonic development results in the arrest of osteoblast differentiation at the level of osteochondrogenic progenitor cells, which instead differentiate into chondrocytes [26]. Furthermore, inactivation of β-catenin in micromass cultures enhances chondrocyte differentiation in vitro [25]. Mice in which ablation of β-catenin is induced in cartilage after birth, develop an osteoarthritis-like phenotype. These studies indicate that β-catenin might be involved in maintaining cartilage in adult indicating that a fine balance of β-catenin is required both for normal chondrogenic differentiation, as well as for the maintenance of cartilage tissue after formation [32, 33].
A number of WNT ligands have been implicated in the regulation of various aspects of endochondral ossification. Although WNT-1 is not endogenously expressed during limb development, overexpression of WNT-1 in chick embryos in vivo was found to cause skeletal abnormalities such as truncation or deletion of skeletal elements [34, 35]. Because retroviral expression of WNT-1 in limb bud micromass cultures resulted in severely inhibited cartilage formation, this might be the underlying mechanism for these skeletal abnormalities in vivo [35]. WNT-3A regulates the expansion of the MSC population, through increasing self-renewal and decreasing apoptosis [36, 37]. Expression of WNT-3A is decreased when chondrogenic differentiation progresses. Furthermore, addition of exogenous WNT-3A blocked the collagen type II expression and suppressed the deposition of sulfated proteoglycans, indicating that down-regulation of WNT-3A is required for chondrogenesis [38]. WNT-4 was found to be involved in joint formation and cartilage development, as it is expressed in developing joints, in the periphery of the joint interzone and in a subset of hypertrophic chondrocytes [39, 40]. Different effects of WNT-4 were found during different stages of skeletal development. Misexpression of WNT-4 resulted in the shortening of long bones and histological examination of developing limbs revealed that cartilage elements in these limbs showed an expanded hypertrophic zone and a thicker osteoid layer of the bone collar, compared to the contralateral control limb [41]. These findings indicate that WNT-4 accelerates chondrocyte maturation, which results in an accumulation of terminally differentiated hypertrophic chondrocytes at the expense of immature round chondrocytes at the ends of the cartilage elements [40]. During skeletal development, WNT-5A is initially expressed in the mesenchyme around the developing condensations, indicating that WNT-5A might be involved in the recruitment of mesenchymal cells into the chondrogenic lineage. At later stages, WNT-5A expression was found in the perichondrium, indicating the involvement of WNT-5A in the formation of bone. Expression of WNT-5B was restricted to prehypertrophic chondrocytes, as well as cells in the outer layer of the perichondrium [39, 40, 42]. WNT-5A and WNT-5B both promote the first steps of chondrogenesis in micromass cultures, whereas cartilage elements in which WNT-5A was misexpressed are smaller in size and show a delay in the maturation of hypertrophic chondrocytes histologically and molecularly [39, 41]. Expression of WNT-7A was found in the dorsal ectoderm in the developing limb [43]. Chondrogenesis was blocked by WNT-7A in micromass cultures in vitro as well as after in vivo overexpression in chick embryos [34]. Furthermore, WNT-7A induces a chondroinhibitory effect, which is mediated by MAP kinase and AP1 signaling [44]. WNT-14 expression was, like WNT-4, found in the developing joint interzone [45]. WNT-14 is implicated in the initial steps of joint development and it was found to arrest and even reverse chondrogenic differentiation. Ectopic expression of WNT-14 in the prechondrogenic region prevents prechondrogenic cells from further differentiating into chondrocytes, with down-regulated expression of chondrogenic markers Collagen II and Sox9 [40]. In the mature joint, WNT-14 continues to be expressed in the synoviocytes and the joint capsule.
The regulation and specificity of WNT signaling is not only dependent on the presence of specific ligands and receptors, but also on the action of endogenous antagonists of WNT signaling. FRZB expression was specifically found in mesenchymal prechondrogenic condensations and at later stages in epiphyseal prearticular chondrocytes [46, 47]. Overexpression of Frzb in vivo blocks chondrocyte maturation at an early hypertrophic stage and prevents endochondral ossification [47]. In addition, Frzb knockout mice, in which a mild activation of WNT/β-catenin signaling was observed, exhibit accelerated hypertrophic chondrocyte maturation [48]. Addition of FRZB to micromass cultures of MSCs promoted glycosaminoglycan synthesis, as well as gene expression and protein expression of SOX9 and collagen type II. DKK-1 was found to have a similar effect, although to a lesser extent [49]. A recent study provided evidence that the WNT antagonists DKK-1 and FRZB in combination with the bone morphogenetic protein (BMP) antagonist GREM1 are significantly higher expressed in articular cartilage compared to growth plate cartilage [50]. The authors provided evidence that these antagonists are natural breaks of hypertrophic differentiation and subsequent endochondral ossification of articular cartilage. WIF-1 expression was found in the mesenchyme surrounding cartilage elements forming in the limb during early skeletal development. In late embryonic and postnatal development, WIF-1 expression was observed in articular cartilage. Moreover, WIF-1 interferes with WNT-3A mediated inhibition of chondrogenesis in micromass cultures [51].
Cartilage Degeneration in Arthritis
Arthritis is a joint disorder that usually involves one or more joints. Osteoarthritis (OA), also known as degenerative arthritis, is the most common form of arthritis. It is a heterogeneous disease characterized by progressive degradation of joint cartilage, typical bone changes and signs of mild synovitis particularly in more advanced stages of the disease. In contrast, rheumatoid arthritis (RA) is a chronic, systemic inflammatory disorder that may affect many tissues and organs, but principally involves synovial joints. In both OA and RA, articular cartilage is the primary target for damage as a result of loss of cartilage homeostasis. Cartilage homeostasis in healthy joints is maintained by the balance of synthesis and degradation of extracellular matrix (ECM). Cartilage undergoes destruction when this balance is lost.
In the adult, articular chondrocytes are fully differentiated cells that play a critical role in the pathogenesis of OA by responding to adverse environmental stimuli by promoting matrix degradation and down-regulating processes essential for cartilage repair. Multiple risk factors have been implicated in the initiation and progression of OA, including mechanical injury, genetics and aging [52]. For instance, in response to traumatic injury chondrocytes activate general gene expression, which results in increased expression of inflammatory mediators, cartilage-degrading proteinases, and stress response factors [53, 54].
Synovial inflammation likely contributes to deregulation of chondrocyte function, amongst others by secreting cytokines that impact chondrocyte activity [55]. Chondrocytes can respond to a number of cytokines and chemokines in the joint tissue and joint fluid. These cytokines and chemokines can be produced by other cells such as fibroblast-like synoviocytes (FLS) which play an important role in the pathogenesis of RA [56]. IL-1β and TNF-α are able to induce the synthesis of ECM degrading enzymes such as MMPs as well as the production of other pro-inflammatory mediators such as prostaglandin E2 (PGE2) and nitric oxide (NO) [57, 58]. In addition, the association of the increased levels of catabolic enzymes and inflammatory mediators such as prostaglandins and NO and the levels of cytokines like IL-1β and TNF-α in synovial fluid and joint tissue has been established [59]. Pro-inflammatory cytokines induce loss of the chondrocytic phenotype of chondrocytes in the matrix and can induce chondrocyte apoptosis [60, 61]. These findings indicate that the pro-inflammatory mediators are crucial mediators of cartilage degeneration.
As was mentioned before, abnormal mechanical loading or synovial inflammation may induce a disequilibrium between the catabolic and anabolic activity of chondrocytes. The relation between increased production of proteinases, including MMPs, MMP-1, MMP-3, MMP-8, MMP-13, and the aggrecanases, particularly ADAMTS-5, with cartilage damage has been documented [62, 63]. FLS in the synovium also produce pro-MMP-3 (precursor form of MMP-3 or stromelysin 1), which in its mature form enhances cartilage degradation [56]. Production and activities of these proteinases are regulated by various mediators such as cytokines, growth factors, prostaglandins, matrix breakdown products, and oxygen species [64, 65]. It has also been shown that expression of the COL2A1 gene is suppressed in upper zones of OA cartilage with progressing matrix destruction, whereas global COL2A1 gene expression is increased in late-stage OA cartilage compared to normal and early degenerative cartilage suggesting a compensatory mechanism [66]. Cessation of cartilage ECM molecule synthesis can be caused by a number of factors such as pro-inflammatory cytokines [60] and NO [61]. Anabolic factors such as BMP-2, activin A and tumor necrosis factor-β (TGF-β) superfamily members might be responsive for the compensatory increase in COL2A1 expression [67, 68]. Importantly, once the cartilage is severely degraded the chondrocyte is unable to reproduce the complex arrangement of collagen laid down during development. Therefore, the imbalance between catabolic and anabolic activities of the chondrocytes is a key contributor to cartilage degeneration.
Phenotypic Modulation of Chondrocyte Function by WNT Signaling
In addition to its function in chondrogenesis and chondrocyte maturation, WNT signaling is also involved in the maintenance of fully differentiated chondrocyte phenotypes and may therefore play a crucial role in cartilage homeostasis throughout adult life. When differentiated chondrocytes are exposed to inflammatory factors such as IL-1 and retinoic acid or when cultured in monolayer, their phenotype is rapidly lost and cells become fibroblast-like. This process is known as dedifferentiation and it is accompanied by increased β-catenin protein expression [69] and differential expression of components of the WNT signaling pathway, such as DKK1, FRZB, WNT-5A, and FZD-4, -8, -9 (unpublished data). Accumulation of β-catenin by ectopic expression or inhibition of its degradation results in a decrease of cartilage-specific ECM molecule synthesis through activation of TCF/LEF transcriptional activity in rabbit chondrocytes [69]. Conditional deletion of the APC gene, which results in up-regulation of β-catenin in mature chondrocytes, also results in a complete loss of the chondrocyte phenotype in vivo [70]. In addition, WNT-3A and WNT-7A caused loss of type II collagen synthesis via stimulation of β-catenin–TCF/LEF transcriptional activity [38]. Moreover, WNT-3A induced the expression of c-Jun and its phosphorylation by c-Jun N-terminal kinase (JNK), resulting in activation of AP-1. AP-1 could suppress the expression of SOX9, a major transcription factor regulating COL2A1 expression [38]. In contrast, WNT-3A inhibited chondrogenesis of mesenchymal cells by stabilizing cell-cell adhesion in a manner independent of β-catenin transcriptional activity [38]. It has also been shown that WNT-7A inhibited NO-induced apoptosis by activating cell survival signaling, such as phosphatidylinositol 3-kinase and AKT, independent of β-catenin transcriptional activity [71]. Together, these results suggest that WNT proteins regulate chondrocyte functions via different mechanisms.
However, all these studies were performed in animal chondrocytes, whereas the function of WNT signaling in human chondrocytes has so far not been well studied. A recent study reported that WNT-3A promoted human articular chondrocyte proliferation through the β-catenin-dependent canonical pathway while simultaneously inducing loss of expression of chondrocyte marker genes via a β-catenin-independent noncanonical pathway (Fig. 3) [11]. Dedifferentiation of human chondrocytes in vitro could not be reversed by inhibition of the canonical WNT pathway either by knockdown of β-catenin or by addition of a TCF/β-catenin inhibitor (unpublished data). Remarkably, during human chondrocyte dedifferentiation the noncanonical WNT-5A is strongly up-regulated which coincided with a down-regulation of COL2A1 expression. Knockdown of WNT-5A reversed COL2A1 expression, again suggesting that dedifferentiation in human chondrocytes appears independent of β-catenin (unpublished data). These findings are in contrast with observations in rabbit chondrocytes, in which β-catenin-TCF/LEF transcriptional activity contributed to chondrocyte dedifferentiation [69]. The controversial findings in human and animal chondrocytes suggest that the exact function of the canonical WNT pathway in articular cartilage may be species-dependent. Such species difference was also observed in the regulation of MMP expression in human and animal chondrocytes [72].

Role of WNT signaling in human chondrocytes WNTs may activate both canonical and noncanonical pathways in a cellular context dependent manner, most likely involving differential expression of FZD receptors at the cell surface. The canonical WNT/β-catenin pathway induces proliferation and expression of target genes such as AXIN2. It represses NF-κB signaling through an inhibitory interaction of β-catenin with NF-κB, consequently inhibiting expression of target genes such as MMPs and IL6. In contrast, TCF4 is able to potentiate NF-κB signaling independent of β-catenin. The noncanonical WNT signaling decreases COL2A1 and SOX9 expression through Ca2+/CaMKII pathway
In contrast to WNT-3A and WNT-7A, WNT-5A and WNT-11 primarily regulate cartilage-specific ECM molecule synthesis through the noncanonical pathway [73]. Stimulation of rabbit chondrocytes with IL-1β up-regulated WNT-5A and down-regulated WNT-11 expression. WNT-5A inhibited COL2A1 expression via the JNK pathway, whereas WNT-11 stimulated COL2A1 expression via the PKC pathway, indicating that WNT-5A and WNT-11 have opposing effects on COL2A1 expression by signaling through distinct noncanonical WNT pathways in rabbit chondrocytes [73]. In human chondrocytes, WNT-5A was also found to block COL2A1 expression, in agreement with its effects in rabbit chondrocytes [72]. Interestingly, WNT-3A is also able to down-regulate COL2A1 and SOX9 expression through the noncanonical Ca2+/CaMKII pathway (Fig. 3) [11]. All these findings substantiate the role of noncanonical cascade in the deregulation of chondrocyte function. Collectively, a direct role of the β-catenin-dependent canonical pathway is unlikely in human chondrocyte dedifferentiation.
WNT Signaling in Cartilage Degeneration
In light of the involvement of WNT signaling in cartilage development and the maintenance of adult chondrocyte phenotype and cartilage homeostasis, dysfunction of the WNT pathway may lead to cartilage tissue disease. Indeed, differential expression of WNT pathway components has been documented in joint disorders such OA and RA. In several genome-wide association studies, the WNT antagonist FRZB has emerged as a candidate gene associated with an increased risk for OA [74–77]. A single-nucleotide polymorphism in FRZB resulting in an Arg324Gly substitution at the carboxyl terminus, which shows diminished ability to antagonize WNT signaling in vitro, was associated with hip OA in females [75]. The correlation of elevated circulating levels of DKK-1, another WNT antagonist, with reduced progression of radiographic hip osteoarthritis (RHOA) in elderly women has also been suggested [78]. This is in line with the proposed role of DKK-1 with FRZB and GREM1 as natural brakes of chondrocyte hypertrophy. Derailed hypertrophic differentiation in articular cartilage has been implemented in the pathogenesis of OA, at least in a subset of patients [50]. Likewise, differential expression of various WNTs and their receptors has been reported in human joint disorders [79]. For example, overexpression of WNT-5A and FZD5 has been found in RA synovial tissues in comparison to a panel of normal adult tissues [79], while the canonical WNT-7B is up-regulated in OA cartilage and RA synovium [80]. In addition, increased expression of the WNT target gene WISP-1 was found in both mouse OA models and in human OA cartilage [81]. A systematic analysis of the WNT signaling pathway revealed up-regulation of WNT-16, down-regulation of FRZB, up-regulation of WNT target genes, and nuclear localization of β-catenin in injured cartilage [82]. In addition, in OA, WNT-16 and β-catenin were barely detectable in preserved cartilage areas, but were dramatically up-regulated in areas of the same joint with moderate to severe OA damage [82]. These findings were subsequently corroborated by observation of increased nuclear β-catenin staining in human OA cartilage compared to control [32]. Therefore, these studies indicate that increased WNT signaling is associated with cartilage degeneration.
To explore the exact function and underlying mechanism of WNT signaling in joint biology and disease, a variety of studies have been conducted in animal models. Although a noteworthy developmental phenotype is not developed, Frzb −/− mice display greater cartilage loss in comparison to wild type controls when exposed to factors known to induce OA, like enzymatic treatment (papain-induced OA), accelerated instability (collagenase-induced ligament and meniscal damage) or inflammation (mBSA-induced monoarthritis) [83, 84]. The mild phenotype might be explained by partial compensation by other antagonists like DKK-1 and GREM1 [50]. Cartilage degradation in the Frzb −/− mice is associated with up-regulation of β-catenin and MMP-9. Interestingly, it was also shown that cartilage injury results in increased WNT activity and decreased expression of FRZB in vitro [85]. In postnatal mouse models, β-catenin signaling has been selectively activated in articular chondrocytes in 3- and 6-month-old mice using an inducible conditional transgenic approach and tissues were analyzed two months later. In 5-month-old mice reduced articular cartilage area and matrix production is observed [32]. In 8-month-old mice cell cloning, surface fibrillation, vertical clefting, and chondrophyte/osteophyte formation is observed. At this age, complete loss of articular cartilage layers and the formation of new woven bone in the subchondral bone area is also found. These findings indicate that activation of β-catenin signaling in articular chondrocytes in adult mice leads to the development of an OA-like phenotype. At both ages, activation of β-catenin accelerates chondrocyte maturation [32]. Because the phenotype 2 months after the inducible conditional activation of β-catenin in 6-month-old mice is more pronounced than in 3-month-old-mice, β-catenin’s role in chondrocytes may be age dependent. This is further underscored by the phenotype of transgenic mouse embryos with conditional activation of β-catenin in collagen type II positive chondrocytes. Constitutive activation of β-catenin in chondrocytes is not compatible with the formation of chondrocytes and the phenotype of these mice resembles the phenotype of embryos lacking Sox9 expression in chondrocytes [86]. A similar phenotype is found in mice with a conditional inactivation of the Apc gene in chondrocytes [70]. Thus, in organogenesis, activation of β-catenin in collagen type II positive chondrocytes arrests further chondrogenesis and induces a complete loss of the chondrocyte phenotype reverting in a more undifferentiated cell type [70, 86], while in adult articular cartilage activation of β-catenin accelerates chondrocyte maturation and induces typical signs of OA [32].
Activation of WNT/β-catenin signaling in rabbit and mouse chondrocytes stimulates the expression of cartilage matrix degrading MMPs [87, 88]. In a spontaneous guinea pig OA model, development of OA is associated with increased β-catenin expression in cartilage [87]. Interestingly, inhibition of β-catenin signaling in articular chondrocytes also causes OA-like cartilage degradation in a Col2a1-ICAT transgenic mouse model [14]. In 6-month-old or elder Col2a1-ICAT transgenic mice, ICAT expression is associated with significant increase in articular chondrocyte apoptosis and cartilage destruction. Consistent with increased TUNEL staining, Bcl-2 and Bcl-x expression are decreased and caspase 9 and caspase 3/7 activity are increased in chondrocytes, suggesting that increased cell apoptosis may contribute to articular cartilage destruction observed in Col2a1-ICAT transgenic mice. Because ICAT may have other cellular targets than β-catenin, it is unclear whether the OA-like phenotype in these mice can solely be attributed to inhibition of β-catenin. Taken together, these findings have led to the hypothesis that in postnatal cartilage low levels of WNT/β-catenin signaling are required for maintenance of normal cartilage function and that deregulation of this pathway may contribute to the development and progression of cartilage degeneration. This hypothesis is supported by strong experimental evidence in animal models. The support for such a role in human OA, however, is less strong and is predominantly based on circumstantial evidence showing associations between increased β-catenin levels and an OA phenotype in cartilage specimens. Evidence for a causal relationship in human is currently lacking.
In contrast to its pro-catabolic role in animal cartilage by inducing ECM degrading enzymes such as MMPs, WNT activation was found to inhibit MMP expression in a TCF/LEF-independent pathway in human articular chondrocytes [72]. In animal cells, the WNT pathway regulates MMP expression through β-catenin-TCF/LEF transcriptional activity [72, 89]. In human chondrocytes, activation of canonical WNT blocks MMP expression through an inhibitory interaction of β-catenin with NF-κB (Fig. 3) [72]. This species difference in the regulation of pro-catabolic MMP expression gives rise to the question whether canonical WNT signaling is a pathogenic factor in human cartilage degeneration. However, similar to findings in animal models, in fibroblast-like synoviocytes from RA patients, activation of canonical WNT signaling by WNT-1 transfection increases expression of MMP-3, while interference with WNT signaling using anti-WNT-1 blocking antibody or the WNT antagonist sFRP-1 decreases MMP-3 expression [90]. WNT signaling may exhibit complicated functions in joint disease by activating multiple cascades and interacting with other pathways, which might also be tissue dependent. In addition, the heterogeneity of cell cultures, age of donors and stages of differentiation of chondrocytes are alternative explanations for the observed differences between human and animal chondrocytes. Furthermore, the cellular context might play a decisive factor in determining how cells respond to β-catenin. Factors that may influence a cell’s response to WNT/β-catenin signaling are amongst others the presence and concentration of specific combinations of Frizzled or LRP5/6 receptors at the cell surface [4] and/or intracellular regulators of canonical WNT signaling that may compete for binding to β-catenin. For example, detailed comparison of Frizzled receptors expression between human and animal chondrocytes as a function of age and cellular differentiation state are lacking. In addition, a number of intracellular targets of β-catenin have been identified such as TCF/LEF transcription factor, NF-κB and SOX9 [3, 72, 86]. These factors may compete for binding to β-catenin and consequently the relative abundance of these factors in a cell may influence β-catenin’s response. Detailed side-by-side analysis of the expression of these factors in chondrocytes from distinct species are lacking. Therefore, the role of WNT signaling in human cartilage degeneration remains elusive and more studies should be focused on extrapolation of knowledge obtained from animal models to the human situation. Our recent study also reveals that TCF4 induces MMP expression and apoptosis, probably through potentiating NF-κB signaling (Fig. 3) and its expression is up-regulated in OA cartilage compared to normal human cartilage (unpublished data). Thus, TCF4 may serve as a potential therapeutic target for OA.
A role of noncanonical WNT signaling in cartilage degeneration is suggested by the induction of MMP expression by WNT-5A in rabbit chondrocytes [91] and repression of chondrocyte marker gene expression by WNT-5A or the WNT-3A-mediated Ca2+ pathway [11]. It has been suggested that canonical and noncanonical pathways reciprocally inhibit each other [11]. Therefore, blocking the canonical/β-catenin pathway will also cause articular chondrocyte dedifferentiation through de-repression of the Ca2+ pathways. Stimuli such as IL-1 [92] and biomechanics [93], which change CaMKII, may significantly influence the outcome of WNT signaling by switching the balance between β-catenin and CaMKII. Thus, IL-1-mediated pro-catabolic activity in cartilage may partially come from its enhancement of the Ca2+ cascade activity initiated by WNT. The opposing induction of the β-catenin pathway by IL-1 serves as a negative feedback to counteract its pro-catabolic activity [72]. Because of the complicated properties of WNT in activating multiple pathways and WNT signaling components showing diverse functions in human chondrocytes, more specific targeted therapy should be developed with respect to treating human joint disease by manipulating WNT signaling.
Conclusion
A remarkable effort has unraveled the stage-dependent regulatory role of WNT signaling in chondrogenesis and cartilage development. Consequently, cumulating evidence has suggested the involvement of WNT signaling in cartilage disease. Although extensive animal studies have indicated that excessive WNT signaling may lead to cartilage destruction, the exact function of WNT signaling in human cartilage is still largely unclear. Species differences in WNT function in chondrocytes have been observed. The future challenge for research is to properly extrapolate our knowledge of WNT signaling in animal models to the human situation. Given the fact that WNT signaling is a complex network, accumulation of our understanding of the involvement of specific WNT components in cartilage degeneration will facilitate the development of more effective and specific treatments for joint disease.
References
Goldring MB, Marcu KB (2009) Cartilage homeostasis in health and rheumatic diseases. Arthritis Res Ther 11:224
Miller JR (2002) The Wnts. Genome Biol 3:REVIEWS3001
Logan CY, Nusse R (2004) The Wnt signaling pathway in development and disease. Annu Rev Cell Dev Biol 20:781–810
van Amerongen R, Mikels A, Nusse R (2008) Alternative wnt signaling is initiated by distinct receptors. Sci Signal 1:re9
Clevers H (2006) Wnt/beta-catenin signaling in development and disease. Cell 127:469–480
Miller JR, Hocking AM, Brown JD, Moon RT (1999) Mechanism and function of signal transduction by the Wnt/beta-catenin and Wnt/Ca2+ pathways. Oncogene 18:7860–7872
Adler PN, Lee H (2001) Frizzled signaling and cell–cell interactions in planar polarity. Curr Opin Cell Biol 13:635–640
Kuhl M, Sheldahl LC, Park M, Miller JR, Moon RT (2000) The Wnt/Ca2+ pathway: a new vertebrate Wnt signaling pathway takes shape. Trends Genet 16:279–283
Kikuchi A, Yamamoto H, Sato A (2009) Selective activation mechanisms of Wnt signaling pathways. Trends Cell Biol 19:119–129
Mikels AJ, Nusse R (2006) Purified Wnt5a protein activates or inhibits beta-catenin-TCF signaling depending on receptor context. PLoS Biol 4:e115
Nalesso G, Sherwood J, Bertrand J, Pap T, Ramachandran M, De Bari C, Pitzalis C, Dell’accio F (2011) WNT-3A modulates articular chondrocyte phenotype by activating both canonical and noncanonical pathways. J Cell Biol 193:551–564
Kawano Y, Kypta R (2003) Secreted antagonists of the Wnt signalling pathway. J Cell Sci 116:2627–2634
Hoeppner LH, Secreto FJ, Westendorf JJ (2009) Wnt signaling as a therapeutic target for bone diseases. Expert Opin Ther Targets 13:485–496
Zhu M, Chen M, Zuscik M, Wu Q, Wang YJ, Rosier RN, O’Keefe RJ, Chen D (2008) Inhibition of beta-catenin signaling in articular chondrocytes results in articular cartilage destruction. Arthritis Rheum 58:2053–2064
Daniels DL, Weis WI (2002) ICAT inhibits beta-catenin binding to Tcf/Lef-family transcription factors and the general coactivator p300 using independent structural modules. Mol Cell 10:573–584
Olsen BR, Reginato AM, Wang W (2000) Bone development. Annu Rev Cell Dev Biol 16:191–220
DeLise AM, Fischer L, Tuan RS (2000) Cellular interactions and signaling in cartilage development. Osteoarthritis Cartilage 8:309–334
Lefebvre V, Behringer RR, de Crombrugghe B (2001) L-Sox5, Sox6 and Sox9 control essential steps of the chondrocyte differentiation pathway. Osteoarthritis Cartilage 9(suppl A):S69–S75
Ng LJ, Wheatley S, Muscat GE, Conway-Campbell J, Bowles J, Wright E, Bell DM, Tam PP, Cheah KS, Koopman P (1997) SOX9 binds DNA, activates transcription, and coexpresses with type II collagen during chondrogenesis in the mouse. Dev Biol 183:108–121
Zelzer E, Olsen BR (2003) The genetic basis for skeletal diseases. Nature 423:343–348
Cohen MM Jr (2006) The new bone biology: pathologic, molecular, and clinical correlates. Am J Med Genet A 140:2646–2706
Karsenty G (1998) Genetics of skeletogenesis. Dev Genet 22:301–313
Karsenty G, Wagner EF (2002) Reaching a genetic and molecular understanding of skeletal development. Dev Cell 2:389–406
Hall BK, Miyake T (2000) All for one and one for all: condensations and the initiation of skeletal development. Bioessays 22:138–147
Day TF, Guo X, Garrett-Beal L, Yang Y (2005) Wnt/beta-catenin signaling in mesenchymal progenitors controls osteoblast and chondrocyte differentiation during vertebrate skeletogenesis. Dev Cell 8:739–750
Hill TP, Spater D, Taketo MM, Birchmeier W, Hartmann C (2005) Canonical Wnt/beta-catenin signaling prevents osteoblasts from differentiating into chondrocytes. Dev Cell 8:727–738
Guo X, Day TF, Jiang X, Garrett-Beal L, Topol L, Yang Y (2004) Wnt/beta-catenin signaling is sufficient and necessary for synovial joint formation. Genes Dev 18:2404–2417
Hens JR, Wilson KM, Dann P, Chen X, Horowitz MC, Wysolmerski JJ (2005) TOPGAL mice show that the canonical Wnt signaling pathway is active during bone development and growth and is activated by mechanical loading in vitro. J Bone Miner Res 20:1103–1113
Hill TP, Taketo MM, Birchmeier W, Hartmann C (2006) Multiple roles of mesenchymal beta-catenin during murine limb patterning. Development 133:1219–1229
Hu H, Hilton MJ, Tu X, Yu K, Ornitz DM, Long F (2005) Sequential roles of Hedgehog and Wnt signaling in osteoblast development. Development 132:49–60
Parr BA, McMahon AP (1995) Dorsalizing signal Wnt-7a required for normal polarity of D-V and A-P axes of mouse limb. Nature 374:350–353
Zhu M, Tang D, Wu Q, Hao S, Chen M, Xie C, Rosier RN, O’Keefe RJ, Zuscik M, Chen D (2009) Activation of beta-catenin signaling in articular chondrocytes leads to osteoarthritis-like phenotype in adult beta-catenin conditional activation mice. J Bone Miner Res 24:12–21
Yuasa T, Kondo N, Yasuhara R, Shimono K, Mackem S, Pacifici M, Iwamoto M, Enomoto-Iwamoto M (2009) Transient activation of Wnt/β-catenin signaling induces abnormal growth plate closure and articular cartilage thickening in postnatal mice. Am J Pathol 175:1993–2003
Rudnicki JA, Brown AM (1997) Inhibition of chondrogenesis by Wnt gene expression in vivo and in vitro. Dev Biol 185:104–118
Parr BA, Shea MJ, Vassileva G, McMahon AP (1993) Mouse Wnt genes exhibit discrete domains of expression in the early embryonic CNS and limb buds. Development 119:247–261
Cho HH, Kim YJ, Kim SJ, Kim JH, Bae YC, Ba B, Jung JS (2006) Endogenous Wnt signaling promotes proliferation and suppresses osteogenic differentiation in human adipose derived stromal cells. Tissue Eng 12:111–121
Boland GM, Perkins G, Hall DJ, Tuan RS (2004) Wnt 3a promotes proliferation and suppresses osteogenic differentiation of adult human mesenchymal stem cells. J Cell Biochem 93:1210–1230
Hwang SG, Yu SS, Lee SW, Chun JS (2005) Wnt-3a regulates chondrocyte differentiation via c-Jun/AP-1 pathway. FEBS Lett 579:4837–4842
Church V, Nohno T, Linker C, Marcelle C, Francis-West P (2002) Wnt regulation of chondrocyte differentiation. J Cell Sci 115:4809–4818
Hartmann C (2002) Wnt-signaling and skeletogenesis. J Musculoskelet Neuronal Interact 2:274–276
Hartmann C, Tabin CJ (2000) Dual roles of Wnt signaling during chondrogenesis in the chicken limb. Development 127:3141–3159
Kawakami Y, Wada N, Nishimatsu SI, Ishikawa T, Noji S, Nohno T (1999) Involvement of Wnt-5a in chondrogenic pattern formation in the chick limb bud. Dev Growth Differ 41:29–40
Akita K, Francis-West P, Vargesson N (1996) The ectodermal control in chick limb development: Wnt-7a, Shh, Bmp-2 and Bmp-4 expression and the effect of FGF-4 on gene expression. Mech Dev 60:127–137
Stott NS, Jiang TX, Chuong CM (1999) Successive formative stages of precartilaginous mesenchymal condensations in vitro: modulation of cell adhesion by Wnt-7A and BMP-2. J Cell Physiol 180:314–324
Hartmann C, Tabin CJ (2001) Wnt-14 plays a pivotal role in inducing synovial joint formation in the developing appendicular skeleton. Cell 104:341–351
Hoang B, Moos M Jr, Vukicevic S, Luyten FP (1996) Primary structure and tissue distribution of FRZB, a novel protein related to Drosophila frizzled, suggest a role in skeletal morphogenesis. J Biol Chem 271:26131–26137
Enomoto-Iwamoto M, Kitagaki J, Koyama E, Tamamura Y, Wu C, Kanatani N, Koike T, Okada H, Komori T, Yoneda T, Church V, Francis-West PH, Kurisu K, Nohno T, Pacifici M, Iwamoto M (2002) The Wnt antagonist Frzb-1 regulates chondrocyte maturation and long bone development during limb skeletogenesis. Dev Biol 251:142–156
Gaur T, Rich L, Lengner CJ, Hussain S, Trevant B, Ayers D, Stein JL, Bodine PV, Komm BS, Stein GS, Lian JB (2006) Secreted frizzled related protein 1 regulates Wnt signaling for BMP2 induced chondrocyte differentiation. J Cell Physiol 208:87–96
Im GI, Quan Z (2010) The effects of Wnt inhibitors on the chondrogenesis of human mesenchymal stem cells. Tissue Eng Part A 16:2405–2413
Leijten JC, van Blitterwijk CA, Karperien M, Emons J, van Gool S, Wit JM, Sticht C, Decker E, Rappold G, Uitterlinden A, Rivadeneira F, van Meurs J, Hofman A, Scherjon S (2012) GREM1, FRZB and DKK1 are key regulators of human articular cartilage homeostasis. Arthritis Rheum 64:3302–3312
Surmann-Schmitt C, Widmann N, Dietz U, Saeger B, Eitzinger N, Nakamura Y, Rattel M, Latham R, Hartmann C, von der Mark H, Schett G, von der Mark K, Stock M (2009) Wif-1 is expressed at cartilage-mesenchyme interfaces and impedes Wnt3a-mediated inhibition of chondrogenesis. J Cell Sci 122:3627–3637
Goldring MB, Goldring SR (2007) Osteoarthritis. J Cell Physiol 213:626–634
Fitzgerald JB, Jin M, Dean D, Wood DJ, Zheng MH, Grodzinsky AJ (2004) Mechanical compression of cartilage explants induces multiple time-dependent gene expression patterns and involves intracellular calcium and cyclic AMP. J Biol Chem 279:19502–19511
Kurz B, Lemke AK, Fay J, Pufe T, Grodzinsky AJ, Schunke M (2005) Pathomechanisms of cartilage destruction by mechanical injury. Ann Anat 187:473–485
Loeser RF (2006) Molecular mechanisms of cartilage destruction: mechanics, inflammatory mediators, and aging collide. Arthritis Rheum 54:1357–1360
Sweeney SE, Firestein GS (2004) Rheumatoid arthritis: regulation of synovial inflammation. Int J Biochem Cell Biol 36:372–378
Goldring MB, Berenbaum F (2004) The regulation of chondrocyte function by proinflammatory mediators: prostaglandins and nitric oxide. Clin Orthop Relat Res (427 suppl):S37–S46
Goldring SR, Goldring MB (2004) The role of cytokines in cartilage matrix degeneration in osteoarthritis. Clin Orthop Relat Res (427 suppl):S27–S36
Tetlow LC, Adlam DJ, Woolley DE (2001) Matrix metalloproteinase and proinflammatory cytokine production by chondrocytes of human osteoarthritic cartilage: associations with degenerative changes. Arthritis Rheum 44:585–594
Hwang SG, Yu SS, Poo H, Chun JS (2005) c-Jun/activator protein-1 mediates interleukin-1beta-induced dedifferentiation but not cyclooxygenase-2 expression in articular chondrocytes. J Biol Chem 280:29780–29787
Kim SJ, Ju JW, Oh CD, Yoon YM, Song WK, Kim JH, Yoo YJ, Bang OS, Kang SS, Chun JS (2002) ERK-1/2 and p38 kinase oppositely regulate nitric oxide-induced apoptosis of chondrocytes in association with p53, caspase-3, and differentiation status. J Biol Chem 277:1332–1339
Cawston TEW, Wilson AJ (2006) Understanding the role of tissue degrading enzymes and their inhibitors in development and disease. Best Pract Res Clin Rheumatol 20:983–1002
Plaas A, Osborn B, Yoshihara Y, Bai Y, Bloom T, Nelson F, Mikecz K, Sandy JD (2007) Aggrecanolysis in human osteoarthritis: confocal localization and biochemical characterization of ADAMTS5-hyaluronan complexes in articular cartilages. Osteoarthritis Cartilage 15:719–734
Burrage PS, Mix KS, Brinckerhoff CE (2006) Matrix metalloproteinases: role in arthritis. Front Biosci 11:529–543
Caterson B, Flannery CR, Hughes CE, Little CB (2000) Mechanisms involved in cartilage proteoglycan catabolism. Matrix Biol 19:333–344
Aigner T, Fundel K, Saas J, Gebhard PM, Haag J, Weiss T, Zien A, Obermayr F, Zimmer R, Bartnik E (2006) Large-scale gene expression profiling reveals major pathogenetic pathways of cartilage degeneration in osteoarthritis. Arthritis Rheum 54:3533–3544
Fukui N, Zhu Y, Maloney WJ, Clohisy J, Sandell LJ (2003) Stimulation of BMP-2 expression by pro-inflammatory cytokines IL-1 and TNF-alpha in normal and osteoarthritic chondrocytes. J Bone Joint Surg Am 85-A(suppl 3):59–66
Hermansson M, Sawaji Y, Bolton M, Alexander S, Wallace A, Begum S, Wait R, Saklatvala J (2004) Proteomic analysis of articular cartilage shows increased type II collagen synthesis in osteoarthritis and expression of inhibin betaA (activin A), a regulatory molecule for chondrocytes. J Biol Chem 279:43514–43521
Ryu JH, Kim SJ, Kim SH, Oh CD, Hwang SG, Chun CH, Oh SH, Seong JK, Huh TL, Chun JS (2002) Regulation of the chondrocyte phenotype by beta-catenin. Development 129:5541–5550
Miclea RL, Karperien M, Bosch CA, van der Horst G, van der Valk MA, Kobayashi T, Kronenberg HM, Rawadi G, Akcakaya P, Lowik CW, Fodde R, Wit JM, Robanus-Maandag EC (2009) Adenomatous polyposis coli–mediated control of beta-catenin is essential for both chondrogenic and osteogenic differentiation of skeletal precursors. BMC Dev Biol 9:26
Hwang SG, Ryu JH, Kim IC, Jho EH, Jung HC, Kim K, Kim SJ, Chun JS (2004) Wnt-7a causes loss of differentiated phenotype and inhibits apoptosis of articular chondrocytes via different mechanisms. J Biol Chem 279:26597–26604
Ma B, van Blitterswijk CA, Karperien M (2012) A Wnt/beta-catenin negative feedback loop inhibits interleukin-1-induced matrix metalloproteinase expression in human articular chondrocytes. Arthritis Rheum 64:2589–2600
Ryu JH, Chun JS (2006) Opposing roles of WNT-5A and WNT-11 in interleukin-1beta regulation of type II collagen expression in articular chondrocytes. J Biol Chem 281:22039–22047
Valdes AM, Loughlin J, Oene MV, Chapman K, Surdulescu GL, Doherty M, Spector TD (2007) Sex and ethnic differences in the association of ASPN, CALM1, COL2A1, COMP, and FRZB with genetic susceptibility to osteoarthritis of the knee. Arthritis Rheum 56:137–146
Loughlin J, Dowling B, Chapman K, Marcelline L, Mustafa Z, Southam L, Ferreira A, Ciesielski C, Carson DA, Corr M (2004) Functional variants within the secreted frizzled-related protein 3 gene are associated with hip osteoarthritis in females. Proc Natl Acad Sci U S A 101:9757–9762
Lane NE, Lian K, Nevitt MC, Zmuda JM, Lui L, Li J, Wang J, Fontecha M, Umblas N, Rosenbach M, de Leon P, Corr M (2006) Frizzled-related protein variants are risk factors for hip osteoarthritis. Arthritis Rheum 54:1246–1254
Min JL, Meulenbelt I, Riyazi N, Kloppenburg M, Houwing-Duistermaat JJ, Seymour AB, Pols HA, van Duijn CM, Slagboom PE (2005) Association of the Frizzled-related protein gene with symptomatic osteoarthritis at multiple sites. Arthritis Rheum 52:1077–1080
Lane NE, Nevitt MC, Lui LY, de Leon P, Corr M (2007) Wnt signaling antagonists are potential prognostic biomarkers for the progression of radiographic hip osteoarthritis in elderly Caucasian women. Arthritis Rheum 56:3319–3325
Sen M, Lauterbach K, El-Gabalawy H, Firestein GS, Corr M, Carson DA (2000) Expression and function of wingless and frizzled homologs in rheumatoid arthritis. Proc Natl Acad Sci U S A 97:2791–2796
Nakamura Y, Nawata M, Wakitani S (2005) Expression profiles and functional analyses of Wnt-related genes in human joint disorders. Am J Pathol 167:97–105
Blom AB, Brockbank SM, van Lent PL, van Beuningen HM, Geurts J, Takahashi N, van der Kraan PM, van de Loo FA, Schreurs BW, Clements K, Newham P, van den Berg WB (2009) Involvement of the Wnt signaling pathway in experimental and human osteoarthritis: prominent role of Wnt-induced signaling protein 1. Arthritis Rheum 60:501–512
Dell’accio F, De Bari C, Eltawil NM, Vanhummelen P, Pitzalis C (2008) Identification of the molecular response of articular cartilage to injury, by microarray screening: Wnt-16 expression and signaling after injury and in osteoarthritis. Arthritis Rheum 58:1410–1421
Bodine PV, Zhao W, Kharode YP, Bex FJ, Lambert AJ, Goad MB, Gaur T, Stein GS, Lian JB, Komm BS (2004) The Wnt antagonist secreted frizzled-related protein-1 is a negative regulator of trabecular bone formation in adult mice. Mol Endocrinol 18:1222–1237
Lories RJ, Peeters J, Bakker A, Tylzanowski P, Derese I, Schrooten J, Thomas JT, Luyten FP (2007) Articular cartilage and biomechanical properties of the long bones in Frzb-knockout mice. Arthritis Rheum 56:4095–4103
Dell’Accio F, De Bari C, El Tawil NM, Barone F, Mitsiadis TA, O’Dowd J, Pitzalis C (2006) Activation of WNT and BMP signaling in adult human articular cartilage following mechanical injury. Arthritis Res Ther 8:R139
Akiyama H, Lyons JP, Mori-Akiyama Y, Yang X, Zhang R, Zhang Z, Deng JM, Taketo MM, Nakamura T, Behringer RR, McCrea PD, de Crombrugghe B (2004) Interactions between Sox9 and beta-catenin control chondrocyte differentiation. Genes Dev 18:1072–1087
Yuasa T, Otani T, Koike T, Iwamoto M, Enomoto-Iwamoto M (2008) Wnt/beta-catenin signaling stimulates matrix catabolic genes and activity in articular chondrocytes: its possible role in joint degeneration. Lab Invest 88:264–274
Yasuhara R, Yuasa T, Williams JA, Byers SW, Shah S, Pacifici M, Iwamoto M, Enomoto-Iwamoto M (2010) Wnt/beta-catenin and retinoic acid receptor signaling pathways interact to regulate chondrocyte function and matrix turnover. J Biol Chem 285:317–327
Yun K, Im SH (2007) Transcriptional regulation of MMP13 by Lef1 in chondrocytes. Biochem Biophys Res Commun 364:1009–1014
Sen M, Reifert J, Lauterbach K, Wolf V, Rubin JS, Corr M, Carson DA (2002) Regulation of fibronectin and metalloproteinase expression by Wnt signaling in rheumatoid arthritis synoviocytes. Arthritis Rheum 46:2867–2877
Ge X, Ma X, Meng J, Zhang C, Ma K, Zhou C (2009) Role of Wnt-5A in interleukin-1beta-induced matrix metalloproteinase expression in rabbit temporomandibular joint condylar chondrocytes. Arthritis Rheum 60:2714–2722
Pritchard S, Guilak F (2006) Effects of interleukin-1 on calcium signaling and the increase of filamentous actin in isolated and in situ articular chondrocytes. Arthritis Rheum 54:2164–2174
Shimazaki A, Wright MO, Elliot K, Salter DM, Millward-Sadler SJ (2006) Calcium/calmodulin-dependent protein kinase II in human articular chondrocytes. Biorheology 43:223–233
Acknowledgments
This research forms part of the Project P2.02 OA control of the research program of the BioMedical Materials institute, cofunded by the Dutch Ministry of Economic Affairs, Agriculture and Innovation. This study has been made possible by an ECTS Career Establishment Award to M.K.
Author information
Authors and Affiliations
Corresponding author
Additional information
The first three authors contributed equally, and all should be considered first author.
The authors report that they have no conflict of interest.
Rights and permissions
About this article
Cite this article
Ma, B., Landman, E.B.M., Miclea, R.L. et al. WNT Signaling and Cartilage: Of Mice and Men. Calcif Tissue Int 92, 399–411 (2013). https://doi.org/10.1007/s00223-012-9675-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00223-012-9675-5