
Abstract
As the cellular component of articular cartilage, chondrocytes are responsible for maintaining in a low-turnover state the unique composition and organization of the matrix that was determined during embryonic and postnatal development. In joint diseases, cartilage homeostasis is disrupted by mechanisms that are driven by combinations of biological mediators that vary according to the disease process, including contributions from other joint tissues. In osteoarthritis (OA), biomechanical stimuli predominate with up-regulation of both catabolic and anabolic cytokines and recapitulation of developmental phenotypes, whereas in rheumatoid arthritis (RA), inflammation and catabolism drive cartilage loss. In vitro studies in chondrocytes have elucidated signaling pathways and transcription factors that orchestrate specific functions that promote cartilage damage in both OA and RA. Thus, understanding how the adult articular chondrocyte functions within its unique environment will aid in the development of rational strategies to protect cartilage from damage resulting from joint disease. This review will cover current knowledge about the specific cellular and biochemical mechanisms that regulate cartilage homeostasis and pathology.
Similar content being viewed by others


Introduction
Adult articular cartilage is an avascular tissue composed of a specialized matrix of collagens, proteoglycans, and non-collagen proteins, in which chondrocytes constitute the unique cellular component. Although chondrocytes in this context do not normally divide, they are assumed to maintain the extracellular matrix (ECM) by low-turnover replacement of certain matrix proteins. During aging and joint disease, this equilibrium is disrupted and the rate of loss of collagens and proteoglycans from the matrix may exceed the rate of deposition of newly synthesized molecules. Originally considered an inert tissue, cartilage is now considered to respond to extrinsic factors that regulate gene expression and protein synthesis in chondrocytes. Numerous studies in vitro and in vivo during the last two decades have confirmed that articular chondrocytes are able to respond to mechanical injury, joint instability due to genetic factors, and biological stimuli such as cytokines and growth and differentiation factors that contribute to structural changes in the surrounding cartilage matrix [1]. Mechanical influences on chondrocyte function are considered to be important in the pathogenesis of osteoarthritis (OA), but chondrocyte responses to molecular signals may vary in different regions, including the calcified cartilage, and also occur at different stages over a long time course (Figure 1). In rheumatoid arthritis (RA), the inflamed synovium is the major source of cytokines and proteinases that mediate cartilage destruction in areas adjacent to the proliferating synovial pannus (Figure 2) [2]. However, the basic cellular mechanisms regulating chondrocyte responses are very different in OA and RA. Moreover, mechanistic insights from in vitro studies ideally should be interpreted in light of direct analysis of human cartilage and other joint tissues and studies in experimental models, including knockout and transgenic mice [3, 4]. The examination of cartilage or chondrocytes from patients undergoing joint replacement has yielded less information in RA patients, in which cartilage damage is extensive, than studies of OA patients. In both, the findings do not reflect early disease. This review will cover current knowledge about the cellular and biochemical mechanisms of cartilage in health and disease derived from studies over the past 10 years.

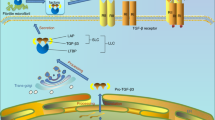
Cellular interactions in cartilage destruction in osteoarthritis. This scheme represents the destruction of the cartilage due to mechanical loading and biological factors. The induction of stress-induced intracellular signals, catabolic cytokines, including interleukin-1 (IL-1) and tumor necrosis factor-alpha (TNF-α), chemokines, and other inflammatory mediators produced by synovial cells and chondrocytes results in the upregulation of cartilage-degrading enzymes of the matrix metalloproteinase (MMP) and ADAMTS families. Matrix degradation products can feedback regulate these cellular events. Anabolic factors, including bone morphogenetic proteins (BMPs) and transforming growth factor-beta (TGF-β), may also be upregulated and participate in osteophyte formation. In addition to matrix loss, evidence of earlier changes, such as chondrocyte proliferation and hypertrophy, increased cartilage calcification with tidemark advancement, and microfractures with angiogenesis from the subchondral bone possibly mediated by vascular endothelial growth factor (VEGF) can be observed in late osteoarthritis samples obtained from patients after total joint replacement. ADAMTS, a disintegrin and metalloproteinase with thrombospondin-1 domains; C/EBP, CCAAT enhancer-binding protein; ESE1, epithelial-specific ETS; ETS, E26 transformation specific; GADD45β, growth arrest and DNA damage 45 beta; HIF-1α, hypoxia-inducible factor-1-alpha; NF-κB, nuclear factor-kappa-B; PA, plasminogen activator; TIMPs, tissue inhibitors of metalloproteinases.

Cellular interactions in cartilage destruction in rheumatoid arthritis. This scheme represents the progressive destruction of the cartilage associated with the invading synovial pannus in rheumatoid arthritis. As a result of immune cell interactions involving T and B lymphocytes, monocytes/macrophages, and dendritic cells, a number of different cytokines are produced in the synovium due to the influx of inflammatory cells from the circulation and synovial cell hyperplasia. The induction of proinflammatory cytokines produced primarily in the synovium, but also by chondrocytes, results in the upregulation of cartilage-degrading enzymes at the cartilage-pannus junction. Chemokines, nitric oxide (NO), and prostaglandins (PGE2) also contribute to the inflammation and tissue catabolism. ADAMTS, a disintegrin and metalloproteinase with thrombospondin-1 domains; IFN-γ, interferon-gamma; IL, interleukin; MMP, matrix metalloproteinase; SDF-1, stromal derived factor 1; TGF-β, transforming growth factor-beta; TNF-α, tumor necrosis factor-alpha; Treg, regulatory T (cell).
Cartilage in health
Cartilage matrix in healthy articular cartilage
Articular cartilage is composed of four distinct regions: (a) the superficial tangential (or gliding) zone, composed of thin collagen fibrils in tangential array and associated with a high concentration of decorin and a low concentration of aggrecan, (b) the middle (or transitional) zone with radial bundles of thicker collagen fibrils, (c) the deep (or radial) zone, in which the collagen bundles are thickest and are arranged in a radial fashion, and (d) the calcified cartilage zone, located immediately below the tidemark and above the subchondral bone [5, 6]. The calcified zone persists after growth plate closure as the 'tidemark' and serves as an important mechanical buffer between the uncalcified articular cartilage and the subchondral bone. From the superficial to the deep zone, cell density progressively decreases, whereas cell volume and the proportion of proteoglycan relative to collagen increase.
The interterritorial cartilage matrix, which is composed of a fibrillar collagen network that bestows tensile strength, differs from the territorial matrix closer to the cell, which contains type VI collagen microfibrils but little or no fibrillar collagen. The interterritorial collagen network consists primarily of type II collagen fibrils with type XI collagen within the fibril and type IX collagen integrated in the fibril surface with the non-collagen domain projecting outward, permitting association with other matrix components and retention of proteoglycans [7]. Collagen XXVII, a novel member of the fibrillar collagen family, also contributes to the formation of a stable cartilage matrix [8].
Compressive resistance is bestowed by the large aggregating proteoglycan aggrecan, which is attached to hyaluronic acid polymers via link protein. The half-life of aggrecan core protein ranges from 3 to 24 years, and the glycosaminoglycan components of aggrecan are synthesized more readily under low-turnover conditions, with more rapid matrix turnover in the pericellular regions. The proteoglycans are essential for protecting the collagen network, which has a half-life of more than 100 years if not subjected to inappropriate degradation. A large number of other noncollagen molecules, including biglycan, decorin, fibromodulin, the matrilins, and cartilage oligomeric matrix protein (COMP), are also present in the matrix. COMP acts as a catalyst in collagen fibrillogenesis [9], and interactions between type IX collagen and COMP or matrilin-3 are essential for proper formation and maintenance of the articular cartilage matrix [10, 11]. Perlecan enhances fibril formation [12], and collagen VI microfibrils connect to collagen II and aggrecan via complexes of matrilin-1 and biglycan or decorin [13].
Chondrocyte physiology and function in healthy articular cartilage
Differences in the morphologies of zonal subpopulations of chondrocytes may reflect matrix composition and are ascribed largely to differences in the mechanical environment [14]. The superficial zone chondrocytes (SZCs) are small and flattened. The middle zone chondrocytes (MZCs) are rounded, and the deep zone chondrocytes (DZCs) are grouped in columns or clusters. In vitro studies with isolated SZCs and DZCs indicate that differences in the expression of molecules, such as lubricin (also known as superficial zone protein or proteoglycan-4) and PTHrP by SZCs and Indian hedgehog (Ihh) and Runx2 by DZCs, may determine the zonal differences in matrix composition and function [15–17].
How chondrocytes maintain their ECM under homeostatic conditions has remained somewhat of a mystery since they do not divide and the matrix isolates them from each other, but gene expression and protein synthesis may be activated by injury. Since the ECM normally shields chondrocytes, they lack access to the vascular system and must rely on facilitated glucose transport via constitutive glucose transporter proteins, GLUT3 and GLUT8 [18], and active membrane transport systems [19]. Chondrocytes exist at low oxygen tension within the cartilage matrix, ranging from 10% at the surface to less than 1% in the deep zones. In vitro, chondrocytes adapt to low oxygen tensions by upregulating hypoxia-inducible factor-1-alpha (HIF-1α), which can stimulate expression of GLUTs [18], and angiogenic factors such as vascular endothelial growth factor (VEGF) [20, 21] as well as a number of genes associated with cartilage anabolism and chondrocyte differentiation [22]. One of our laboratories has identified growth arrest and DNA damage 45 beta (GADD45β), which previously was implicated as an anti-apoptotic factor during genotoxic stress and cell cycle arrest in other cell types as a survival factor in healthy articular chondrocytes [23]. Thus, by modulating the intracellular expression of survival factors, including HIF-1α and GADD45β, chondrocytes survive efficiently in the avascular cartilage matrix and respond to environmental changes.
The aging process may affect the material properties of healthy cartilage by altering the content, composition, and structural organization of collagen and proteoglycan [24–26]. This has been attributed to overall decreased anabolism and to the accumulation of advanced glycation end products (AGEs) that enhance collagen cross-linking [27]. Unless perturbed, healthy chondrocytes remain in a postmitotic quiescent state throughout life, with their decreasing proliferative potential being attributed to replicative senescence associated with erosion of telomere length [28]. The accumulation of cartilage matrix proteins in the endoplasmic reticulum and Golgi of chondrocytes, which have been modified by oxidative stress during aging, may lead to decreased synthesis of cartilage matrix proteins and diminished cell survival [29].
Cartilage in joint disease
The loss of balance between cartilage anabolism and catabolism
Although the etiologies of OA and RA are different, both diseases present states of inappropriate articular cartilage destruction, which is largely the result of elevated expression and activities of proteolytic enzymes. Whereas these enzymes normally are involved in the formation, remodeling, and repair of connective tissues, a shift in equilibrium between anabolic and catabolic activities occurs in OA as a response to abnormal mechanical loading in conjunction with genetic abnormalities or injury to the cartilage and surrounding joint tissues. In RA, the inflamed synovium is the major source of cytokine-induced proteinases, although the episodic intra-articular inflammation with synovitis indicates that the synovium may also be a source of cytokines and cartilage-degrading proteinases in OA [30, 31]. However, in OA, these degradative enzymes are produced primarily by chondrocytes due to inductive stimuli, including mechanical stress, injury with attendant destabilization, oxidative stress, cell-matrix interactions, and changes in growth factor responses and matrix during aging.
Of the proteinases that degrade cartilage collagens and proteoglycans in joint disease, matrix metalloproteinases (MMPs) and aggrecanases have been given the greatest attention because they degrade native collagens and proteoglycans [32–34]. These include the collagenases (MMP-1, MMP-8, and MMP-13), the gelatinases (MMP-2 and MMP-9), stromelysin-1 (MMP-3), and membrane type I (MT1) MMP (MMP-14) [35]. MMP-10, similar to MMP-3, activates pro-collagenases, is detectable in OA and RA synovial fluids and joint tissues, and is produced in vitro by both the synovium and chondrocytes in response to inflammatory cytokines [36]. MMP-14, produced principally by RA synovial tissue, is important for synovial invasiveness [37], whereas the MMP-14 produced by OA chondrocytes activates pro-MMP-13, which in turn cleaves pro-MMP-9 [38]. Other MMPs, including MMP-16 and MMP-28 [32, 39], and many members of the reprolysin-related proteinases of the ADAM (a disintegrin and metalloproteinase) family, including ADAM-17/TACE (tumor necrosis factor-alpha [TNF-α]-converting enzyme), are expressed in cartilage, but their specific roles in cartilage damage in either OA or RA have yet to be defined [40–42]. Although several of the MMPs, including MMP-3, MMP-8, and MMP-14, are capable of degrading proteoglycans, ADAMTS (ADAM with thrombospondin-1 domains)-4 and ADAMTS-5 are now regarded as the principal aggrecan-degrading enzymes in cartilage [43, 44]. Aggrecanase inhibitors that target ADAMTS-5 have been developed and are awaiting opportunities for clinical trials in OA [45].
OA and RA differ with respect to the sites as well as the origins of disrupted matrix homeostasis. In OA, proteoglycan loss and type II collagen cleavage initially occur at the cartilage surface, with evidence of pericellular damage in deeper zones as the lesion progresses [46]. In RA, intrinsic chondrocyte-derived chondrolytic activity is present at the cartilage-pannus junction, as well as in deeper zones of cartilage matrix [47], although elevated levels of MMPs in RA synovial fluids likely originate from the synovium. There are also differences in matrix synthetic responses in OA and RA. Whereas type II collagen synthesis is reduced in early RA [48], there is evidence of compensatory increases in type II collagen synthesis in deeper regions of OA cartilage [14].
This is in agreement with findings of enhanced global synthesis and gene expression of aggrecan and type II collagen in human OA compared with healthy cartilage [49–51]. Importantly, microarray studies using full-thickness cartilage have also shown that many collagen genes, including collagen, type II, alpha 1 (COL2A1), are upregulated in late-stage OA [23, 51]. The latter applies mainly to MZCs and DZCs, as revealed by laser capture microdissection, whereas this anabolic phenotype is less obvious in the degenerated areas of the upper regions [52].
Inflammation and cartilage destruction
In vivo and in vitro studies have shown that chondrocytes produce a number of inflammatory mediators, such as interleukin-1-beta (IL-1β) and TNF-α, which are present in RA or OA joint tissues and fluids. Chondrocytes respond to these proinflammatory cytokines by increasing the production of proteinases, prostaglandins, and nitric oxide (NO) [2, 25]. The first recognition of IL-1 as a regulator of chondrocyte function stems largely from work in in vitro culture models showing that activities derived from synovium or monocyte macrophages induce the production of cartilage-degrading proteinases (reviewed in [2, 53]).
IL-1, TNF-α, MMP-1, MMP-3, MMP-8, and MMP-13, and type II collagen cleavage epitopes have been shown to colocalize in matrix-depleted regions of RA cartilage [48, 54] and OA cartilage [46, 55]. In addition, chondrocytes express several chemokines as well as chemokine receptors that may participate in cartilage catabolism [56, 57]. IL-1β also induces other proinflammatory cytokines such as IL-17, which has similar effects on chondrocytes [58, 59]. IL-32, a recently discovered cytokine that induces TNF-α, IL-1β, IL-6, and chemokines, is also expressed in the synovia of RA patients and contributes to TNF-α-dependent inflammation and cartilage proteoglycan loss [60]. The importance of synergisms between IL-1 and TNF-α and with other cytokines, such as IL-17, IL-6, and oncostatin M, in RA or OA joints has been inferred primarily from culture models [61–63]. The up-regulation of cyclooxygenase-2 (COX-2), MMP13, and NOS2 gene expression by IL-1β in chondrocytes and other cell types is mediated by the induction and activation of a number of transcription factors, including nuclear factor-kappa-B (NF-κB), CCAAT enhancer-binding protein (C/EBP), activator protein 1 (AP-1), and E26 transformation specific family members, which regulate stress- and inflammation-induced signaling [64]. IL-1β also uses these mechanisms to suppress the expression of a number of genes associated with the differentiated chondrocyte phenotype, including COL2A1 and cartilage-derived retinoic acid-sensitive protein/melanoma inhibitory activity (CD-RAP/MIA) [64–66]. The role of epigenetics in regulating these cellular events in cartilage is under current consideration [67].
The IL-1R/Toll-like receptor (TLR) superfamily of receptors, which has a key role in innate immunity and inflammation, has received recent attention with respect to cartilage pathology. Human articular chondrocytes can express TLR1, TLR2, and TLR4, and the activation of TLR2 by IL-1, TNF-α, peptidoglycans, lipopolysaccharide, or fibronectin fragments increases the production of MMPs, NO, prostaglandin E (PGE), and VEGF [68–73]. In immune complex-mediated arthritis, TLR4 regulates early-onset inflammation and cartilage destruction by IL-10-mediated upregulation of Fcγ receptor expression and enhanced cytokine production [74]. The IL-18 receptor shares homology with IL-1RI and has a TLR signaling domain. IL-18 has effects similar to IL-1 in human chondrocytes and stimulates chondrocyte apoptosis, although studies do not suggest a pivotal role in cartilage destruction in RA [75, 76]. IL-33, an ST2-TLR ligand, is associated with endothelial cells in RA synovium, but its role in cartilage destruction has not been examined [77]. Of recent interest are the suppressor of cytokine signaling (SOCS) molecules, including SOCS3, which is induced by IL-1 and acts as a negative feedback regulator during insulin-like growth factor 1 (IGF-1) desensitization in the absence of NO by inhibiting insulin receptor substrate 1 (IRS-1) phosphorylation [78].
The increased production of prostaglandins by inflammatory cytokines is mediated via induction of the expression of not only COX-2 but also microsomal PGE synthase 1 (mPGES-1) [79, 80]. In addition to opposing the induction of COX-2, inducible nitric oxide synthetase (iNOS), and MMPs and the suppression of aggrecan synthesis by IL-1, activators of the peroxisome proliferator-activated receptor gamma (PPARγ), including the endogenous ligand 15-deoxy-Δ12,14 prosta-glandin J2 (PGJ2), inhibit IL-1-induced expression of mPGES-1 [81, 82]. Recent evidence indicates that PPARα agonists may protect chondrocytes against IL-1-induced responses by increasing the expression of IL-1Ra [83].
White adipose tissue has been proposed as a major source of both pro- and anti-inflammatory cytokines, including IL-1Ra and IL-10 [84]. Roles for adipokines, identified originally as products of adipocytes, have received recent attention, not only because of their relationship to obesity, but also because they can have pro- or anti-inflammatory effects in joint tissues and may serve as a link between the neuroendocrine and immune systems [85]. Leptin expression is enhanced during acute inflammation, correlating negatively with inflammatory markers in RA sera [86]. The expression of leptin is elevated in OA cartilage and in osteophytes and it stimulates IGF-1 and transforming growth factor-beta-1 (TGF-β1) synthesis in chondrocytes [87]. Leptin synergizes with IL-1 or interferon-gamma to increase NO production in chondrocytes [88], and leptin deficiency attenuates inflammatory processes in experimental arthritis [89]. It has been proposed that the dys-regulated balance between leptin and other adipokines, such as adiponectin, promotes destructive inflammatory processes [90]. Recent studies indicate that resistin plays a role in early stages of trauma-induced OA and in RA at local sites of inflammation and that serum resistin reflects inflammation and disease activity [91, 92].
Effects of mechanical loading
In young individuals without genetic abnormalities, biomechanical factors due to trauma are strongly implicated in initiating the OA lesion. Mechanical disruption of cell-matrix interactions may lead to aberrant chondrocyte behavior, contributing to fibrillations, cell clusters, and changes in quantity, distribution, or composition of matrix proteins [93, 94]. In the early stages of OA, transient increases in chondrocyte proliferation and increased metabolic activity are associated with a localized loss of proteoglycans at the cartilage surface followed by cleavage of type II collagen (reviewed in [95, 96]). These events result in increased water content and decreased tensile strength of the matrix as the lesion progresses.
Chondrocytes can respond to direct biomechanical perturbation by upregulating synthetic activity or by increasing the production of inflammatory cytokines, which are also produced by other joint tissues. In vitro mechanical loading experiments have revealed that injurious static compression stimulates proteoglycan loss, damages the collagen network, and reduces synthesis of cartilage matrix proteins, whereas dynamic compression increases matrix synthetic activity [97]. In response to traumatic injury, global gene expression is activated, resulting in increased expression of inflammatory mediators, cartilage-degrading proteinases, and stress response factors [98, 99]. Neuronal signaling molecules, such as substance P and its receptor, NK1, and N-methyl-D-aspartic acid receptors (NMDARs), which require glutamate and glycine binding for activation, have been implicated in mechanotransduction in chondrocytes in a recent study [100].
Chondrocytes have receptors for responding to mechanical stimulation, many of which are also receptors for ECM components [101]. Among these are several of the integrins that serve as receptors for fibronectin and type II collagen fragments, which upon activation stimulate the production of proteinases, cytokines, and chemokines [102]. Discoidin domain receptor 2 (DDR-2), a receptor for native type II collagen fibrils, is activated on chondrocytes via Ras/Raf/Mek signaling and preferentially induces MMP-13 via p38 mitogen-activated protein kinase (MAPK); this is a universal mechanism that occurs after loss of proteoglycans, not only in genetic models, but also in surgical mouse OA and human OA [103]. On the other hand, in RA the cell-cell adhesion molecule, cadherin-11, is expressed at the interface between the RA synovial pannus and cartilage and facilitates cartilage invasion and erosion in mouse models in vivo and in human RA tissues in vitro and ex vivo [104] in a TNF-α-dependent manner [105]. Recent studies indicate that lubricin is an important secreted product of chondrocytes, synovial cells, and other joint tissues which is downregulated in OA and RA and modulated by cytokines and growth factors [91, 92].
Stress responses in cartilage
Injurious mechanical stress and cartilage matrix degradation products are capable of stimulating the same signaling pathways as those induced by inflammatory cytokines [98, 106–109]. Along with extracellular signal-regulated kinase 1/2 (ERK1/2), the key protein kinases in the c-jun N-terminal kinase (JNK), p38 MAPK, and NF-κB signaling cascades are activated, particularly in the upper zones of OA cartilage [110]. Furthermore, the engagement of integrin receptors by fibronectin or collagen fragments activates focal adhesion kinase signaling and transmits signals intersecting with ERK, JNK, and p38 pathways [111, 112]. Cascades of multiple protein kinases are involved in these responses, including protein kinase Cζ, which is upregulated in OA cartilage and is required for activation of NF-κB by IL-1 and TNF-α [113]. However, it remains controversial whether inflammatory cytokines are primary or secondary effectors of cartilage damage and defective repair mechanisms in OA since these same pathways also induce or amplify the expression of cytokine genes. Interestingly, physiological loading may protect against cartilage loss by inhibiting IκB kinase-beta (IKKβ) activity in the canonical NF-κB cascade and attenuating NF-κB transcriptional activity [114] as well as by inhibiting TAK1 (TGF-β-activated kinase 1) phosphorylation [115]. In addition, genetic factors that cause disruption of chondrocyte differentiation and function and influence the composition and structure of the cartilage matrix may contribute to abnormal biomechanics, independently of the influence of inflammation.
Reactive oxygen species (ROS) play a critical role in chondrocyte homeostasis, but during aging, trauma, and OA, partial oxygen variations and mechanical stress as well as inflammation induce abnormal ROS production, which exceeds the antioxidant capacity leading to oxidative stress. ROS and attendant oxidative stress impair growth factor responses, enhance senescence through telomere shortening, and impair mitochondrial function [28, 116, 117]. ROS levels are also induced by activation of RAGE, the receptor for AGEs, which regulates chondrocyte and synovial responses in OA [118]. In chondrocytes, interaction of RAGE with S100A4, a member of the S100 family of calcium-binding proteins, stimulates MMP-13 production via phosphorylation of Pyk2, MAPKs, and NF-κB signaling [119]. RAGE expression and S100A1 release are stimulated in chondrocytes in vitro and increased in OA cartilage. Transglutaminase 1, which is induced by inflammation and stress, transforms S100A1 into a procatabolic cytokine that signals through RAGE and the p38 MAPK pathway to induce chondrocyte hypertrophy and aggrecan degradation [120]. In experimental murine arthritis models, S100A8 and S100A9 are involved in the upregulation and activation of MMPs and aggrecanases [121, 122]. In addition, high-mobility group protein 1 (HMGB1), another important RAGE ligand and also a chromatin architectural protein, is produced by inflamed synovium and thus acts as a RAGE-dependent proinflammatory cytokine in RA [123]. The differential regulation and expression of GLUT isoforms by hypoxia, growth factors, and inflammatory cytokines may contribute to intracellular stress responses [124]. COX-2 is also involved in the chondrocyte response to high shear stress, associated with reduced antioxidant capacity and increased apoptosis [125]. Modulation of such intracellular stress response mechanisms may provide strategies for novel therapies.
Biomarkers of cartilage pathology
The recent development of assays for specific biological markers, which reflect quantitative and dynamic changes in the synthetic and degradation products of cartilage and bone matrix components, has provided a means of identifying patients at risk for rapid joint damage and also for early monitoring of the efficacy of disease-modifying therapies. Molecules originating from the articular cartilage, including aggrecan fragments, which contain chondroitin sulfate and keratan sulfate, type II collagen fragments, and collagen pyridinoline cross-links, are usually released as degradation products as a result of catabolic processes. Specific antibodies that detect either synthetic or cleavage epitopes have been developed to study biological markers of cartilage metabolism in synovial fluids, sera, and urine of patients with OA or RA (reviewed in [126–129]). Aggrecan degradation products are assayed using antibodies 846, 3B3(-), and 7D4 that detect chondroitin sulfate neoepitopes, 5D4 that detects keratan sulfate epitopes, and the VIDIPEN and NITEGE antibodies that recognize aggrecanase and MMP cleavage sites, respectively, within the interglobular G1 domain of aggrecan [33]. Similarly, the C2C antibody (previously known as Col2-3/4CLong mono) has been used to detect specific cleavage of the triple helix of type II collagen [48, 129]. Increased ratios of C2C to the synthetic marker, CPII, are associated with a greater likelihood of radiological progression in OA patients [130]. Other markers included COMP [131]; YKL-40/HC-gp39, or chitinase 3-like protein 1 (CH3L1), which is induced in chondrocytes by inflammatory cytokines [132]; and CD-RAP, also known as MIA [133, 134]. Such biomarker assays have been used as research tools and are currently under evaluation for monitoring cartilage degradation or repair in patient populations. C-reactive protein, IL-6, and MMP-3 have also been identified as potential biomarkers in both RA and OA patient populations. A single marker has not proven to be sufficient, however, and the major challenge will be to apply such biomarkers to the diagnosis and monitoring of disease in individual patients and to correlate them with structural changes in cartilage identified by magnetic resonance imaging techniques [135].
The genetics of cartilage pathology
Results of epidemiological studies, analysis of patterns of familial clustering, twin studies, and the characterization of rare genetic disorders suggest that genetic abnormalities can result in early onset of OA and increased susceptibility to RA. For example, twin studies have shown that the influence of genetic factors may approach 70% in OA that affects certain joints. Candidate gene studies and genome-wide linkage analyses have revealed polymorphisms or mutations in genes encoding ECM and signaling molecules that may determine OA susceptibility [136–138]. Gender differences have been noted and gene defects may appear more prominently in different joints [136, 139]. Gene defects associated with congenital cartilage dysplasias that affect the formation of cartilage matrix and patterning of skeletal elements may adversely affect joint alignment and congruity and thus contribute to early onset of OA in these individuals [140]. Although whole-genome linkage analyses of RA patients have not addressed cartilage specifically, this work has pointed to immunological pathways and inflammatory signals that may modulate cartilage destruction [141].
Genomic and proteomic analyses, which have been performed in cytokine-treated chondrocytes, in cartilage from patients with OA, and in rheumatoid synovium, have provided some insights into novel mechanisms that might govern chondrocyte responses in both OA and RA [57, 63, 102, 142]. When coupled with biological analyses that address candidate genes, gene profiling studies of cartilage derived from patients with OA have also begun to yield new information about mediators and pathways [23, 51, 143, 144]. Similarly, microarray analysis of cocultures of synovial fibroblasts with chondrocytes in alginate has identified markers of inflammation and cartilage destruction associated with RA pathogenesis [145].
Lessons from mouse models
Insight into cartilage pathology in RA has been gleaned from the examination of type II collagen-induced arthritis and other types of inflammatory arthritis in mice with transgenic over-expression or knockout of genes encoding cytokines, their receptors, or activators. These studies have led in part to the conclusion that TNF-α drives acute inflammation whereas IL-1 has a pivotal role in sustaining cartilage erosion [146]. In support of this concept, crossing arthritic human TNF transgenic (hTNFtg) mice with IL-1α- and β-deficient strains protected against cartilage erosion without affecting synovial inflammation [147]. The success of anti-TNF-α therapy in most but not all patients highlights the importance of inflammation in joint destruction.
In vivo studies have also shown that alterations in cartilage matrix molecules or in regulators of chondrocyte differentiation can lead to OA pathology. The importance of the fine protein network and ECM structural integrity in postnatal cartilage health is well documented in studies of deficiencies or mutations in cartilage matrix genes, including Col2a1, Col9a1, Col11a1, aggrecan, matrilin-3, or fibromodulin alone or together with biglycan, which lead to age-dependent cartilage degeneration similar to that in OA patients [140, 148, 149]. Deficiency of Timp3 (tissue inhibitor of metalloproteinases 3) or postnatal overexpression of constitutively active Mmp13 also promotes OA-like pathology [150, 151].
Importantly, surgically induced OA disease models in mutant mice have also implicated ADAMTS5 [152, 153], DDR-2 [103], and Runx2 [154] as contributors to the onset and/or severity of OA joint disease. Knockout of IL-1β is also protective against OA induced by destabilization of the medial meniscus [155]. Although single gene defects do not model all aspects of human OA, the loss or mutation of a gene that is involved in the synthesis or remodeling of the cartilage matrix may lead to the disruption of other gene functions in chondrocytes, thus resulting in joint instability and OA-like pathology. Thus, novel mechanistic insights into the initiation or progression of OA may be discovered by identifying intracellular effectors of ECM homeostasis and remodelling in vitro and evaluating their functions in animal models of OA disease.
Chondrogenesis, chondrocyte hypertrophy, calcified cartilage, and bone in cartilage pathology
During skeletal development, the chondrocytes arise from mesenchymal progenitors to synthesize the templates, or cartilage anlagen, for the developing limbs in a process known as chondrogenesis [156]. Following mesenchymal condensation and chondroprogenitor cell differentiation, chondrocytes undergo proliferation, terminal differentiation to hypertrophy, and apoptosis, whereby hypertrophic cartilage is replaced by bone in endochondral ossification. A number of signaling pathways and transcription factors play stage-specific roles in chondrogenesis and a similar sequence of events occurs in the postnatal growth plate, leading to rapid growth of the skeleton [64, 156–158].
Chondrogenesis is orchestrated in part by Sox9 and Runx2, two pivotal transcriptional regulators that determine the fate of chondrocytes to remain within cartilage or undergo hypertrophic maturation prior to ossification and is also subject to complex regulation by interplay of the fibroblast growth factor, TGF-β, BMP, and Wnt signaling pathways [159–162]. Differential signaling during chondrocyte maturation occurs via TGF-β-regulated signal-transducing mothers against decapentaplegic (Smads) 2 and 3 that act to maintain articular chondrocytes in an arrested state and BMP-regulated Smads 1 and 5 that accelerate their differentiation. Sox9, which is essential for type II collagen (COL2A1) gene expression, is most highly expressed in proliferating chondrocytes and has opposing positive and negative effects on the early and late stages of chondrogenesis, respectively. Sox9 cooperates with two related proteins, L-Sox5 and Sox6, which are targets of Sox9 itself and function as architectural HMG-like chromatin modifiers. Moreover, BMP signaling, through the type I Bmpr1a and Bmpr1b receptors, redundantly drives chondrogenesis via Sox9, Sox5, and Sox6. In addition, Runx2, which drives the terminal phase of chondrogenesis [163], is subject to direct inhibition by Sox9 [164]. In cooperation with BMP-induced Smads, Runx2 also upregulates GADD45β, a positive regulator of the terminal hypertrophic phase of chondrogenesis which drives the expression of Mmp13 and Col10a1 in the mouse embryonic growth plate [165]. More recently, the findings of our groups suggest that GADD45β contributes to the homeostasis of healthy and early OA articular chondrocytes as an effector of cell survival and as one of the factors induced by NF-κB that contributes to the imbalance in matrix remodelling in OA cartilage by suppressing COL2A1 gene expression [23] and that the NF-κB activating kinases, IKKα and IKKβ, differentially contribute to OA pathology by also regulating matrix remodelling in conjunction with chondrocyte differentiation [166].
Endochondral ossification, in which the hypertrophic chondrocyte undergoes a stress response associated with ECM remodelling, has been proposed as a 'developmental model' to understand the contribution of exacerbated environmental stresses to OA pathology [167–170]. Changes in the mineral content and thickness of the calcified cartilage and the associated tidemark advancement may be related to recapitulation of the hypertrophic phenotype, including COL10A1, MMP-13, and Runx2 gene expression, observed in the deep zone of OA cartilage [167, 171]. In addition to COL10A1 and MMP-13, other chondrocyte terminal differentiation-related genes, such as MMP-9 and Ihh, are detected in the vicinity of early OA lesions along with decreased levels of Sox9 mRNA [172]. However, Sox9 expression does not always localize with COL2A1 mRNA in adult articular cartilage [52, 173]. Apoptosis is a rare event in OA cartilage but may be a consequence of the chondrocyte stress response associated with hypertrophy [174]. Interestingly, one of our recent studies indicates that intracellular stress response genes are upregulated in early OA, whereas a number of genes encoding cartilage-specific and nonspecific collagens and other matrix proteins are upregulated in late-stage OA cartilage [23]. Moreover, articular chondrocytes in micromass culture show 'phenotypic plasticity' comparable to mesenchymal stem cells (MSCs) undergoing chondrogenesis, by recapitulating processes akin to chondrocyte hypertrophy [175], which one of our labs recently has shown to be subject to differential control by canonical NF-κB signaling and IKKα [166]. This process may also be modulated by Src kinases [176, 177].
Additional supporting evidence for dysregulation of endochondral ossification as a factor in OA pathology comes from genetic association studies identifying OA susceptibility genes across different populations [138, 170, 178]. These include the genes encoding asporin (ASPN), a TGF-β-binding protein with biglycan and decorin sequence homology [179], secreted frizzled-related protein 3 (FRZB), a WNT/β-catenin signaling antagonist [180, 181], and deiodinase 2 (DIO2), an enzyme that converts inactive thyroid hormone, T4, to active T3 [182]. The activation of WNT/β-catenin in mature postnatal growth plate chondrocytes stimulates hypertrophy, matrix mineralization, and expression of VEGF, ADAMTS5, MMP-13, and several other MMPs [183]. Findings from microarray analyses of bone from OA patients [184] and in Frzb knockout mice [185] also suggest that signaling modifications in the calcified cartilage could contribute to increased subchondral plate thickness accompanying tidemark advancement at the border with the articular cartilage and the angiogenesis observed at the osteochondral junction [186]. Moreover, endochondral ossification also contributes to the formation of osteophytes [187–189]. Interestingly, HMGB1 released by hypertrophic cartilage, prior to the onset of programmed cell death, contributes to endochondral ossification by acting as a chemotactic factor for osteoclasts at the growth plate [190], and HMGB1-induced NF-κB signaling is also required for cellular chemotaxis in response to HMGB1-RAGE engagement [191]. Thus, IKK-mediated NF-κB signaling not only may intrinsically influence the differentiation of chondrocytes toward a hypertrophy-like state [166], but also could subsequently drive aspects of intercellular communication culminating in endochondral ossification [190].
Changes in the periarticular and subchondral bone also occur in both RA and OA and may contribute to cartilage pathology. Receptor activator of NFκB (RANK), a member of the TNF receptor family, RANK ligand (RANKL), and the soluble receptor osteoprotegerin regulate osteoclast differentiation and activity and are important mediators of bone destruction in RA. IKKβ-mediated, but not IKKα-mediated, NF-κB signaling is associated with inflammation-induced bone loss [192] and is also critical for the survival of osteoclast precursors by suppressing JNK-dependent apoptosis in response to RANKL signaling [193]. IL-17 induces RANKL, inducing bone destruction independently of IL-1 and bypassing the requirement for TNF in inflammatory arthritis [58]. Although RANK and RANKL are expressed in adult articular chondrocytes, a direct action in cartilage has not been identified [194]. Since cartilage destruction is not blocked directly by the inhibition of RANKL, at least in inflammatory models, indirect effects may occur through protection of the bone [195, 196], as suggested by recent studies in experimental models [197, 198]. A link between RANKL and WNT has been suggested by findings in hTNFtg mice and RA tissues, in which decreased β-catenin and high DKK-1, a WNT inhibitor, were demonstrated in synovium and in cartilage adjacent to inflammatory tissue [199] (reviewed in [200]). In contrast, increased β-catenin was observed in OA cartilage and conditional overexpression in mouse cartilage leads to premature chondrocyte differentiation and development of OA-like phenotype [201]. Interestingly, Runx2-dependent expression of RANKL occurs in hypertrophic chondrocytes at the boundary next to the calcifying cartilage in the developing growth plate [202].
Mesenchymal progenitor cells in cartilage and their use in tissue engineering
MSCs from bone marrow and other adult tissues, including muscle, adipose tissue, and synovium or other tissue sites, which have the capacity to differentiate into cartilage, bone, fat, and muscle cells, are under investigation as sources of cartilage progenitor cells for cartilage tissue engineering [203–206]. Studies in vitro indicate that the same growth and differentiation factors that regulate different stages of cartilage development may be able to promote cartilage repair [207–209]. IGF-1 is a potent stimulator of proteoglycan synthesis, particularly when combined with other anabolic factors, including BMPs [210, 211]. Moreover, ex vivo gene transfer of anabolic factors such as BMPs, TGF-β, and IGF-1 has been explored as an approach to promote differentiation of autologous chondrocytes or MSCs before implantation [212, 213]. Recently, endochondral ossification has been achieved with murine embryonic stem cells in tissue-engineered constructs implanted in cranial bone of rats [214].
BMP-2 and BMP-7 (osteogenic protein 1) are currently approved for multiple indications in the area of bone fracture repair and spinal fusion, but the capacity of BMPs and TGF-β to induce chondrocyte hypertrophy in cartilage repair models and to promote osteophyte formation may prevent controlled repair of articular cartilage in vivo [207]. Since the injection of free TGF-β or adenovirus-mediated delivery of TGF-β promotes fibrosis and osteophyte formation, while stimulating proteoglycan synthesis in cartilage, the local application of molecules that block endogenous TGF-β signaling, such as the soluble form of TGF-βRII, inhibitory SMADs, or the physiological antagonist latency-associated peptide 1 (LAP-1), has been proposed as a more effective strategy [188]. Additional strategies include gene transfer of Sox9, alone or together with L-Sox5 and Sox6, into MSCs ex vivo or into joint tissues in vivo to more directly promote the expression of cartilage matrix genes [215, 216]. Strategies to stably express interfering RNAs in vivo could also provide a means of blocking dysregulated ECM remodelling or inappropriate endochondral ossification of articular chondrocytes.
Despite intensive investigation of cartilage repair strategies and the increased understanding of the cellular mechanisms involved, many issues remain to be resolved. These include the fabrication and maintenance of the repair tissue in the same zonal composition as the original cartilage, the recruitment and maintenance of cells with an appropriate chondrocyte phenotype, and integration of the repair construct with the surrounding cartilage matrix [217]. These issues are also compounded when cartilage loss is severe or when chronic inflammation exists, as in RA.
Conclusion
Laboratory investigations in vitro and in vivo regarding the role of the chondrocyte in remodeling the cartilage matrix in the RA and OA joint have identified novel molecules and mechanisms and provided new understanding of the contributions of known mediators. In RA, mediators involved in immunomodulation and synovial cell function, including cytokines, chemokines, and adhesion molecules, have primary roles in the inflammatory and catabolic processes in the joint, but they may also, directly or indirectly, promote cartilage damage. Despite our increasing knowledge of the mechanisms regulating the responses of chondrocytes to anabolic and catabolic factors involved in developing and adult cartilage, the development of disease-modifying therapies for OA patients has been elusive. In RA, in which significant advances have been achieved in our understanding of the cellular interactions in the RA joint involving macrophages, T and B lymphocytes, and synovial fibroblasts, there is still a need for therapeutic strategies that prevent the extensive cartilage and bone loss, despite the clinical success of anti-TNF therapy for RA. Further work using the principles of cell and molecular biology, such as those described in this review, will be necessary for uncovering new therapies for targeting cartilage destruction in both degenerative and inflammatory joint disease.
Note
The Scientific Basis of Rheumatology: A Decade of Progress
This article is part of a special collection of reviews, The Scientific Basis of Rheumatology: A Decade of Progress, published to mark Arthritis Research & Therapy's 10th anniversary.
Other articles in this series can be found at: http://arthritis-research.com/sbr
Abbreviations
- ADAM:
-
a disintegrin and metalloproteinase
- ADAMTS:
-
a disintegrin and metalloproteinase with thrombospondin-1 domains
- AGE:
-
advanced glycation end product
- CD-RAP:
-
cartilage-derived retinoic acid-sensitive protein
- COL2A1:
-
collagen, type II, alpha 1
- COMP:
-
cartilage oligomeric matrix protein
- COX-2:
-
cyclooxygenase 2
- DDR-2:
-
discoidin domain receptor 2
- DZC:
-
deep zone chondrocyte
- ECM:
-
extracellular matrix
- ERK:
-
extracellular signal-regulated kinase
- FRZB:
-
frizzled-related protein 3
- GADD45β:
-
growth arrest and DNA damage 45 beta
- GLUT:
-
glucose transporter protein
- HIF-1α:
-
hypoxia-inducible factor-1-alpha
- HMGB1:
-
high-mobility group protein 1
- hTNFtg:
-
human tumor necrosis factor transgenic
- IGF-1:
-
insulin-like growth factor 1
- Ihh:
-
Indian hedgehog
- IKK:
-
IκB kinase
- IL:
-
interleukin
- JNK:
-
c-jun N-terminal kinase
- MAPK:
-
mitogen-activated protein kinase
- MIA:
-
melanoma inhibitory activity
- MMP:
-
matrix metalloproteinase
- mPGES-1:
-
microsomal prostaglandin E synthase 1
- MSC:
-
mesenchymal stem cell
- MZC:
-
middle zone chondrocyte
- NF-κB:
-
nuclear factor-kappa-B
- NO:
-
nitric oxide
- OA:
-
osteoarthritis
- PGE:
-
prostaglandin E
- PPAR:
-
peroxisome proliferator-activated receptor
- RA:
-
rheumatoid arthritis
- RAGE:
-
receptor for advanced glycation end products
- RANK:
-
receptor activator of nuclear factor-kappa-B
- RANKL:
-
receptor activator of nuclear factor-kappa-B ligand
- ROS:
-
reactive oxygen species
- SMAD:
-
signal-transducing mothers against decapentaplegic
- SOCS:
-
suppressor of cytokine signaling
- SZC:
-
superficial zone chondrocyte
- TGF-β:
-
transforming growth factor-beta
- TLR:
-
Toll-like receptor
- TNF-α:
-
tumor necrosis factor-alpha
- VEGF:
-
vascular endothelial growth factor.
References
Goldring MB, Goldring SR: Osteoarthritis. J Cell Physiol. 2007, 213: 626-634.
Dayer JM: The process of identifying and understanding cytokines: from basic studies to treating rheumatic diseases. Best Pract Res Clin Rheumatol. 2004, 18: 31-45.
Loo van de FA, Geurts J, Berg van den WB: Gene therapy works in animal models of rheumatoid arthritis...so what!. Curr Rheumatol Rep. 2006, 8: 386-393.
Berg van den WB: Lessons from animal models of osteoarthritis. Curr Rheumatol Rep. 2008, 10: 26-29.
Poole AR: Cartilage in health and disease. Arthritis and Allied Conditions: A Textbook of Rheumatology. Edited by: Koopman WS. 2005, Philadelphia: Lippincott, Williams, and Wilkins, 223-269. 15
Goldring MB: Chapter 3: cartilage and chondrocytes. Kelley's Textbook of Rheumatology. Edited by: Firestein GS, Budd RC, McInnes IB, Sergent JS, Harris ED, Ruddy S. 2008, Philadelphia: WB Saunders, an imprint of Elsevier Inc, 37-69. 8
Eyre DR, Weis MA, Wu JJ: Articular cartilage collagen: an irreplaceable framework?. Eur Cell Mater. 2006, 12: 57-63.
Plumb DA, Dhir V, Mironov A, Ferrara L, Poulsom R, Kadler KE, Thornton DJ, Briggs MD, Boot-Handford RP: Collagen XXVII is developmentally regulated and forms thin fibrillar structures distinct from those of classical vertebrate fibrillar collagens. J Biol Chem. 2007, 282: 12791-12795.
Halasz K, Kassner A, Morgelin M, Heinegard D: COMP acts as a catalyst in collagen fibrillogenesis. J Biol Chem. 2007, 282: 31166-31173.
Leighton MP, Nundlall S, Starborg T, Meadows RS, Suleman F, Knowles L, Wagener R, Thornton DJ, Kadler KE, Boot-Handford RP, Briggs MD: Decreased chondrocyte proliferation and dys-regulated apoptosis in the cartilage growth plate are key features of a murine model of epiphyseal dysplasia caused by a matn3 mutation. Hum Mol Genet. 2007, 16: 1728-1741.
Pirog-Garcia KA, Meadows RS, Knowles L, Heinegard D, Thornton DJ, Kadler KE, Boot-Handford RP, Briggs MD: Reduced cell proliferation and increased apoptosis are significant pathological mechanisms in a murine model of mild pseudoachondroplasia resulting from a mutation in the C-terminal domain of COMP. Hum Mol Genet. 2007, 16: 2072-2088.
Kvist AJ, Johnson AE, Mörgelin M, Gustafsson E, Bengtsson E, Lindblom K, Aszódi A, Fässler R, Sasaki T, Timpl R, Aspberg A: Chondroitin sulfate perlecan enhances collagen fibril formation. Implications for perlecan chondrodysplasias. J Biol Chem. 2006, 281: 33127-33139.
Wiberg C, Klatt AR, Wagener R, Paulsson M, Bateman JF, Heinegard D, Morgelin M: Complexes of matrilin-1 and biglycan or decorin connect collagen VI microfibrils to both collagen II and aggrecan. J Biol Chem. 2003, 278: 37698-37704.
Poole AR, Guilak F, Abramson SB: Etiopathogenesis of osteoarthritis. Osteoarthritis: Diagnosis and Medical/Surgical Management. Edited by: Moskowitz RW, Altman RW, Hochberg MC, Buckwalter JA, Goldberg VM. 2007, Philadelphia: Lippincott, Williams, and Wilkins, 27-49. 4
Cheng C, Conte E, Pleshko-Camacho N, Hidaka C: Differences in matrix accumulation and hypertrophy in superficial and deep zone chondrocytes are controlled by bone morphogenetic protein. Matrix Biol. 2007, 26: 541-553.
Eleswarapu SV, Leipzig ND, Athanasiou KA: Gene expression of single articular chondrocytes. Cell Tissue Res. 2007, 327: 43-54.
Chen X, Macica CM, Nasiri A, Broadus AE: Regulation of articular chondrocyte proliferation and differentiation by indian hedgehog and parathyroid hormone-related protein in mice. Arthritis Rheum. 2008, 58: 3788-3797.
Mobasheri A, Richardson S, Mobasheri R, Shakibaei M, Hoyland JA: Hypoxia inducible factor-1 and facilitative glucose transporters GLUT1 and GLUT3: putative molecular components of the oxygen and glucose sensing apparatus in articular chondrocytes. Histol Histopathol. 2005, 20: 1327-1338.
Wilkins RJ, Browning JA, Ellory JC: Surviving in a matrix: membrane transport in articular chondrocytes. J Membr Biol. 2000, 177: 95-108.
Lin C, McGough R, Aswad B, Block JA, Terek R: Hypoxia induces HIF-1a and VEGF expression in chondrosarcoma cells and chondrocytes. J Orthop Res. 2004, 22: 1175-1181.
Pufe T, Lemke A, Kurz B, Petersen W, Tillmann B, Grodzinsky AJ, Mentlein R: Mechanical overload induces VEGF in cartilage discs via hypoxia-inducible factor. Am J Pathol. 2004, 164: 185-192.
Robins JC, Akeno N, Mukherjee A, Dalal RR, Aronow BJ, Koopman P, Clemens TL: Hypoxia induces chondrocyte-specific gene expression in mesenchymal cells in association with transcriptional activation of Sox9. Bone. 2005, 37: 313-322.
Ijiri K, Zerbini LF, Peng H, Otu HH, Tsuchimochi K, Otero M, Dragomir C, Walsh N, Bierbaum BE, Mattingly D, van Flandern G, Komiya S, Aigner T, Libermann TA, Goldring MB: Differential expression of GADD45b in normal and osteoarthritic cartilage: Potential role in homeostasis of articular chondrocytes. Arthritis Rheum. 2008, 58: 2075-2087.
Dudhia J: Aggrecan, aging and assembly in articular cartilage. Cell Mol Life Sci. 2005, 62: 2241-2256.
Loeser RF: Molecular mechanisms of cartilage destruction: mechanics, inflammatory mediators, and aging collide. Arthritis Rheum. 2006, 54: 1357-1360.
Aigner T, Haag J, Martin J, Buckwalter J: Osteoarthritis: aging of matrix and cells – going for a remedy. Curr Drug Targets. 2007, 8: 325-331.
Verzijl N, Bank RA, TeKoppele JM, DeGroot J: AGEing and osteoarthritis: a different perspective. Curr Opin Rheumatol. 2003, 15: 616-622.
Martin JA, Brown TD, Heiner AD, Buckwalter JA: Chondrocyte senescence, joint loading and osteoarthritis. Clin Orthop Relat Res. 2004, 427 (Suppl): S96-103.
Yang L, Carlson SG, McBurney D, Horton WE: Multiple signals induce endoplasmic reticulum stress in both primary and immortalized chondrocytes resulting in loss of differentiation, impaired cell growth, and apoptosis. J Biol Chem. 2005, 280: 31156-31165.
Benito MJ, Veale DJ, FitzGerald O, Berg van den WB, Bresnihan B: Synovial tissue inflammation in early and late osteoarthritis. Ann Rheum Dis. 2005, 64: 1263-1267.
Pearle AD, Scanzello CR, George S, Mandl LA, DiCarlo EF, Peterson M, Sculco TP, Crow MK: Elevated high-sensitivity C-reactive protein levels are associated with local inflammatory findings in patients with osteoarthritis. Osteoarthritis Cartilage. 2007, 15: 516-523.
Cawston TE, Wilson AJ: Understanding the role of tissue degrading enzymes and their inhibitors in development and disease. Best Pract Res Clin Rheumatol. 2006, 20: 983-1002.
Sandy JD: A contentious issue finds some clarity: on the independent and complementary roles of aggrecanase activity and MMP activity in human joint aggrecanolysis. Osteoarthritis Cartilage. 2006, 14: 95-100.
Rengel Y, Ospelt C, Gay S: Proteinases in the joint: clinical relevance of proteinases in joint destruction. Arthritis Res Ther. 2007, 9: 221-
Murphy G, Nagase H: Reappraising metalloproteinases in rheumatoid arthritis and osteoarthritis: destruction or repair?. Nat Clin Pract Rheumatol. 2008, 4: 128-135.
Barksby HE, Milner JM, Patterson AM, Peake NJ, Hui W, Robson T, Lakey R, Middleton J, Cawston TE, Richards CD, Rowan AD: Matrix metalloproteinase 10 promotion of collagenolysis via procollagenase activation: implications for cartilage degradation in arthritis. Arthritis Rheum. 2006, 54: 3244-3253.
Rutkauskaite E, Volkmer D, Shigeyama Y, Schedel J, Pap G, Muller-Ladner U, Meinecke I, Alexander D, Gay RE, Drynda S, Neumann W, Michel BA, Aicher WK, Gay S, Pap T: Retroviral gene transfer of an antisense construct against membrane type 1 matrix metalloproteinase reduces the invasiveness of rheumatoid arthritis synovial fibroblasts. Arthritis Rheum. 2005, 52: 2010-2014.
Dreier R, Grassel S, Fuchs S, Schaumburger J, Bruckner P: Pro-MMP-9 is a specific macrophage product and is activated by osteoarthritic chondrocytes via MMP-3 or a MT1-MMP/MMP-13 cascade. Exp Cell Res. 2004, 297: 303-312.
Kevorkian L, Young DA, Darrah C, Donell ST, Shepstone L, Porter S, Brockbank SM, Edwards DR, Parker AE, Clark IM: Expression profiling of metalloproteinases and their inhibitors in cartilage. Arthritis Rheum. 2004, 50: 131-141.
Burrage PS, Huntington JT, Sporn MB, Brinckerhoff CE: Regulation of matrix metalloproteinase gene expression by a retinoid × receptor-specific ligand. Arthritis Rheum. 2007, 56: 892-904.
Murphy G, Lee MH: What are the roles of metalloproteinases in cartilage and bone damage?. Ann Rheum Dis. 2005, 64 (Suppl 4): iv44-47.
Overall CM, Blobel CP: In search of partners: linking extracellular proteases to substrates. Nat Rev Mol Cell Biol. 2007, 8: 245-257.
Arner EC: Aggrecanase-mediated cartilage degradation. Curr Opin Pharmacol. 2002, 2: 322-329.
Plaas A, Osborn B, Yoshihara Y, Bai Y, Bloom T, Nelson F, Mikecz K, Sandy JD: Aggrecanolysis in human osteoarthritis: confocal localization and biochemical characterization of ADAMTS5-hyaluronan complexes in articular cartilages. Osteoarthritis Cartilage. 2007, 15: 719-734.
Gilbert AM, Bursavich MG, Lombardi S, Georgiadis KE, Reifenberg E, Flannery CR, Morris EA: N-((8-hydroxy-5-substituted-quinolin-7-yl)(phenyl)methyl)-2-phenyloxy/amin o-acetamide inhibitors of ADAMTS-5 (Aggrecanase-2). Bioorg Med Chem Lett. 2008, 18: 6454-6457.
Wu W, Billinghurst RC, Pidoux I, Antoniou J, Zukor D, Tanzer M, Poole AR: Sites of collagenase cleavage and denaturation of type II collagen in aging and osteoarthritic articular cartilage and their relationship to the distribution of matrix metalloproteinase 1 and matrix metalloproteinase 13. Arthritis Rheum. 2002, 46: 2087-2094.
Kane D, Jensen LE, Grehan S, Whitehead AS, Bresnihan B, Fitzgerald O: Quantitation of metalloproteinase gene expression in rheumatoid and psoriatic arthritis synovial tissue distal and proximal to the cartilage-pannus junction. J Rheumatol. 2004, 31: 1274-1280.
Fraser A, Fearon U, Billinghurst RC, Ionescu M, Reece R, Barwick T, Emery P, Poole AR, Veale DJ: Turnover of type II collagen and aggrecan in cartilage matrix at the onset of inflammatory arthritis in humans: relationship to mediators of systemic and local inflammation. Arthritis Rheum. 2003, 48: 3085-3095.
Bau B, Gebhard PM, Haag J, Knorr T, Bartnik E, Aigner T: Relative messenger RNA expression profiling of collagenases and aggrecanases in human articular chondrocytes in vivo and in vitro. Arthritis Rheum. 2002, 46: 2648-2657.
Hermansson M, Sawaji Y, Bolton M, Alexander S, Wallace A, Begum S, Wait R, Saklatvala J: Proteomic analysis of articular cartilage shows increased type II collagen synthesis in osteoarthritis and expression of inhibin bA (activin A), a regulatory molecule for chondrocytes. J Biol Chem. 2004, 279: 43514-43521.
Aigner T, Fundel K, Saas J, Gebhard PM, Haag J, Weiss T, Zien A, Obermayr F, Zimmer R, Bartnik E: Large-scale gene expression profiling reveals major pathogenetic pathways of cartilage degeneration in osteoarthritis. Arthritis Rheum. 2006, 54: 3533-3544.
Fukui N, Ikeda Y, Ohnuki T, Tanaka N, Hikita A, Mitomi H, Mori T, Juji T, Katsuragawa Y, Yamamoto S, Sawabe M, Yamane S, Suzuki R, Sandell LJ, Ochi T: Regional differences in chondrocyte metabolism in osteoarthritis: a detailed analysis by laser capture microdissection. Arthritis Rheum. 2008, 58: 154-163.
Arend WP, Goldring MB: The development of anticytokine therapeutics for rheumatic diseases. Arthritis Rheum. 2008, 58: S102-109.
Tetlow LC, Woolley DE: Comparative immunolocalization studies of collagenase 1 and collagenase 3 production in the rheumatoid lesion, and by human chondrocytes and synoviocytes in vitro. Br J Rheumatol. 1998, 37: 64-70.
Tetlow LC, Adlam DJ, Woolley DE: Matrix metalloproteinase and proinflammatory cytokine production by chondrocytes of human osteoarthritic cartilage. Arthritis Rheum. 2001, 44: 585-594.
Borzi RM, Mazzetti I, Marcu KB, Facchini A: Chemokines in cartilage degradation. Clin Orthop Relat Res. 2004, 427 (Suppl): S53-61.
Sandell LJ, Xing X, Franz C, Davies S, Chang LW, Patra D: Exuberant expression of chemokine genes by adult human articular chondrocytes in response to IL-1b. Osteoarthritis Cartilage. 2008, 16: 1560-1571.
Lubberts E, Koenders MI, Berg van den WB: The role of T cell interleukin-17 in conducting destructive arthritis: lessons from animal models. Arthritis Res Ther. 2005, 7: 29-37.
Koenders MI, Joosten LA, Berg van den WB: Potential new targets in arthritis therapy: interleukin (IL)-17 and its relation to tumour necrosis factor and IL-1 in experimental arthritis. Ann Rheum Dis. 2006, 65 (Suppl 3): iii29-33.
Joosten LA, Netea MG, Kim SH, Yoon DY, Oppers-Walgreen B, Radstake TR, Barrera P, Loo van de FA, Dinarello CA, Berg van den WB: IL-32, a proinflammatory cytokine in rheumatoid arthritis. Proc Natl Acad Sci USA. 2006, 103: 3298-3303.
Rowan AD, Koshy PJ, Shingleton WD, Degnan BA, Heath JK, Vernallis AB, Spaull JR, Life PF, Hudson K, Cawston TE: Synergistic effects of glycoprotein 130 binding cytokines in combination with interleukin-1 on cartilage collagen breakdown. Arthritis Rheum. 2001, 44: 1620-1632.
Koshy PJ, Henderson N, Logan C, Life PF, Cawston TE, Rowan AD: Interleukin 17 induces cartilage collagen breakdown: novel synergistic effects in combination with proinflammatory cytokines. Ann Rheum Dis. 2002, 61: 704-713.
Barksby HE, Hui W, Wappler I, Peters HH, Milner JM, Richards CD, Cawston TE, Rowan AD: Interleukin-1 in combination with oncostatin M up-regulates multiple genes in chondrocytes: implications for cartilage destruction and repair. Arthritis Rheum. 2006, 54: 540-550.
Goldring MB, Sandell LJ: Transcriptional control of chondrocyte gene expression. OA, Inflammation and Degradation: A Continuum. Edited by: Buckwalter J, Lotz M, Stoltz JF. 2007, Amsterdam: IOS Press, 118-142.
Imamura T, Imamura C, Iwamoto Y, Sandell LJ: Transcriptional Co-activators CREB-binding protein/p300 increase chondrocyte Cd-rap gene expression by multiple mechanisms including sequestration of the repressor CCAAT/enhancer-binding protein. J Biol Chem. 2005, 280: 16625-16634.
Peng H, Tan L, Osaki M, Zhan Y, Ijiri K, Tsuchimochi K, Otero M, Wang H, Choy BK, Grall FT, Gu X, Libermann TA, Oettgen P, Goldring MB: ESE-1 is a potent repressor of type II collagen gene (COL2A1) transcription in human chondrocytes. J Cell Physiol. 2008, 215: 562-573.
Roach HI, Yamada N, Cheung KS, Tilley S, Clarke NM, Oreffo RO, Kokubun S, Bronner F: Association between the abnormal expression of matrix-degrading enzymes by human osteoarthritic chondrocytes and demethylation of specific CpG sites in the promoter regions. Arthritis Rheum. 2005, 52: 3110-3124.
Kim HA, Cho ML, Choi HY, Yoon CS, Jhun JY, Oh HJ, Kim HY: The catabolic pathway mediated by Toll-like receptors in human osteoarthritic chondrocytes. Arthritis Rheum. 2006, 54: 2152-2163.
Su SL, Tsai CD, Lee CH, Salter DM, Lee HS: Expression and regulation of Toll-like receptor 2 by IL-1b and fibronectin fragments in human articular chondrocytes. Osteoarthritis Cartilage. 2005, 13: 879-886.
Varoga D, Paulsen F, Mentlein R, Fay J, Kurz B, Schutz R, Wruck C, Goldring MB, Pufe T: TLR-2-mediated induction of vascular endothelial growth factor (VEGF) in cartilage in septic joint disease. J Pathol. 2006, 210: 315-324.
Bobacz K, Sunk IG, Hofstaetter JG, Amoyo L, Toma CD, Akira S, Weichhart T, Saemann M, Smolen JS: Toll-like receptors and chondrocytes: the lipopolysaccharide-induced decrease in cartilage matrix synthesis is dependent on the presence of toll-like receptor 4 and antagonized by bone morphogenetic protein 7. Arthritis Rheum. 2007, 56: 1880-1893.
Haglund L, Bernier SM, Onnerfjord P, Recklies AD: Proteomic analysis of the LPS-induced stress response in rat chondrocytes reveals induction of innate immune response components in articular cartilage. Matrix Biol. 2008, 27: 107-118.
Zhang Q, Hui W, Litherland GJ, Barter MJ, Davidson R, Darrah C, Donell ST, Clark IM, Cawston TE, Robinson JH, Rowan AD, Young DA: Differential Toll-like receptor-dependent collagenase expression in chondrocytes. Ann Rheum Dis. 2008, 67: 1633-1641.
van Lent PL, Blom AB, Grevers L, Sloetjes A, Berg van den WB: Toll-like receptor 4 induced FcgR expression potentiates early onset of joint inflammation and cartilage destruction during immune complex arthritis: Toll-like receptor 4 largely regulates FcgR expression by interleukin 10. Ann Rheum Dis. 2007, 66: 334-340.
Dai SM, Shan ZZ, Nishioka K, Yudoh K: Implication of interleukin 18 in production of matrix metalloproteinases in articular chondrocytes in arthritis: direct effect on chondrocytes may not be pivotal. Ann Rheum Dis. 2005, 64: 735-742.
John T, Kohl B, Mobasheri A, Ertel W, Shakibaei M: Interleukin-18 induces apoptosis in human articular chondrocytes. Histol Histopathol. 2007, 22: 469-482.
Barksby HE, Lea SR, Preshaw PM, Taylor JJ: The expanding family of interleukin-1 cytokines and their role in destructive inflammatory disorders. Clin Exp Immunol. 2007, 149: 217-225.
Smeets RL, Veenbergen S, Arntz OJ, Bennink MB, Joosten LA, Berg van den WB, Loo van de FA: A novel role for suppressor of cytokine signaling 3 in cartilage destruction via induction of chondrocyte desensitization toward insulin-like growth factor. Arthritis Rheum. 2006, 54: 1518-1528.
Masuko-Hongo K, Berenbaum F, Humbert L, Salvat C, Goldring MB, Thirion S: Up-regulation of microsomal prostaglandin E synthase 1 in osteoarthritic human cartilage: critical roles of the ERK-1/2 and p38 signaling pathways. Arthritis Rheum. 2004, 50: 2829-2838.
Whiteman M, Spencer JP, Zhu YZ, Armstrong JS, Schantz JT: Peroxynitrite-modified collagen-II induces p38/ERK and NF-kB-dependent synthesis of prostaglandin E2 and nitric oxide in chondrogenically differentiated mesenchymal progenitor cells. Osteoarthritis Cartilage. 2006, 14: 460-470.
Cheng S, Afif H, Martel-Pelletier J, Pelletier JP, Li X, Farrajota K, Lavigne M, Fahmi H: Activation of peroxisome proliferator-activated receptor g inhibits interleukin-1b-induced membrane-associated prostaglandin E2 synthase-1 expression in human synovial fibroblasts by interfering with Egr-1. J Biol Chem. 2004, 279: 22057-22065.
Li X, Afif H, Cheng S, Martel-Pelletier J, Pelletier JP, Ranger P, Fahmi H: Expression and regulation of microsomal prostaglandin E synthase-1 in human osteoarthritic cartilage and chondrocytes. J Rheumatol. 2005, 32: 887-895.
Francois M, Richette P, Tsagris L, Fitting C, Lemay C, Benallaoua M, Tahiri K, Corvol MT: Activation of the peroxisome proliferator-activated receptor a pathway potentiates interleukin-1 receptor antagonist production in cytokine-treated chondrocytes. Arthritis Rheum. 2006, 54: 1233-1245.
Dayer JM, Chicheportiche R, Juge-Aubry C, Meier C: Adipose tissue has anti-inflammatory properties: focus on IL-1 receptor antagonist (IL-1Ra). Ann N Y Acad Sci. 2006, 1069: 444-453.
Otero M, Lago R, Gomez R, Dieguez C, Lago F, Gomez-Reino J, Gualillo O: Towards a pro-inflammatory and immunomodulatory emerging role of leptin. Rheumatology (Oxford). 2006, 45: 944-950.
Popa C, Netea MG, Radstake TR, van Riel PL, Barrera P, Meer van der JW: Markers of inflammation are negatively correlated with serum leptin in rheumatoid arthritis. Ann Rheum Dis. 2005, 64: 1195-1198.
Dumond H, Presle N, Terlain B, Mainard D, Loeuille D, Netter P, Pottie P: Evidence for a key role of leptin in osteoarthritis. Arthritis Rheum. 2003, 48: 3118-3129.
Otero M, Lago R, Lago F, Reino JJ, Gualillo O: Signalling pathway involved in nitric oxide synthase type II activation in chondrocytes: synergistic effect of leptin with interleukin-1. Arthritis Res Ther. 2005, 7: R581-591.
Palmer G, Aurrand-Lions M, Contassot E, Talabot-Ayer D, Ducrest-Gay D, Vesin C, Chobaz-Peclat V, Busso N, Gabay C: Indirect effects of leptin receptor deficiency on lymphocyte populations and immune response in db/db mice. J Immunol. 2006, 177: 2899-2907.
Lago F, Dieguez C, Gómez-Reino J, Gualillo O: The emerging role of adipokines as mediators of inflammation and immune responses. Cytokine Growth Factor Rev. 2007, 18: 313-325.
Lee JH, Ort T, Ma K, Picha K, Carton J, Marsters PA, Lohmander LS, Baribaud F, Song XY, Blake S: Resistin is elevated following traumatic joint injury and causes matrix degradation and release of inflammatory cytokines from articular cartilage in vitro. Osteoarthritis Cartilage. 2008,
Senolt L, Housa D, Vernerova Z, Jirasek T, Svobodova R, Veigl D, Anderlova K, Muller-Ladner U, Pavelka K, Haluzik M: Resistin in rheumatoid arthritis synovial tissue, synovial fluid and serum. Ann Rheum Dis. 2007, 66: 458-463.
Lee JH, Fitzgerald JB, Dimicco MA, Grodzinsky AJ: Mechanical injury of cartilage explants causes specific time-dependent changes in chondrocyte gene expression. Arthritis Rheum. 2005, 52: 2386-2395.
Alexopoulos LG, Williams GM, Upton ML, Setton LA, Guilak F: Osteoarthritic changes in the biphasic mechanical properties of the chondrocyte pericellular matrix in articular cartilage. J Biomech. 2005, 38: 509-517.
Roach HI, Aigner T, Soder S, Haag J, Welkerling H: Pathobiology of osteoarthritis: pathomechanisms and potential therapeutic targets. Curr Drug Targets. 2007, 8: 271-282.
Sandell LJ: Anabolic factors in degenerative joint disease. Curr Drug Targets. 2007, 8: 359-365.
Guilak F, Fermor B, Keefe FJ, Kraus VB, Olson SA, Pisetsky DS, Setton LA, Weinberg JB: The role of biomechanics and inflammation in cartilage injury and repair. Clin Orthop Relat Res. 2004, 17-26. 423
Fitzgerald JB, Jin M, Dean D, Wood DJ, Zheng MH, Grodzinsky AJ: Mechanical compression of cartilage explants induces multiple time-dependent gene expression patterns and involves intracellular calcium and cyclic AMP. J Biol Chem. 2004, 279: 19502-19511.
Kurz B, Lemke AK, Fay J, Pufe T, Grodzinsky AJ, Schunke M: Pathomechanisms of cartilage destruction by mechanical injury. Ann Anat. 2005, 187: 473-485.
Ramage L, Martel MA, Hardingham GE, Salter DM: NMDA receptor expression and activity in osteoarthritic human articular chondrocytes. Osteoarthritis Cartilage. 2008, 16: 1576-1584.
Millward-Sadler SJ, Salter DM: Integrin-dependent signal cascades in chondrocyte mechanotransduction. Ann Biomed Eng. 2004, 32: 435-446.
Pulai JI, Chen H, Im HJ, Kumar S, Hanning C, Hegde PS, Loeser RF: NF-k B mediates the stimulation of cytokine and chemokine expression by human articular chondrocytes in response to fibronectin fragments. J Immunol. 2005, 174: 5781-5788.
Xu L, Peng H, Glasson S, Lee PL, Hu K, Ijiri K, Olsen BR, Goldring MB, Li Y: Increased expression of the collagen receptor discoidin domain receptor 2 in articular cartilage as a key event in the pathogenesis of osteoarthritis. Arthritis Rheum. 2007, 56: 2663-2673.
Lee DM, Kiener HP, Agarwal SK, Noss EH, Watts GF, Chisaka O, Takeichi M, Brenner MB: Cadherin-11 in synovial lining formation and pathology in arthritis. Science. 2007, 315: 1006-1010.
Vandooren B, Cantaert T, ter Borg M, Noordenbos T, Kuhlman R, Gerlag D, Bongartz T, Reedquist K, Tak PP, Baeten D: Tumor necrosis factor a drives cadherin 11 expression in rheumatoid inflammation. Arthritis Rheum. 2008, 58: 3051-3062.
Fanning PJ, Emkey G, Smith RJ, Grodzinsky AJ, Szasz N, Trippel SB: Mechanical regulation of mitogen-activated protein kinase signaling in articular cartilage. J Biol Chem. 2003, 278: 50940-50948.
Deschner J, Hofman CR, Piesco NP, Agarwal S: Signal transduction by mechanical strain in chondrocytes. Curr Opin Clin Nutr Metab Care. 2003, 6: 289-293.
Zhou Y, Millward-Sadler SJ, Lin H, Robinson H, Goldring M, Salter DM, Nuki G: Evidence for JNK-dependent up-regulation of proteoglycan synthesis and for activation of JNK1 following cyclical mechanical stimulation in a human chondrocyte culture model. Osteoarthritis Cartilage. 2007, 15: 884-893.
Knobloch TJ, Madhavan S, Nam J, Agarwal S, Agarwal S: Regulation of chondrocytic gene expression by biomechanical signals. Crit Rev Eukaryot Gene Expr. 2008, 18: 139-150.
Fan Z, Soder S, Oehler S, Fundel K, Aigner T: Activation of interleukin-1 signaling cascades in normal and osteoarthritic articular cartilage. Am J Pathol. 2007, 171: 938-946.
Ronziere MC, Aubert-Foucher E, Gouttenoire J, Bernaud J, Herbage D, Mallein-Gerin F: Integrin a1b1 mediates collagen induction of MMP-13 expression in MC615 chondrocytes. Biochim Biophys Acta. 2005, 1746: 55-64.
Loeser RF, Forsyth CB, Samarel AM, Im HJ: Fibronectin fragment activation of proline-rich tyrosine kinase PYK2 mediates integrin signals regulating collagenase-3 expression by human chondrocytes through a protein kinase C-dependent pathway. J Biol Chem. 2003, 278: 24577-24585.
LaVallie ER, Chockalingam PS, Collins-Racie LA, Freeman BA, Keohan CC, Leitges M, Dorner AJ, Morris EA, Majumdar MK, Arai M: Protein kinase Cz is up-regulated in osteoarthritic cartilage and is required for activation of NF-kB by tumor necrosis factor and interleukin-1 in articular chondrocytes. J Biol Chem. 2006, 281: 24124-24137.
Dossumbekova A, Anghelina M, Madhavan S, He L, Quan N, Knobloch T, Agarwal S: Biomechanical signals inhibit IKK activity to attenuate NF-kB transcription activity in inflamed chondrocytes. Arthritis Rheum. 2007, 56: 3284-3296.
Madhavan S, Anghelina M, Sjostrom D, Dossumbekova A, Guttridge DC, Agarwal S: Biomechanical signals suppress TAK1 activation to inhibit NF-kB transcriptional activation in fibrochondrocytes. J Immunol. 2007, 179: 6246-6254.
Grishko VI, Ho R, Wilson GL, Pearsall AW: Diminished mitochondrial DNA integrity and repair capacity in OA chondrocytes. Osteoarthritis Cartilage. 2009, 17: 107-113.
Ruiz-Romero C, Calamia V, Mateos J, Carreira V, Martínez-Gomariz M, Fernández M, Blanco FJ: Mitochondrial dysregulation of osteoarthritic human articular chondrocytes analyzed by proteomics: a decrease in mitochondrial superoxide dismutase points to a redox imbalance. Mol Cell Proteomics. 2009, 8: 172-189.
Steenvoorden MM, Huizinga TW, Verzijl N, Bank RA, Ronday HK, Luning HA, Lafeber FP, Toes RE, DeGroot J: Activation of receptor for advanced glycation end products in osteoarthritis leads to increased stimulation of chondrocytes and synoviocytes. Arthritis Rheum. 2006, 54: 253-263.
Yammani RR, Carlson CS, Bresnick AR, Loeser RF: Increase in production of matrix metalloproteinase 13 by human articular chondrocytes due to stimulation with S100A4: role of the receptor for advanced glycation end products. Arthritis Rheum. 2006, 54: 2901-2911.
Cecil DL, Terkeltaub R: Transamidation by transglutaminase 2 transforms S100A11 calgranulin into a procatabolic cytokine for chondrocytes. J Immunol. 2008, 180: 8378-8385.
van Lent PL, Grevers L, Blom AB, Sloetjes A, Mort JS, Vogl T, Nacken W, Berg van den WB, Roth J: Myeloid-related proteins S100A8/S100A9 regulate joint inflammation and cartilage destruction during antigen-induced arthritis. Ann Rheum Dis. 2008, 67: 1750-1758.
van Lent PL, Grevers LC, Blom AB, Arntz OJ, Loo van de FA, Kraan van der P, Abdollahi-Roodsaz S, Srikrishna G, Freeze H, Sloetjes A, Nacken W, Vogl T, Roth J, Berg van den WB: Stimulation of chondrocyte-mediated cartilage destruction by S100A8 in experimental murine arthritis. Arthritis Rheum. 2008, 58: 3776-3787.
Taniguchi N, Kawahara K, Yone K, Hashiguchi T, Yamakuchi M, Goto M, Inoue K, Yamada S, Ijiri K, Matsunaga S, Nakajima T, Komiya S, Maruyama I: High mobility group box chromosomal protein 1 plays a role in the pathogenesis of rheumatoid arthritis as a novel cytokine. Arthritis Rheum. 2003, 48: 971-981.
Mobasheri A, Bondy CA, Moley K, Mendes AF, Rosa SC, Richardson SM, Hoyland JA, Barrett-Jolley R, Shakibaei M: Facilitative glucose transporters in articular chondrocytes. Expression, distribution and functional regulation of GLUT isoforms by hypoxia, hypoxia mimetics, growth factors and pro-inflammatory cytokines. Adv Anat Embryol Cell Biol. 2008, 200: 1-84.
Healy ZR, Lee NH, Gao X, Goldring MB, Talalay P, Kensler TW, Konstantopoulos K: Divergent responses of chondrocytes and endothelial cells to shear stress: cross-talk among COX-2, the phase 2 response, and apoptosis. Proc Natl Acad Sci USA. 2005, 102: 14010-14015.
Charni-Ben Tabassi N, Garnero P: Monitoring cartilage turnover. Curr Rheumatol Rep. 2007, 9: 16-24.
Rousseau JC, Delmas PD: Biological markers in osteoarthritis. Nat Clin Pract Rheumatol. 2007, 3: 346-356.
Dayer E, Dayer JM, Roux-Lombard P: Primer: the practical use of biological markers of rheumatic and systemic inflammatory diseases. Nat Clin Pract Rheumatol. 2007, 3: 512-520.
Henrotin Y, Addison S, Kraus V, Deberg M: Type II collagen markers in osteoarthritis: what do they indicate?. Curr Opin Rheumatol. 2007, 19: 444-450.
Cahue S, Sharma L, Dunlop D, Ionescu M, Song J, Lobanok T, King L, Poole AR: The ratio of type II collagen breakdown to synthesis and its relationship with the progression of knee osteoarthritis. Osteoarthritis Cartilage. 2007, 15: 819-823.
Lindqvist E, Eberhardt K, Bendtzen K, Heinegard D, Saxne T: Prognostic laboratory markers of joint damage in rheumatoid arthritis. Ann Rheum Dis. 2005, 64: 196-201.
Baeten D, Steenbakkers PG, Rijnders AM, Boots AM, Veys EM, De Keyser F: Detection of major histocompatibility complex/human cartilage gp-39 complexes in rheumatoid arthritis synovitis as a specific and independent histologic marker. Arthritis Rheum. 2004, 50: 444-451.
Saito S, Kondo S, Mishima S, Ishiguro N, Hasegawa Y, Sandell LJ, Iwata H: Analysis of cartilage-derived retinoic-acid-sensitive protein (CD-RAP) in synovial fluid from patients with osteoarthritis and rheumatoid arthritis. J Bone Joint Surg Br. 2002, 84: 1066-1069.
Vandooren B, Cantaert T, van Lierop MJ, Bos E, De Rycke L, Veys EM, De Keyser F, Bresnihan B, Luyten FP, Verdonk PC, Tak PP, Boots AH, Baeten D: Melanoma inhibitory activity, a biomarker related to chondrocyte anabolism, is reversibly suppressed by proinflammatory cytokines in rheumatoid arthritis. Ann Rheum Dis. 2008
Hunter DJ, Li J, LaValley M, Bauer DC, Nevitt M, DeGroot J, Poole R, Eyre D, Guermazi A, Gale D, Felson DT: Cartilage markers and their association with cartilage loss on magnetic resonance imaging in knee osteoarthritis: the Boston Osteoarthritis Knee Study. Arthritis Res Ther. 2007, 9: R108-
Valdes AM, Loughlin J, Oene MV, Chapman K, Surdulescu GL, Doherty M, Spector TD: Sex and ethnic differences in the association of ASPN, CALM1, COL2A1, COMP, and FRZB with genetic susceptibility to osteoarthritis of the knee. Arthritis Rheum. 2007, 56: 137-146.
Valdes AM, Van Oene M, Hart DJ, Surdulescu GL, Loughlin J, Doherty M, Spector TD: Reproducible genetic associations between candidate genes and clinical knee osteoarthritis in men and women. Arthritis Rheum. 2006, 54: 533-539.
Loughlin J: Polymorphism in signal transduction is a major route through which osteoarthritis susceptibility is acting. Curr Opin Rheumatol. 2005, 17: 629-633.
Bukulmez H, Matthews AL, Sullivan CM, Chen C, Kraay MJ, Elston RC, Moskowitz RW, Goldberg VM, Warman ML: Hip joint replacement surgery for idiopathic osteoarthritis aggregates in families. Arthritis Res Ther. 2006, 8: R25-
Li Y, Xu L, Olsen BR: Lessons from genetic forms of osteoarthritis for the pathogenesis of the disease. Osteoarthritis Cartilage. 2007, 15: 1101-1105.
Gregersen PK, Behrens TW: Genetics of autoimmune diseases – disorders of immune homeostasis. Nat Rev Genet. 2006, 7: 917-928.
Vincenti MP, Brinckerhoff CE: Early response genes induced in chondrocytes stimulated with the inflammatory cytokine interleukin-1b. Arthritis Res. 2001, 3: 381-388.
Attur MG, Dave MN, Akamatsu M, Katoh M, Amin AR: A system biology approach to bioinformatics and functional genomics in complex human diseases: arthritis. Curr Issues Mol Biol. 2002, 4: 129-146.
Sato T, Konomi K, Yamasaki S, Aratani S, Tsuchimochi K, Yokouchi M, Masuko-Hongo K, Yagishita N, Nakamura H, Komiya S, Beppu M, Aoki H, Nishioka K, Nakajima T: Comparative analysis of gene expression profiles in intact and damaged regions of human osteoarthritic cartilage. Arthritis Rheum. 2006, 54: 808-817.
Andreas K, Lubke C, Haupl T, Dehne T, Morawietz L, Ringe J, Kaps C, Sittinger M: Key regulatory molecules of cartilage destruction in rheumatoid arthritis: an in vitro study. Arthritis Res Ther. 2008, 10: R9-
Berg van den WB, van Riel PL: Uncoupling of inflammation and destruction in rheumatoid arthritis: myth or reality?. Arthritis Rheum. 2005, 52: 995-999.
Zwerina J, Redlich K, Polzer K, Joosten L, Krönke G, Distler J, Hess A, Pundt N, Pap T, Hoffmann O, Gasser J, Scheinecker C, Smolen JS, Berg van den W, Schett G: TNF-induced structural joint damage is mediated by IL-1. Proc Natl Acad Sci USA. 2007, 104: 11742-11747.
Helminen HJ, Saamanen AM, Salminen H, Hyttinen MM: Transgenic mouse models for studying the role of cartilage macromolecules in osteoarthritis. Rheumatology (Oxford). 2002, 41: 848-856.
Ameye LG, Young MF: Animal models of osteoarthritis: lessons learned while seeking the 'Holy Grail'. Curr Opin Rheumatol. 2006, 18: 537-547.
Sahebjam S, Khokha R, Mort JS: Increased collagen and aggrecan degradation with age in the joints of Timp3(-/-) mice. Arthritis Rheum. 2007, 56: 905-909.
Neuhold LA, Killar L, Zhao W, Sung ML, Warner L, Kulik J, Turner J, Wu W, Billinghurst C, Meijers T, Poole AR, Babij P, DeGennaro LJ: Postnatal expression in hyaline cartilage of constitutively active human collagenase-3 (MMP-13) induces osteoarthritis in mice. J Clin Invest. 2001, 107: 35-44.
Glasson SS, Askew R, Sheppard B, Carito B, Blanchet T, Ma HL, Flannery CR, Peluso D, Kanki K, Yang Z, Majumdar MK, Morris EA: Deletion of active ADAMTS5 prevents cartilage degradation in a murine model of osteoarthritis. Nature. 2005, 434: 644-648.
Stanton H, Rogerson FM, East CJ, Golub SB, Lawlor KE, Meeker CT, Little CB, Last K, Farmer PJ, Campbell IK, Fourie AM, Fosang AJ: ADAMTS5 is the major aggrecanase in mouse cartilage in vivo and in vitro. Nature. 2005, 434: 648-652.
Kamekura S, Kawasaki Y, Hoshi K, Shimoaka T, Chikuda H, Maruyama Z, Komori T, Sato S, Takeda S, Karsenty G, Nakamura K, Chung UI, Kawaguchi H: Contribution of runt-related transcription factor 2 to the pathogenesis of osteoarthritis in mice after induction of knee joint instability. Arthritis Rheum. 2006, 54: 2462-2470.
Glasson SS, Blanchet TJ, Morris EA: The surgical destabilization of the medial meniscus (DMM) model of osteoarthritis in the 129/SvEv mouse. Osteoarthritis Cartilage. 2007, 15: 1061-1069.
Goldring MB, Tsuchimochi K, Ijiri K: The control of chondrogenesis. J Cell Biochem. 2006, 97: 33-44.
Lefebvre V, Smits P: Transcriptional control of chondrocyte fate and differentiation. Birth Defects Res C Embryo Today. 2005, 75: 200-212.
Hidaka C, Goldring MB: Regulatory mechanisms of chondrogenesis and implications for understanding articular cartilage homeostasis. Curr Rheumatol Rev. 2008, 4: 136-147.
Haque T, Nakada S, Hamdy RC: A review of FGF18: its expression, signaling pathways and possible functions during embryogenesis and post-natal development. Histol Histopathol. 2007, 22: 97-105.
Wu X, Shi W, Cao X: Multiplicity of BMP signaling in skeletal development. Ann N Y Acad Sci. 2007, 1116: 29-49.
Yoon BS, Pogue R, Ovchinnikov DA, Yoshii I, Mishina Y, Behringer RR, Lyons KM: BMPs regulate multiple aspects of growthplate chondrogenesis through opposing actions on FGF pathways. Development. 2006, 133: 4667-4678.
Zhong N, Gersch RP, Hadjiargyrou M: Wnt signaling activation during bone regeneration and the role of Dishevelled in chondrocyte proliferation and differentiation. Bone. 2006, 39: 5-16.
Dong YF, Soung do Y, Schwarz EM, O'Keefe RJ, Drissi H: Wnt induction of chondrocyte hypertrophy through the Runx2 transcription factor. J Cell Physiol. 2006, 208: 77-86.
Zhou G, Zheng Q, Engin F, Munivez E, Chen Y, Sebald E, Krakow D, Lee B: Dominance of SOX9 function over RUNX2 during skeletogenesis. Proc Natl Acad Sci USA. 2006, 103: 19004-19009.
Ijiri K, Zerbini LF, Peng H, Correa RG, Lu B, Walsh N, Zhao Y, Taniguchi N, Huang XL, Otu H, Wang H, Wang JF, Komiya S, Ducy P, Rahman MU, Flavell RA, Gravallese EM, Oettgen P, Libermann TA, Goldring MB: A novel role for GADD45b as a mediator of MMP-13 gene expression during chondrocyte terminal differentiation. J Biol Chem. 2005, 280: 38544-38555.
Olivotto E, Borzi RM, Vitellozzi R, Pagani S, Facchini A, Battistelli M, Penzo M, Li X, Flamigni F, Li J, Falcieri E, Facchini A, Marcu KB: Differential requirements for IKKa and IKKb in the differentiation of primary human osteoarthritic chondrocytes. Arthritis Rheum. 2008, 58: 227-239.
Aigner T, Bartnik E, Sohler F, Zimmer R: Functional genomics of osteoarthritis: on the way to evaluate disease hypotheses. Clin Orthop Relat Res. 2004, 427 (Suppl): S138-143.
Drissi H, Zuscik M, Rosier R, O'Keefe R: Transcriptional regulation of chondrocyte maturation: potential involvement of transcription factors in OA pathogenesis. Mol Aspects Med. 2005, 26: 169-179.
Terkeltaub RA: Aging, inflammation, and altered chondrocyte differentiation in articular cartilage calcification and osteoarthritis. 2007, Amsterdam: IOS Press
Bos SD, Slagboom PE, Meulenbelt I: New insights into osteoarthritis: early developmental features of an ageing-related disease. Curr Opin Rheumatol. 2008, 20: 553-559.
Wang X, Manner PA, Horner A, Shum L, Tuan RS, Nuckolls GH: Regulation of MMP-13 expression by RUNX2 and FGF2 in osteoarthritic cartilage. Osteoarthritis Cartilage. 2004, 12: 963-973.
Tchetina EV, Squires G, Poole AR: Increased type II collagen degradation and very early focal cartilage degeneration is associated with upregulation of chondrocyte differentiation related genes in early human articular cartilage lesions. J Rheumatol. 2005, 32: 876-886.
Aigner T, Gebhard PM, Schmid E, Bau B, Harley V, Poschl E: SOX9 expression does not correlate with type II collagen expression in adult articular chondrocytes. Matrix Biol. 2003, 22: 363-372.
Aigner T, Kim HA, Roach HI: Apoptosis in osteoarthritis. Rheum Dis Clin North Am. 2004, 30: 639-653. xi.
Tallheden T, Dennis JE, Lennon DP, Sjogren-Jansson E, Caplan AI, Lindahl A: Phenotypic plasticity of human articular chondrocytes. J Bone Joint Surg Am. 2003, 85-A (Suppl 2): 93-100.
Bursell L, Woods A, James CG, Pala D, Leask A, Beier F: Src kinase inhibition promotes the chondrocyte phenotype. Arthritis Res Ther. 2007, 9: R105-
Pala D, Kapoor M, Woods A, Kennedy L, Liu S, Chen S, Bursell L, Lyons KM, Carter DE, Beier F, Leask A: Focal adhesion kinase/Src suppresses early chondrogenesis: central role of CCN2. J Biol Chem. 2008, 283: 9239-9247.
Valdes AM, Spector TD: The contribution of genes to osteoarthritis. Rheum Dis Clin North Am. 2008, 34: 581-603.
Kizawa H, Kou I, Iida A, Sudo A, Miyamoto Y, Fukuda A, Mabuchi A, Kotani A, Kawakami A, Yamamoto S, Uchida A, Nakamura K, Notoya K, Nakamura Y, Ikegawa S: An aspartic acid repeat polymorphism in asporin inhibits chondrogenesis and increases susceptibility to osteoarthritis. Nat Genet. 2005, 37: 138-144.
Lane NE, Lian K, Nevitt MC, Zmuda JM, Lui L, Li J, Wang J, Fontecha M, Umblas N, Rosenbach M, de Leon P, Corr M: Frizzled-related protein variants are risk factors for hip osteoarthritis. Arthritis Rheum. 2006, 54: 1246-1254.
Loughlin J, Dowling B, Chapman K, Marcelline L, Mustafa Z, Southam L, Ferreira A, Ciesielski C, Carson DA, Corr M: Functional variants within the secreted frizzled-related protein 3 gene are associated with hip osteoarthritis in females. Proc Natl Acad Sci USA. 2004, 101: 9757-9762.
Meulenbelt I, Min JL, Bos S, Riyazi N, Houwing-Duistermaat JJ, Wijk van der HJ, Kroon HM, Nakajima M, Ikegawa S, Uitterlinden AG, van Meurs JB, Deure van der WM, Visser TJ, Seymour AB, Lakenberg N, Breggen van der R, Kremer D, van Duijn CM, Kloppenburg M, Loughlin J, Slagboom PE: Identification of DIO2 as a new susceptibility locus for symptomatic osteoarthritis. Hum Mol Genet. 2008, 17: 1867-1875.
Tamamura Y, Otani T, Kanatani N, Koyama E, Kitagaki J, Komori T, Yamada Y, Costantini F, Wakisaka S, Pacifici M, Iwamoto M, EnomotoIwamoto M: Developmental regulation of Wnt/b-catenin signals is required for growth plate assembly, cartilage integrity, and endochondral ossification. J Biol Chem. 2005, 280: 19185-19195.
Hopwood B, Tsykin A, Findlay DM, Fazzalari NL: Microarray gene expression profiling of osteoarthritic bone suggests altered bone remodelling, WNT and transforming growth factor-b/bone morphogenic protein signalling. Arthritis Res Ther. 2007, 9: R100-
Lories RJ, Peeters J, Bakker A, Tylzanowski P, Derese I, Schrooten J, Thomas JT, Luyten FP: Articular cartilage and biomechanical properties of the long bones in Frzb-knockout mice. Arthritis Rheum. 2007, 56: 4095-4103.
Walsh DA, Bonnet CS, Turner EL, Wilson D, Situ M, McWilliams DF: Angiogenesis in the synovium and at the osteochondral junction in osteoarthritis. Osteoarthritis Cartilage. 2007, 15: 743-751.
Burr DB: Anatomy and physiology of the mineralized tissues: role in the pathogenesis of osteoarthrosis. Osteoarthritis Cartilage. 2004, 12 (Suppl A): S20-30.
Blaney Davidson EN, Kraan van der PM, Berg van den WB: TGF-b and osteoarthritis. Osteoarthritis Cartilage. 2007, 15: 597-604.
Kraan van der PM, Berg van den WB: Osteophytes: relevance and biology. Osteoarthritis Cartilage. 2007, 15: 237-244.
Taniguchi N, Yoshida K, Ito T, Tsuda M, Mishima Y, Furumatsu T, Ronfani L, Abeyama K, Kawahara K, Komiya S, Maruyama I, Lotz M, Bianchi ME, Asahara H: Stage-specific secretion of HMGB1 in cartilage regulates endochondral ossification. Mol Cell Biol. 2007, 27: 5650-5663.
Palumbo R, Galvez BG, Pusterla T, De Marchis F, Cossu G, Marcu KB, Bianchi ME: Cells migrating to sites of tissue damage in response to the danger signal HMGB1 require NF-kB activation. J Cell Biol. 2007, 179: 33-40.
Ruocco MG, Maeda S, Park JM, Lawrence T, Hsu LC, Cao Y, Schett G, Wagner EF, Karin M: I{k}B kinase (IKK){b}, but not IKK{a}, is a critical mediator of osteoclast survival and is required for inflammation-induced bone loss. J Exp Med. 2005, 201: 1677-1687.
Otero JE, Dai S, Foglia D, Alhawagri M, Vacher J, Pasparakis M, Abu-Amer Y: Defective osteoclastogenesis by IKKb-null precursors is a result of receptor activator of NF-kB ligand (RANKL)-induced JNK-dependent apoptosis and impaired differentiation. J Biol Chem. 2008, 283: 24546-24553.
Komuro H, Olee T, Kuhn K, Quach J, Brinson DC, Shikhman A, Valbracht J, Creighton-Achermann L, Lotz M: The osteoprotegerin/receptor activator of nuclear factor kB/receptor activator of nuclear factor kB ligand system in cartilage. Arthritis Rheum. 2001, 44: 2768-2776.
Pettit AR, Ji H, von Stechow D, Muller R, Goldring SR, Choi Y, Benoist C, Gravallese EM: TRANCE/RANKL knockout mice are protected from bone erosion in a serum transfer model of arthritis. Am J Pathol. 2001, 159: 1689-1699.
Schett G, Hayer S, Zwerina J, Redlich K, Smolen JS: Mechanisms of Disease: the link between RANKL and arthritic bone disease. Nat Clin Pract Rheumatol. 2005, 1: 47-54.
Kadri A, Ea HK, Bazille C, Hannouche D, Liote F, Cohen-Solal ME: Osteoprotegerin inhibits cartilage degradation through an effect on trabecular bone in murine experimental osteoarthritis. Arthritis Rheum. 2008, 58: 2379-2386.
Kwan Tat S, Pelletier JP, Lajeunesse D, Fahmi H, Lavigne M, Martel-Pelletier J: The differential expression of osteoprotegerin (OPG) and receptor activator of nuclear factor kB ligand (RANKL) in human osteoarthritic subchondral bone osteoblasts is an indicator of the metabolic state of these disease cells. Clin Exp Rheumatol. 2008, 26: 295-304.
Diarra D, Stolina M, Polzer K, Zwerina J, Ominsky MS, Dwyer D, Korb A, Smolen J, Hoffmann M, Scheinecker C, Heide van der D, Landewe R, Lacey D, Richards WG, Schett G: Dickkopf-1 is a master regulator of joint remodeling. Nat Med. 2007, 13: 156-163.
Goldring SR, Goldring MB: Eating bone or adding it: the Wnt pathway decides. Nat Med. 2007, 13: 133-134.
Zhu M, Tang D, Wu Q, Hao S, Chen M, Xie C, Rosier R, O'Keefe R, Zuscik M, Chen D: Activation of beta-catenin signaling in articular chondrocytes leads to osteoarthritis-like phenotype in adult beta-catenin conditional activation mice. J Bone Miner Res. 2009, 24: 12-21.
Kishimoto K, Kitazawa R, Kurosaka M, Maeda S, Kitazawa S: Expression profile of genes related to osteoclastogenesis in mouse growth plate and articular cartilage. Histochem Cell Biol. 2006, 125: 593-602.
Barry FP, Murphy JM: Mesenchymal stem cells: clinical applications and biological characterization. Int J Biochem Cell Biol. 2004, 36: 568-584.
Caplan AI: Review: mesenchymal stem cells: cell-based reconstructive therapy in orthopedics. Tissue Eng. 2005, 11: 1198-1211.
Huang JI, Kazmi N, Durbhakula MM, Hering TM, Yoo JU, Johnstone B: Chondrogenic potential of progenitor cells derived from human bone marrow and adipose tissue: A patient-matched comparison. J Orthop Res. 2005, 23: 1383-1389.
Palmer GD, Steinert A, Pascher A, Gouze E, Gouze JN, Betz O, Johnstone B, Evans CH, Ghivizzani SC: Gene-induced chondrogenesis of primary mesenchymal stem cells in vitro. Mol Ther. 2005, 12: 219-228.
Goldring MB: Are bone morphogenetic proteins effective inducers of cartilage repair? Ex vivo transduction of muscle-derived stem cells. Arthritis Rheum. 2006, 54: 387-389.
Kuroda R, Usas A, Kubo S, Corsi K, Peng H, Rose T, Cummins J, Fu FH, Huard J: Cartilage repair using bone morphogenetic protein 4 and muscle-derived stem cells. Arthritis Rheum. 2006, 54: 433-442.
Magne D, Vinatier C, Julien M, Weiss P, Guicheux J: Mesenchymal stem cell therapy to rebuild cartilage. Trends Mol Med. 2005, 11: 519-526.
Loeser RF, Chubinskaya S, Pacione C, Im HJ: Basic fibroblast growth factor inhibits the anabolic activity of insulin-like growth factor 1 and osteogenic protein 1 in adult human articular chondrocytes. Arthritis Rheum. 2005, 52: 3910-3917.
Goodrich LR, Hidaka C, Robbins PD, Evans CH, Nixon AJ: Genetic modification of chondrocytes with insulin-like growth factor-1 enhances cartilage healing in an equine model. J Bone Joint Surg Br. 2007, 89: 672-685.
Evans CH: Novel biological approaches to the intra-articular treatment of osteoarthritis. BioDrugs. 2005, 19: 355-362.
Lories RJ, Luyten FP: Bone morphogenetic protein signaling in joint homeostasis and disease. Cytokine Growth Factor Rev. 2005, 16: 287-298.
Jukes JM, Both SK, Leusink A, Sterk LMT, van Blitterswijk CA, de Boer J: Endochondral bone tissue engineering using embryonic stem cells. Proc Natl Acad Sci USA. 2008, 105: 6840-6845.
Ikeda T, Kamekura S, Mabuchi A, Kou I, Seki S, Takato T, Nakamura K, Kawaguchi H, Ikegawa S, Chung UI: The combination of SOX5, SOX6, and SOX9 (the SOX trio) provides signals sufficient for induction of permanent cartilage. Arthritis Rheum. 2004, 50: 3561-3573.
Tew SR, Li Y, Pothacharoen P, Tweats LM, Hawkins RE, Hardingham TE: Retroviral transduction with SOX9 enhances re-expression of the chondrocyte phenotype in passaged osteoarthritic human articular chondrocytes. Osteoarthritis Cartilage. 2005, 13: 80-89.
Kuo CK, Li WJ, Mauck RL, Tuan RS: Cartilage tissue engineering: its potential and uses. Curr Opin Rheumatol. 2006, 18: 64-73.
Acknowledgements
Research relating to this review was supported by National Institutes of Health (NIH) (Bethesda, MD, USA) grant AG022021 and by the Arthritis Foundation. KBM greatly acknowledges his collaborators in the Laboratorio di Immunologia e Genetica, Istituti Ortopedici Rizzoli (Bologna, Italy), in particular Rosa Maria Borzi, Eleonora Olivotto, Stefania Pagani, and Andrea Facchini. The research of KBM was supported in part by the Rizzoli Institute, the Carisbo Foundation of Bologna, a Rientro dei Cervelli award, the MAIN EU FPVI Network of Excellence, and NIH grant GM066882.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
About this article
Cite this article
Goldring, M.B., Marcu, K.B. Cartilage homeostasis in health and rheumatic diseases. Arthritis Res Ther 11, 224 (2009). https://doi.org/10.1186/ar2592
Published:
DOI: https://doi.org/10.1186/ar2592