
Abstract
Temporal lobe epilepsy (TLE) is the most common type of epilepsy in humans. Cognitive impairment and memory consolidation problems are common among TLE patients. To understand the changes in the cellular process of memory in TLE, we studied the long-term depression (LTD) in Schaffer-collateral (Sc) CA1 synapses in an epilepsy model. Long-term potentiation (LTP) was investigated in patient samples and animal models by several groups, but LTD was not studied with the same interest in epilepsy research. Here we induced epileptiform activity in rat hippocampal slices using magnesium-free high-potassium (7.5 mM K +) artificial cerebrospinal fluid (HK-ACSF) and characterized the LTD in Sc-CA1 synapses. We found that epileptiform activity abolished/impaired LTD and depotentiation in the Sc-CA1 synapses. In control slices, application of NMDA (30 μM for 3 min) induced chemical LTD (c-LTD) in Sc-CA1 synapses, whereas epileptiform activity induced slices showed slow onset potentiation. Induction of LTD using 1 Hz, 900 pulses yielded a similar outcome as c-LTD. Both forms of LTD were NMDA receptor dependent. In addition, we found that the polarity changes in the synaptic plasticity in epileptiform-induced slices were blocked by GluN2B antagonists ifenprodil and Ro 25–6981. Our data suggest that epileptiform-induced metaplasticity inhibits LTD in Sc-CA1 synapses. We provide new insight into the cellular mechanism of memory formation during epilepsy.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Epilepsy is one of the most common brain disorders, which is estimated to affect about 1% of the world population (Tripathi et al. 2012; Stafstrom and Carmant 2015; Devinsky et al. 2018). This neurological disorder is caused by hyper-synchronisation of neuronal activity in the brain and is characterized by spontaneous recurrent seizures. The process that leads to epilepsy (epileptogenesis) involves spatial and temporal changes in the structure and function of a neuron and/or neuronal networks (Heinemann 2004; Antonio et al. 2016; Klein et al. 2018). Epilepsy patients and experimental models exhibited impairment in memory and cognitive tasks (Baker et al. 2011; Hermann et al. 2021). The real cause of memory and cognitive impairment in epilepsy is still unknown. The impairments have a multidimensional aspect; recurrent seizures, antiepileptic drugs, surgical therapy, and psychosocial matters (Aldenkamp and Bodde 2005; Hoppe et al. 2007; Klein et al. 2018). Many studies show that cognitive impairment already exists at the time of a new diagnosis of epilepsy, which imparts that seizures itself might have generated cognitive impairment (Baker et al. 2011). Neuropathological investigation by many groups revealed neuronal cell loss, astrogliosis, and mossy fiber sprouting in the hippocampus of experimental models as well as in TLE patient brain samples (Comper et al. 2017; Devinsky et al. 2018). But neurodegeneration was not observed in the animal model of febrile seizure, which points out that epileptogenesis can also happen without neuronal loss.
Understanding the cellular and molecular mechanism of impaired memory formation in epilepsy is the key for treating the comorbidities of the disease. At the cellular level, memory is encoded via the changes in synaptic plasticity. The changes in the synaptic plasticity in the neuron and/or neuronal networks attribute to changes in the function of the brain. Activity-dependent synaptic plasticity long-term potentiation (LTP) and long-term depression (LTD) are the extensively studied cellular models of learning and memory (Malenka and Bear 2004; Kemp and Manahan-Vaughan 2007; Collingridge et al. 2010; Sheng and Ertürk 2014; Vose and Stanton 2017; Nicoll 2017) in the central nervous system,(CNS). Impairment in LTP is well documented in epilepsy but the role of LTD is not investigated at the same level in epileptic tissue. This is due to the fact that LTP was regarded as the mechanism behind memory formation and storage. The role of LTD in learning and memory and bidirectional synaptic plasticity is becoming clearer that it is necessary for memory formation and storage in the hippocampus (Kemp and Manahan-Vaughan 2007; Dietz and Manahan-Vaughan 2017). Bidirectional plasticity is essential for tuning plasticity in the brain. How the synaptic changes happening during epileptiform activity contribute to bidirectional plasticity can be an important aspect of investigation. Here using an in vitro epilepsy model, we studied LTD in Sc-CA1 synapses. Investigating LTD in the epileptic model will bring new molecular/synaptic information regarding cognitive impairments in patients with epilepsy.
Methods
All animal experimental procedures were in accordance with the institutional animal ethics committee, SCTIMST, Trivandrum, Kerala, India-SCT/ABS/IAEC-83/18, dated 11/03/2014 and PGIMER, Chandigarh, India-106/IAEC/730 dated 06/02/2020. We used Wistar rats of both sexes aged between P12–P28. The rats were anesthetized with isoflurane, and brains were removed carefully after decapitation. 250–500 µm-thick transverse hippocampal slices were prepared from the brain in ice-cold ACSF continuously bubbled with 95% O2 and 5% CO2 mixture (carbogen) using the vibratome (Tedpella, USA). The slices were incubated in an incubation chamber for 30 min at 32 °C in ACSF containing (in mM): 125 NaCl, 25 NaHCO3, 2.5 KCl, 1.21 NaH2PO4.2H2O, 1 MgSO4, 10 D-glucose, 2 CaCl2, bubbled with carbogen (pH 7.4) (Punnakkal and Dominic 2018). After 30 min of incubation, the slices were kept at room temperature in ACSF and used for the experiments.
High potassium (HK)-ACSF containing (in mM): 125 NaCl, 25 NaHCO3, 7.5 KCl, 1.21 NaH2PO4.2H2O, 10 D-glucose, 2 CaCl2 (pH 7.4) was used to induce epileptiform activities in the brain slices as described earlier (Punnakkal and Dominic 2018). Drugs (AP5, NMDA, TCN-201, Ro 25-6981, and Ifenprodil) were dissolved in ACSF before perfusion. All chemicals used for ACSF were purchased from Merck. TCN 201 was from Sigma. The rest of the drugs were from Abcam. The antagonists AP5, TCN201 (Edman et al. 2012) Ro 25-6981and ifenprodil (Fischer et al. 1997) were perfused with ACSF.
The slices were placed in a recording chamber for the experiments and were kept alive by continuous perfusion of carbogen bubbled ACSF. The flow rate of the perfusion system was maintained at ~ 1.5–2 ml/min. Glass pipettes were made from borosilicate glass capillaries using a PC-10 puller from Narishige, Japan. The glass pipettes filled with ACSF were used for stimulating Sc-pathway and recorded field potentials using AM systems microelectrode amplifier (model 1800). Schaffer collateral pathway was stimulated using AM systems isolated pulse stimulator (model 2100). The LTP was induced by the application of high-frequency stimulus (HFS) (100 Hz stimulus, 3 pulses, 10 s interval) (Malenka and Bear 2004; Müller et al. 2013). The LTD was induced by chemical method (30 µM NMDA application for 3 min, cLTD) or electrically by delivering low-frequency stimulation (LFS) (900 pulses of 1 Hz stimulus) (Kamal et al. 1999; Malenka and Bear 2004; Lanté et al. 2006; Collingridge et al. 2010). The degree of LTD/LTP/depotentiation was measured as an average of the peak amplitude of the last 3 min of the recording post-LTD/LTP/depotentiation protocol. The signals were filtered between 1 Hz to 5 kHz and then acquired using NI PCI 6221 data card. The data were recorded and analyzed using WinWCP V5.5.5 (Strathclyde Electrophysiology Software).
Whole cell patch clamp experiments were done in CA1 pyramidal neurons. Patch pipettes had a resistance of 4–6 MΩ and were filled internal solution containing: (in mM) 130 Cs-methanesulfonate, 5 CsCl, 5 EGTA, 1 Mg-ATP, 0.1 Na-GTP, 10 HEPES, 5 QX-314 (pH 7.4, adjusted with CsOH, 280–300 mOsm). Spontaneous excitatory postsynaptic currents (EPSCs) and inhibitory postsynaptic currents (IPSCs) were recorded from CA1 pyramidal neurons in both control and epileptiform-induced slices. The cells were held at − 70 mV for recording spontaneous EPSCs and at 0 mV for recording spontaneous IPSCs for at least 5 min. A minimum 2 min recording duration was used for analysis. The recordings were made using HEKA double patch, EPC10 amplifier and Patch master software. The signals were filtered at 2.9 kHz and sampled at 10 kHz. The peaks were analyzed using Minianalysis software (Synaptosoft, USA).
In vitro epilepsy model
Hippocampal slices were transferred to the recording chamber, perfused with ACSF bubbled with carbogen. Glass electrode filled with ACSF was placed in the CA1 region and recorded baseline activity. After recording the control activity, epileptiform activity was induced in the slices by perfusing 7.5 mM KCl and zero magnesium containing ACSF (HK-ACSF). We used this model, because the increase in potassium during epilepsy is a natural process and to avoid interference of drugs during plasticity experiments. Epileptiform activity was observed in 8–12 min after the perfusion of HK-ACSF (Fig. 2A) as reported earlier (Punnakkal and Dominic 2018). After confirming a stable epileptiform activity in slices for 15 min, it was shifted to normal ACSF then the slices were studied for synaptic plasticity in the Sc-CA1 synapses.
Statistical analysis
All data were pooled across animals and were presented as mean ± SEM. Statistical significance was evaluated using a two-tailed Student’s t test or ANOVA. ***p < 0.0001, **p < 0.005, *p < 0.05. N represents the number of animals and n the number of slices/experiments.
Results
Epileptiform activity impaired chemical LTD in hippocampal Sc-CA1 synapses
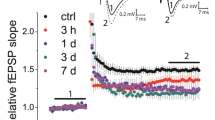
We recorded excitatory postsynaptic field potentials (EPSPs) from stratum radiatum of CA1 region by stimulating the Schaffer collaterals with glass electrodes filled with ACSF. After recording a stable base line EPSPs for 5 min, LTD was induced in the slices by applying NMDA 30 μM for 3 min (Kamal et al. 1999; Malenka and Bear 2004; Huang and Kandel 2007; Collingridge et al. 2010). The application of NMDA induced a stable LTD (Fig. 1, filled circle) (0.81 ± 0.010, n = 7, N = 5, P < 0.001) in control slices and no change in EPSPs was observed in the slices without NMDA application (Fig. 1, open circle) (0.98 ± 0.011, (n = 4, N = 2). We repeated the experiments in slices after observing the epileptiform activity (Punnakkal and Dominic 2018). In epileptiform-induced slices, application of NMDA showed an impaired LTD (Fig. 2C, open circle). Interestingly, we observed a slow onset potentiation lasted during the entire recording period. The LTD-induced in control slices (Fig. 2C, filled circles, LTD data from Fig. 1 for comparison) and epileptiform-induced slices were significantly different (Fig. 2C) (0.81 ± 0.010, n = 7, N = 5 vs 1.21 ± 0.014, n = 7, N = 5, p < 0.0001). Instead of inducing LTD, the synaptic plasticity changed its sign and induced LTP.

Chemical LTD induced in Sc-CA1 synapses in hippocampal slices. Summary of the cLTD induced in Sc-CA1 synapses. Application of 30 μM NMDA for 3 min induced LTD in Sc-CA1 synapses in hippocampal slices (filled circles), whereas no LTD was observed in controls (open circles) without NMDA application. Representative traces (black—before LTD induction, grey—after LTD 25–30 min, averaged traces) show the LTD induction in NMDA perfused slices, but no LTD was observed in control slices. Scale bars 0.2 mV and 5 ms. The bar diagram summarizes the LTD induced in Sc-CA1 synapses. Error bars indicate the standard error of the mean. Values are means ± SEM and statistically significant differences (unpaired t test, two-tailed) are indicated as *** means p < 0.0001

Protocol and summary of cLTD induced in epileptiform-induced slices. A Schematic diagram of the protocol. B Example traces of field recordings, base line and epileptiform activity induced in slices by perfusing HK-ACSF. C cLTD induced in control slices (filled circles). cLTD protocol induced LTP in epileptiform-induced slices (open circles). Representative traces (black—before LTD induction, grey—after LTD induction 25–30 min, averaged traces) show the LTD induction in control slices and LTP in epileptiform-induced slices. Error bars indicate the standard error of the mean. Scale bars 0.2 mV and 5 ms. Bar diagram summarizes the LTD in control and LTP induced in epileptiform induced slices. Values are means ± SEM and statistically significant differences (unpaired t test, two-tailed) are indicated as *** means p < 0.0001
Epileptiform activity impaired depotentiation in the hippocampal slices
Hippocampal Sc-CA1 synapses are known for their bi-directional plasticity (Wagner and Alger 1996). LTP and LTD are shown to exist in the same synapses (Park et al. 2019). Here we induced LTP in Sc-CA1 synapses using high frequency stimulation (100 Hz, 3 times, 10 s interval) in the control slices as well as in epileptiform induced slices (Fig. 3A, filled and open circles, respectively) (1.29 ± 0.010, n = 6, N = 4 vs 1.44 ± 0.042, n = 6, N = 5, p < 0.005) and summary of LTP was shown in Fig. 3B. The LTP induced in epileptic slices showed a significant increase compared to control slices, which was matching with previous reports (Müller et al., 2013). After inducing LTP in control slices for 25 min, we depotentiated the synapses by bath applying 30 μm NMDA for 3 min into the slices. Application of NMDA, depotentiated the synapses to the base line value (Fig. 3A, filled circles) (1.06 + 0.016, n = 6, N = 4) in control slices, but in epileptiform-induced slices NMDA application depotentiated the synapses (for about 10–15 min), but gradually started increasing the amplitude and reversed to the initial potentiation by 25–30 min (Fig. 3A, open circle) (1.23 ± 0.013, n = 6, N = 5, p < 0.0001) and the summary is shown in Fig. 3C. These experiments showed that the epileptiform activity determines the direction of synaptic plasticity.

Summary of the depotentiation in control and epileptiform induced slices. A HFS-induced LTP in control slices, following NMDA application induced complete depotentiation, in control slices (filled circles). In epileptiform-induced slices, HFS showed an increase in LTP, but NMDA application-induced LTD soon potentiated to LTP (open circles). Representative traces (black—before LTP induction, grey—after depotentiation 25–30 min, averaged traces). Error bars indicate the standard error of the mean. Scale bars 0.2 mV and 5 ms. B Bar diagrams summarize the LTP in control slices and epileptiform-induced slices. C Bar diagrams summarize the depotentiation in control slices and impaired depotentiation in epileptiform-induced slices. Values are means ± SEM and statistically significant differences (unpaired t test, two-tailed) are indicated as **p < 0.005, *** means p < 0.0001
LTD induced with 1 Hz, 900 pulses stimulation was abolished in epileptic hippocampal slices
Next, we induced LTD in hippocampal Sc-CA1 using the protocol 1 Hz, 900 pulses (Sajikumar and Frey 2003; Lanté et al. 2006; Collingridge et al. 2010). This protocol applied for 15 min induced comparable LTD in control slices (Fig. 4A, filled circles) (0.77 ± 0.010; n = 5, N = 4). But in slices with epileptiform activity, 1 Hz LFS protocol induced LTP, instead of LTD (Fig. 4A, open circles) (1.28 ± 0.013, n = 10, N = 8, p < 0.0001). These experiments confirm the change in the property of the Sc-CA1 synapses after epileptiform activity (Rajasekaran et al. 2013). This pointed out that epileptiform activity might have changed the excitability of the synapses. To characterize the 1 Hz LTD in Sc-CA1 synapses, we applied NMDA receptor antagonist MK801/AP5, which completely blocked the induction of LTD in the control as well as in the epileptic slices (Fig. 4A, open squares, and 4D) (1.05 ± 0.00, n = 8, N = 5; 1.08 ± 0.01, n = 5, N = 3 for MK801 and AP5, respectively). This has been confirmed that the LTD induced in Sc–CA1 synapses was NMDA receptor dependent (Malenka and Bear 2004; Collingridge et al. 2010).

NMDA receptor subtype-dependent effect on metaplasticity in epileptiform-induced slices. A 1 Hz LFS-induced LTD in control slices (filled circles), LTD induction was blocked in the presence of MK 801 (open squares) and epileptiform-induced slices showed impaired plasticity/LTP (open circles). B Application of TCN 201 (GluN2A antagonist) failed to block the impaired plasticity in epileptiform-induced slices (open triangles) and epileptiform induced change in synaptic plasticity without TCN 201(filled circles). C Perfusion of Ifenprodil (GluN2B antagonist) inhibited/reduced the plasticity change in epileptiform induced slices (open squares) and epileptiform induced change in synaptic plasticity without ifenprodil (closed circles). Representative traces (black— before LTD induction, grey—after LTD 25–30 min, averaged traces). Scale bars 0.2 mV and 5 ms. D Summary of the action of NMDA receptor subtype specific antagonist on epileptiform induced metaplasticity. AP5 pan NMDA receptor antagonist, TCN 201 (TCN) antagonist of GluN2A NMDA receptor subtype. Ifenprodil (IFEN) and Ro 25–6981 (RO) antagonists of GluN2B NMDA receptor subtype. Values are means ± SEM and statistically significant differences (ANOVA) are indicated as *** means p < 0.0001
Change in synaptic plasticity in the epileptiform-activated Sc-CA1 synapses is GluN2B subtype dependent
We next asked the question whether NMDAR subtypes have any role in determining the direction of plasticity. For this purpose, we performed experiments using NMDA receptor subtype specific antagonists TCN 201(GluN2A specific), Ifenprodil and Ro 25-6981 (GluN2B specific) in epileptiform-induced slices. Induction of LTD in the presence of GluN2A-specific antagonist (TCN-201) produced LTP (Fig. 4B, open triangle) (1.24 ± 0.02, n = 4, N = 4). No difference was found when compared with the LTP induced in epileptiform-induced slices via LTD protocol Fig. 4B (closed circle, data from 4A for comparison). This result suggests that the change in plasticity is not caused by GluN2A receptors. To study the contribution of GluN2B, we induced LTD in the presence of Ifenprodil and Ro25-6981. Both the GluN2B antagonists inhibited the LTP induction (Fig. 4C, open squares ifenprodil; closed circle epi-data from 4A for comparison and Fig. 4D) (1.28 ± 0.013 vs 1.11 ± 0.01, n = 5, N = 3, p < 0.0001; 1.28 ± 0.01 vs 1.11 ± 0.02, n = 6, N = 3 p < 0.0001) and it was comparable with AP5 (1.08 ± 0.01, n = 5, N = 3, p < 0.0001) effect in epileptiform-induced slices (Fig. 4D). Our experiments confirmed that GluN2B contributes to the change in the sign of the plasticity.
Epileptiform activity changed the inhibitory synaptic transmission in Sc-CA1 synapses
To study whether the epileptiform activity changed the excitatory and inhibitory synaptic transmission, we conducted single cell patch clamp experiments (Banerjee et al. 2013). Spontaneous excitatory postsynaptic currents (sEPSC) were recorded from CA1 pyramidal neurons at − 70 mv. We found no change in the amplitude (13.7 ± 0.42 pA vs. 13.5 ± 0.35pA, n = 6, N = 5) and inter event interval (1.54 ± 0.122 s vs 1.47 ± 0.115 s, n = 6, N = 5) of the sEPSCs of control and epileptiform-induced slices (Fig. 5A). But when we studied the spontaneous inhibitory postsynaptic currents (sIPSCs) at 0 mV in CA1pyramidal neurons there was a significant reduction in the peak amplitude (25.7 ± 1.20 pA vs 20.3 ± 0.97 pA, n = 5, N = 5, p < 0.005) as well as inter event interval (0.56 ± 0.05 s vs 0.36 ± 0.03 s, n = 5, N = 5, p < 0.005) (Fig. 5B). Picrotoxin perfusion confirmed the IPSCs were GABA A receptor currents. The cumulative probability distribution also showed a significant difference in amplitude (Kolmogorov–Smirnov test, p < 0.005), and inter event interval (Kolmogorov–Smirnov test, p < 0.05) of sIPSCs. These experiments suggested a transformation of inhibitory GABA A currents in the epileptiform activity-induced slices.

Change in the inhibitory currents in CA1 pyramidal neurons after epileptiform activity. A) Spontaneous excitatory postsynaptic currents recorded at − 70 mv (control—black; epileptiform activity-induced slices—grey, shown above). Below, the summary and cumulative distribution of the amplitude and inter-event interval of control (black) and epileptiform activity-induced slices (grey). B) Spontaneous inhibitory postsynaptic currents recorded at 0 mv (control—black; epileptiform activity-induced slices—grey, shown above). Below, the summary and cumulative distribution of the amplitude and inter-event interval of control (black) and epileptiform activity-induced slices (grey) Values are means ± SEM and statistically significant differences (paired t test) are indicated as **p < 0.005, *p < 0.05. The cumulative probability distribution significance is measured as the Kolmogorov–Smirnov test
Discussion
In the present study, we unraveled a new aspect of LTD in the epilepsy model. LTD is a less explored area of research in epilepsy. Our results showed that LTD and depotentiation were abolished/impaired in the hippocampal Sc-CA1 synapses after the epileptiform activity. The data imply that metaplasticity was induced in the synapses during epileptiform activity and inhibited LTD in the Sc-CA1 synapses. The LTD induced in these synapses was NMDA receptor-dependent. Moreover, NMDA receptor subtype GluN2B was responsible for the impaired synaptic plasticity. So, targeting the GluN2B receptors may have a therapeutic value in treating cognitive impairments in patients with epilepsy.
In many epilepsy patients, memory impairments were documented in the first diagnosis itself (Baker et al. 2011). But how the seizures caused this impairment is still not clear. Studies mainly focused on LTP as a cellular memory model, neglecting LTD. Now the involvement of LTD in spatial memory and novelty detection is well recognized (Kemp and Manahan-Vaughan 2007). Here using LTD as a cellular memory model, we studied how epileptiform activity modulated the synaptic plasticity in Sc-CA1 synapses. In our study, LTD was abolished /impaired in epileptiform activity induced slices. In c-LTD, NMDA perfusion in the epileptiform activity-induced slices reduced the amplitude of EPSPs at the beginning but slowly potentiated to LTP at 25–30 min. To confirm the changes in synaptic plasticity in Sc-CA1 synapses, we also studied LFS protocol 1 Hz, 900 pulses, which also induced stable LTD in control slices (Huang and Kandel 2007; Collingridge et al. 2010), but LTD was abolished in epileptiform activity induced slices. These two experiments confirmed that LTD was impaired in Sc-CA1 synapses after epileptiform activity. These experiments clearly showed that how the seizures itself can impair synaptic plasticity and explains the memory impairment in patients with epilepsy.
NMDA receptors are important for Sc-CA1 LTP and LTD (Malenka and Bear 2004; Huang and Kandel 2007). In Sc-CA1 synapses, the polarity of the plasticity depends on the frequency, strength of the stimulations, and amount of calcium passed through the glutamate receptors (Deisseroth et al. 1995; Huang and Kandel 2007). In Sc-CA1 synapses, HFS stimulation produced LTP, whereas LFS produced LTD (Malenka and Bear 2004; Huang and Kandel 2007). Coan et al. 1989 found that a low concentration of magnesium solution blocked LTP using HFS, which is due to the tonic activation of NMDA receptors in low magnesium. In our studies, we used low magnesium to induce epileptiform activity but LTP and LTD experiments were carried out in normal ACSF. In the present study, we found an increase in LTP in epileptiform-induced slices as reported earlier (Müller et al. 2013). The reduced inhibitory GABA A currents explain the increase in LTP. Conditions during mild hypoxia, hypoglycemia, and increased ammonia caused tonic activation of NMDA receptors and impaired LTP (Zorumski and Izumi 2012). We are providing the data regarding LTD inhibition due to epileptiform activity. We have previously shown that the application of NMDA antagonists reduced epileptiform events in Sc-CA1 synapses (Punnakkal and Dominic 2018). The change in plasticity may point to the fact that a change in the excitability of the cells due to epileptiform activity, if so then LTP studies should show an increased LTP in epilepsy models and human studies. But that is not the case. LTP in epilepsy models showed varying results. In many cases, epilepsy reduced the LTP in brain areas (Kryukov et al. 2016; Lenz et al. 2017), and studies also showed an increase or impairment in LTP after epileptic activity (Zhou et al. 2007; Müller et al. 2013). In a study, an organotypic culture model showed impaired LTP and increased LTD after incubating the culture overnight with GABA receptor blockers (Abegg et al. 2004). These variations might be due to the complexity of the disease and to a lesser extent due to the experimental models and protocols used to study LTP. More detailed studies are needed to explain these differences in synaptic plasticity in epilepsy models.
Many studies found that the prior state of the synapses determines the synaptic plasticity (Coan et al. 1989; Huang et al. 1992; Christie and Abraham 1992; Abraham 2008). The previous history of NMDA receptor activation determines the LTP threshold in the hippocampus (Coan et al. 1989). When a weak tetanus (30 Hz) was followed by a high frequency LTP stimulation, LTP was inhibited in the hippocampus (Huang et al. 1992; Abraham 2008). The priming dependent inhibition of LTP was caused by NMDA receptors, adenosine receptors, P38 mitogen activated protein kinases, and phosphatases (Abraham 2008). Increasing the cell excitability via beta adrenergic receptor activation primed the synapses and increased LTP induction in hippocampal synapses (Cohen et al. 1999). We also found that epileptiform activity increased the magnitude of LTP compared to the control slices. In the present study, the increase in LTP might be due to the reduction in the inhibitory GABA A currents (Fig. 5B). LTD studies are comparatively less in metaplasticity. A previous study showed that priming a low frequency stimulation (LFS) followed by LFS protocol in CA1 synapses enhanced LTD in hippocampal synapses (Mockett et al. 2002). In the present study, epileptiform activity impaired LTD in cLTD and abolished LTD in LFS protocol. Instead of inducing stable LTD, both protocols produced LTP. LTP is considered as a model for learning and memory. However, NMDA receptor activation and protein synthesis are some of the prerequisites for considering LTP as a physiological model for memory. In our study, the LTD protocol induced LTP in epileptiform-induced slices, here the LTP induced might not be activating the synapses in a physiological way and thus the observed LTP may not be linked to memory formation. Moreover, it might also interfere with the real mechanisms of synaptic plasticity. Another mechanism that may contribute to the memory impairment was the loss of bidirectional plasticity in epileptiform-induced slices. The change in inhibitory GABA A currents in epileptiform-induced slices was also a factor needed to be counted for the impaired synaptic plasticity. The reduction in the amplitude of sIPSCs refers to a change in presynaptic and/or postsynaptic function. The change in presynaptic quantal release and/or reduced expression of GABA A receptors in postsynaptic membrane contribute to this reduction. Internalization of GABA A is one of the main causes of drug resistant epilepsy (Ragozzino et al. 2005; Goodkin et al. 2007). The presynaptic effect might be due to cannabinoid or opioid receptors. In hippocampus, not all interneurons (GABAergic) are modulated by cannabinoid receptors. The increase in quantal release frequency (inter-event interval) may be an act of compensatory mechanism to protect the cell from over excitation and further damage. More detailed studies are needed to characterize the molecular pathways of LTD in epilepsy models.
The importance of NMDA receptors in learning and memory is well documented in the hippocampus (Sprengel and Single 1999; Tang et al. 1999; Malenka and Bear 2004; Wang et al. 2021). We investigated the role of NMDA receptors in LTD of the in vitro epilepsy model. In control and epileptiform-induced slices, both LFS- and NMDA-induced LTD was blocked by NMDA receptor antagonists MK801/AP5, which confirms the LTD induced was NMDA receptor dependent. GluN1 knock out mice showed impaired synaptic plasticity and memory formation (Tsien et al. 1996). GluN2B overexpression enhanced learning and memory in mice. The GluN2B over expression induced enhanced LTP, but no change in LTD was observed in Sc-CA1 synapses (Tang et al. 1999). To study the impaired synaptic plasticity in epileptiform induced slices, we perfused NMDA receptor subtype specific antagonists. The GluN2B-specific antagonists ifenprodil and Ro 25–6981 blocked the LTP induced by LFS protocol in epileptiform-induced slices. Whereas, the GluN2A specific antagonist TCN 201 failed to block the LTP in epileptiform activity induced slices. These results indicate that the changes in the plasticity in epileptiform activity induced slices were due to the change in the subtype composition of NMDA receptors. Change in the NMDA receptor subtype was reported in epilepsy models (Müller et al. 2013). Our previous study showed that the GluN2B antagonist reduced the epileptiform activity in neonatal hippocampal slices (Punnakkal and Dominic 2018). Many studies explored the role of NMDA receptor subtype activation during synaptic plasticity and found that there is no subtype-dependent contribution to LTP in the hippocampus (Berberich et al. 2005; Weitlauf et al. 2005). LTD was also not found to be NMDA receptor subtype dependent in the hippocampus (Morishita et al. 2007). But studies in the cortex show a subtype dependence on synaptic plasticity (Massey et al. 2004). If LTP and LTD are subtype dependent then increased expression of GluN2B will produce an enhanced LTD in GluN2B overexpressed mice. But in GluN2B overexpressed mice, LTP was increased and no change in LTD was observed (Tang et al. 1999). In our studies, GluN2A antagonist did not block the LTP induced in epileptiform activity induced slices, but GluN2B antagonists blocked the impaired synaptic plasticity, which implies that the GluN2B receptor was responsible for the reversal of synaptic plasticity in the epilepsy model. Our results also confirm that LTD is not subtype specific as reported previously (Morishita et al. 2007). Histopathological and immunohistochemistry of the amygdala of patients with epilepsy failed to show any change in glutamate receptors including NMDA receptor subtypes (Jafarian et al. 2019). Cortical dysplasia (CD) is regarded as one of the reasons behind drug-resistant epilepsy. In cortical slices from CD patients, a study found an increased expression of GluN2B, and the application of GluN2B-specific antagonist suppressed the epileptiform activity in dysplastic slices (Möddel et al. 2005). Animal studies also reported a change in GluN2B expression in epilepsy models. Müller et al. (2013) reported that LTP was increased in Sc-CA1 synapses due to overexpression of GluN2B in the pilocarpine rat model of epilepsy. Epilepsy with depression rats showed a higher level of GluN2B phosphorylation compared to the epilepsy without depression group and treatment with GluN2B blockers rescued the depressive behaviour (Peng et al. 2016). In PTZ kindling model showed an upregulation of GluN2B receptors and increased astrocytosis. Treatment with GluN2B antagonist significantly reduced the PTZ kindled astrocytosis (Zhu et al. 2015).
Impairment in synaptic plasticity was reported in many neuro disorders. LTP was impaired in epilepsy models and patients (Beck et al. 2000). In the disease model of Parkinson s disease and patients, LTD and depotentiation were impaired (Picconi et al. 2003; Huang et al. 2011). The same was true in the present study, depotentiation was induced successfully in control slices, which means that the potentiated EPSPs came back to the base line potentials after the protocol. Epileptiform-induced slices also induced depotentiation in the synapses but potentiated to LTP by 25–30 min. In the human dentate gyrus, a previous study reported impaired depotentiation (Beck et al. 2000), which is in line with our findings. The reduction in inhibitory GABA A currents in epileptiform-induced slices may also contribute to the impairment in depotentiation, but more studies are needed to find out the signaling pathways.
Conclusions
Our results show that metaplasticity was induced in the Sc-CA1 synapses during epileptiform activity. This plasticity inhibited the LTD in Sc-CA1 synapses in epileptiform-induced slices. LTD in control slices and epileptiform-induced slices was NMDA receptor dependent. Moreover, the GluN2B subtype of NMDA receptors was responsible for the impaired synaptic plasticity in epileptiform-induced slices. The present investigation will trigger more studies in the direction of metaplasticity to explain the molecular mechanism in the impairment of memory in patients with epilepsy. This study identified the GluN2B subtype NMDA receptor as a new target for memory impairments in epilepsy.
References
Abegg MH, Savic N, Ehrengruber MU et al (2004) Epileptiform activity in rat hippocampus strengthens excitatory synapses. J Physiol 554:439–448. https://doi.org/10.1113/jphysiol.2003.052662
Abraham WC (2008) Metaplasticity: tuning synapses and networks for plasticity. Nat Rev Neurosci 9:387. https://doi.org/10.1038/nrn2356
Aldenkamp AP, Bodde N (2005) Behaviour, cognition and epilepsy. Acta Neurol Scand Suppl 182:19–25. https://doi.org/10.1111/j.1600-0404.2005.00523.x
Antonio LL, Anderson ML, Angamo EA et al (2016) In vitro seizure like events and changes in ionic concentration. J Neurosci Methods 260:33–44. https://doi.org/10.1016/j.jneumeth.2015.08.014
Baker GA, Taylor J, Aldenkamp AP, SANAD group, (2011) Newly diagnosed epilepsy: cognitive outcome after 12 months. Epilepsia 52:1084–1091. https://doi.org/10.1111/j.1528-1167.2011.03043.x
Banerjee J, Alkondon M, Albuquerque EX, Pereira EFR (2013) Contribution of CA3 and CA1 pyramidal neurons to the tonic α7 nAChR-dependent glutamatergic input to CA1 pyramidal neurons. Neurosci Lett 554:167–171. https://doi.org/10.1016/j.neulet.2013.08.025
Beck H, Goussakov IV, Lie A et al (2000) Synaptic plasticity in the human dentate gyrus. J Neurosci off J Soc Neurosci 20:7080–7086
Berberich S, Punnakkal P, Jensen V et al (2005) Lack of NMDA receptor subtype selectivity for hippocampal long-term potentiation. J Neurosci off J Soc Neurosci 25:6907–6910. https://doi.org/10.1523/JNEUROSCI.1905-05.2005
Christie BR, Abraham WC (1992) Priming of associative long-term depression in the dentate gyrus by θ frequency synaptic activity. Neuron 9:79–84. https://doi.org/10.1016/0896-6273(92)90222-Y
Coan EJ, Irving AJ, Collingridge GL (1989) Low-frequency activation of the NMDA receptor system can prevent the induction of LTP. Neurosci Lett 105:205–210. https://doi.org/10.1016/0304-3940(89)90038-4
Cohen AS, Coussens CM, Raymond CR, Abraham WC (1999) Long-lasting increase in cellular excitability associated with the priming of LTP induction in rat hippocampus. J Neurophysiol 82:3139–3148. https://doi.org/10.1152/jn.1999.82.6.3139
Collingridge GL, Peineau S, Howland JG, Wang YT (2010) Long-term depression in the CNS. Nat Rev Neurosci 11:459–473. https://doi.org/10.1038/nrn2867
Comper SM, Jardim AP, Corso JT et al (2017) Impact of hippocampal subfield histopathology in episodic memory impairment in mesial temporal lobe epilepsy and hippocampal sclerosis. Epilepsy Behav EB 75:183–189. https://doi.org/10.1016/j.yebeh.2017.08.013
Deisseroth K, Bito H, Schulman H, Tsien RW (1995) Synaptic plasticity: a molecular mechanism for metaplasticity. Curr Biol 5:1334–1338. https://doi.org/10.1016/S0960-9822(95)00262-4
Devinsky et al., 2018 Devinsky O, Vezzani A, O’Brien TJ, et al (2018) Epilepsy. Nat Rev Dis Primer 4:18024. https://doi.org/10.1038/nrdp.2018.24
Dietz B, Manahan-Vaughan D (2017) Hippocampal long-term depression is facilitated by the acquisition and updating of memory of spatial auditory content and requires mGlu5 activation. Neuropharmacology 115:30–41. https://doi.org/10.1016/j.neuropharm.2016.02.026
Edman S, McKay S, Macdonald LJ et al (2012) TCN 201 selectively blocks GluN2A-containing NMDARs in a GluN1 co-agonist dependent but non-competitive manner. Neuropharmacology 63:441–449. https://doi.org/10.1016/j.neuropharm.2012.04.027
Fischer G, Mutel V, Trube G et al (1997) Ro 25–6981, a highly potent and selective blocker of N-methyl-D-aspartate receptors containing the NR2B subunit. characterization in vitro. J Pharmacol Exp Ther 283:1285–1292
Goodkin HP, Sun C, Yeh J-L et al (2007) GABAA receptor internalization during seizures. Epilepsia 48:109–113. https://doi.org/10.1111/j.1528-1167.2007.01297.x
Heinemann U (2004) Basic mechanisms of partial epilepsies. Curr Opin Neurol 17:155–159. https://doi.org/10.1097/00019052-200404000-00012
Hermann BP, Struck AF, Busch RM et al (2021) Neurobehavioural comorbidities of epilepsy: towards a network-based precision taxonomy. Nat Rev Neurol. https://doi.org/10.1038/s41582-021-00555-z
Hoppe C, Elger CE, Helmstaedter C (2007) Long-term memory impairment in patients with focal epilepsy. Epilepsia 48(Suppl 9):26–29. https://doi.org/10.1111/j.1528-1167.2007.01397.x
Huang YY, Colino A, Selig DK, Malenka RC (1992) The influence of prior synaptic activity on the induction of long-term potentiation. Science 255:730–733. https://doi.org/10.1126/science.1346729
Huang Y-Y, Kandel ER (2007) Low-frequency stimulation induces a pathway-specific late phase of LTP in the amygdala that is mediated by PKA and dependent on protein synthesis. Learn Mem 14:497–503. https://doi.org/10.1101/lm.593407
Huang Y-Z, Rothwell JC, Lu C-S et al (2011) Abnormal bidirectional plasticity-like effects in parkinson’s disease. Brain J Neurol 134:2312–2320. https://doi.org/10.1093/brain/awr158
Jafarian M, Modarres Mousavi SM, Alipour F et al (2019) Cell injury and receptor expression in the epileptic human amygdala. Neurobiol Dis 124:416–427. https://doi.org/10.1016/j.nbd.2018.12.017
Kamal A, Ramakers GM, Urban IJ et al (1999) Chemical LTD in the CA1 field of the hippocampus from young and mature rats. Eur J Neurosci 11:3512–3516. https://doi.org/10.1046/j.1460-9568.1999.00769.x
Kemp A, Manahan-Vaughan D (2007) Hippocampal long-term depression: master or minion in declarative memory processes? Trends Neurosci 30:111–118. https://doi.org/10.1016/j.tins.2007.01.002
Klein P, Dingledine R, Aronica E et al (2018) Commonalities in epileptogenic processes from different acute brain insults: do they translate? Epilepsia 59:37–66. https://doi.org/10.1111/epi.13965
Kryukov KA, Kim KK, Magazanik LG, Zaitsev AV (2016) Status epilepticus alters hippocampal long-term synaptic potentiation in a rat lithium-pilocarpine model. NeuroReport 27:1191–1195. https://doi.org/10.1097/WNR.0000000000000656
Lanté F, Cavalier M, Cohen-Solal C et al (2006) Developmental switch from LTD to LTP in low frequency-induced plasticity. Hippocampus 16:981–989. https://doi.org/10.1002/hipo.20228
Lenz M, Ben Shimon M, Deller T et al (2017) Pilocarpine-induced status epilepticus is associated with changes in the actin-modulating protein synaptopodin and alterations in long-term potentiation in the mouse hippocampus. Neural Plast 2017:2652560. https://doi.org/10.1155/2017/2652560
Malenka RC, Bear MF (2004) LTP and LTD: an embarrassment of riches. Neuron 44:5–21. https://doi.org/10.1016/j.neuron.2004.09.012
Massey PV, Johnson BE, Moult PR et al (2004) Differential roles of NR2A and NR2B-containing NMDA receptors in cortical long-term potentiation and long-term depression. J Neurosci off J Soc Neurosci 24:7821–7828. https://doi.org/10.1523/JNEUROSCI.1697-04.2004
Mockett B, Coussens C, Abraham WC (2002) NMDA receptor-mediated metaplasticity during the induction of long-term depression by low-frequency stimulation. Eur J Neurosci 15:1819–1826. https://doi.org/10.1046/j.1460-9568.2002.02008.x
Möddel G, Jacobson B, Ying Z et al (2005) The NMDA receptor NR2B subunit contributes to epileptogenesis in human cortical dysplasia. Brain Res 1046:10–23. https://doi.org/10.1016/j.brainres.2005.03.042
Morishita W, Lu W, Smith GB et al (2007) Activation of NR2B-containing NMDA receptors is not required for NMDA receptor-dependent long-term depression. Neuropharmacology 52:71–76. https://doi.org/10.1016/j.neuropharm.2006.07.005
Müller L, Tokay T, Porath K et al (2013) Enhanced NMDA receptor-dependent LTP in the epileptic CA1 area via upregulation of NR2B. Neurobiol Dis 54:183–193. https://doi.org/10.1016/j.nbd.2012.12.011
Nicoll RA (2017) A brief history of long-term Potentiation. Neuron 93:281–290. https://doi.org/10.1016/j.neuron.2016.12.015
Park P, Sanderson TM, Bortolotto ZA et al (2019) Differential sensitivity of three forms of hippocampal synaptic potentiation to depotentiation. Mol Brain 12:30. https://doi.org/10.1186/s13041-019-0451-6
Peng W-F, Ding J, Li X et al (2016) N-methyl-D-aspartate receptor NR2B subunit involved in depression-like behaviours in lithium chloride-pilocarpine chronic rat epilepsy model. Epilepsy Res 119:77–85. https://doi.org/10.1016/j.eplepsyres.2015.09.013
Picconi B, Centonze D, Håkansson K et al (2003) Loss of bidirectional striatal synaptic plasticity in L-DOPA-induced dyskinesia. Nat Neurosci 6:501–506. https://doi.org/10.1038/nn1040
Punnakkal P, Dominic D (2018) NMDA receptor GluN2 subtypes control epileptiform events in the hippocampus. Neuromolecular Med 20:90–96. https://doi.org/10.1007/s12017-018-8477-y
Ragozzino D, Palma E, Di Angelantonio S et al (2005) Rundown of GABA type a receptors is a dysfunction associated with human drug-resistant mesial temporal lobe epilepsy. Proc Natl Acad Sci U S A 102:15219–15223. https://doi.org/10.1073/pnas.0507339102
Rajasekaran K, Joshi S, Kozhemyakin M et al (2013) Receptor trafficking hypothesis revisited: plasticity of AMPA receptors during established status epilepticus. Epilepsia 54:14–16. https://doi.org/10.1111/epi.12266
Sajikumar S, Frey JU (2003) Anisomycin inhibits the late maintenance of long-term depression in rat hippocampal slices in vitro. Neurosci Lett 338:147–150. https://doi.org/10.1016/S0304-3940(02)01400-3
Sheng M, Ertürk A (2014) Long-term depression: a cell biological view. Philos Trans R Soc Lond B Biol Sci 369:20130138. https://doi.org/10.1098/rstb.2013.0138
Sprengel R, Single FN (1999) Mice with genetically modified NMDA and AMPA receptors. Ann N Y Acad Sci 868:494–501. https://doi.org/10.1111/j.1749-6632.1999.tb11318.x
Stafstrom CE, Carmant L (2015) Seizures and epilepsy: an overview for neuroscientists. Cold Spring Harb Perspect Med 5:a022426. https://doi.org/10.1101/cshperspect.a022426
Tang YP, Shimizu E, Dube GR et al (1999) Genetic enhancement of learning and memory in mice. Nature 401:63–69. https://doi.org/10.1038/43432
Tripathi M, Jain DC, Devi MG et al (2012) Need for a national epilepsy control program. Ann Indian Acad Neurol 15:89–93. https://doi.org/10.4103/0972-2327.94989
Tsien JZ, Huerta PT, Tonegawa S (1996) The Essential role of hippocampal CA1 NMDA receptor-dependent synaptic plasticity in spatial memory. Cell 87:1327–1338. https://doi.org/10.1016/S0092-8674(00)81827-9
Vose LR, Stanton PK (2017) Synaptic plasticity, metaplasticity and depression. Curr Neuropharmacol 15:71–86. https://doi.org/10.2174/1570159X14666160202121111
Wagner JJ, Alger BE (1996) Homosynaptic LTD and depotentiation: do they differ in name only? Hippocampus 6:24–29. https://doi.org/10.1002/(SICI)1098-1063(1996)6:1%3c24::AID-HIPO5%3e3.0.CO;2-7
Wang J, Han J, Wang S et al (2021) Forebrain GluN2A overexpression impairs fear extinction and NMDAR-dependent long-term depression in the lateral amygdala. Brain Res Bull. https://doi.org/10.1016/j.brainresbull.2021.05.023
Weitlauf C, Honse Y, Auberson YP et al (2005) Activation of NR2A-containing NMDA receptors is not obligatory for NMDA receptor-dependent long-term potentiation. J Neurosci off J Soc Neurosci 25:8386–8390. https://doi.org/10.1523/JNEUROSCI.2388-05.2005
Zhou J-L, Shatskikh TN, Liu X, Holmes GL (2007) Impaired single cell firing and long-term potentiation parallels memory impairment following recurrent seizures. Eur J Neurosci 25:3667–3677. https://doi.org/10.1111/j.1460-9568.2007.05598.x
Zhu X, Dong J, Shen K et al (2015) NMDA receptor NR2B subunits contribute to PTZ-kindling-induced hippocampal astrocytosis and oxidative stress. Brain Res Bull 114:70–78. https://doi.org/10.1016/j.brainresbull.2015.04.002
Zorumski CF, Izumi Y (2012) NMDA receptors and metaplasticity: mechanisms and possible roles in neuropsychiatric disorders. Neurosci Biobehav Rev 36:989–1000. https://doi.org/10.1016/j.neubiorev.2011.12.011
Acknowledgements
We thank Dr. Shobi Veleri and Ms. Chahat Pathania for critically reading the manuscript. Dr. John Dempster (University of Strathclyde, Glasgow) for the software. This work was supported by the Department of Biotechnology, Govt. of India (Ramalingaswami Fellowship to PP, No. BT/RLF/Re-entry/04/2012).
Author information
Authors and Affiliations
Contributions
PP conceived, planned the experiments, analyzed data, and wrote the manuscript. AS, RN, and ZSR, carried out the experiments and analyzed the data. All authors read the manuscript and commented on it.
Corresponding author
Ethics declarations
Conflict of interest
The authors have no conflicts of interest. Data of the manuscript will be made available on request.
Additional information
Communicated by Sreedharan Sajikumar.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Nasarudeen, R., Singh, A., Rana, Z.S. et al. Epileptiform activity induced metaplasticity impairs bidirectional plasticity in the hippocampal CA1 synapses via GluN2B NMDA receptors. Exp Brain Res 240, 3339–3349 (2022). https://doi.org/10.1007/s00221-022-06486-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00221-022-06486-5