
Abstract
The in vitro antioxidant activity and the protective effect against human low density lipoprotein oxidation of coffees prepared using different degrees of roasting was evaluated. Coffees with the highest amount of brown pigments (dark coffee) showed the highest peroxyl radical scavenging activity. These coffees also protected human low-density lipoprotein (LDL) against oxidation, although green coffee extracts showed more protection. In a different experiment, coffee extracts were incubated with human plasma prior to isolation of LDL particles. This showed, for the first time, that incubation of plasma with dark, but not green coffee extracts protected the LDL against oxidation by copper or by the thermolabile azo compound AAPH. Antioxidants in the dark coffee extracts must therefore have become associated with the LDL particles. Brown compounds, especially those derived from the Maillard reaction, are the compounds most likely to be responsible for this activity.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Coffee is one of the most widely consumed beverages in the world. Two species have commercial importance: Coffea arabica (commonly known as arabica coffee) and Coffea canephora (commonly known as robusta coffee). Green coffee beans contains important amounts of phenolic compounds, mainly chlorogenic acid (CGA), accounting for up to 10% of the weight of green coffee [1]. During the roasting process, which is necessary to develop the typical sensory properties of coffee, the beans undergo profound changes in chemical composition, mostly as a result of the Maillard reaction [2, 3]. The major compositional changes occurring are the decrease of phenolic compounds and the formation of brown, water-soluble polymers called melanoidins, although decrease in protein, amino acids and other compounds is also described [1].
Traditionally, coffee has been consumed throughout the world for its pleasant flavor and aroma as well as its stimulatory properties due to its caffeine content. In addition, in recent years there has been an increasing interest in the possible positive implications of coffee consumption for human health [4]. It is well known that brewed coffee has very strong antioxidant activity due to the presence of phenolic compounds and melanoidins [5–9]. Depending on the intensity of the roasting process, the influence of one or other group of compounds on the overall antioxidant activity is different. While phenolic compounds are more important under milder roasting conditions, a more intense treatment decreases the concentration of these compounds and Maillard reaction products (MRPs), especially melanoidins, become the most important contributors [5–9].
Despite the above cited studies reporting the antioxidant activity of coffee, there are only two studies investigating the protective effect of coffee brews against oxidation of human low-density lipoprotein (LDL) [10, 11]. Oxidation of LDL increases the risk of atherosclerosis, the underlying cause of cardiovascular diseases, including coronary heart disease and stroke [12, 13]. Cardiovascular diseases accounts for almost half (48 %) of deaths in Europe and is thus a major health issue (http://www.dphpc.ox.ac.uk/bhfhprg). Therefore, better understanding of the mechanisms related with the defense of lipoprotein oxidation including those involving dietary components is of great interest. Coffee compounds may act as free radical scavengers in plasma or interstitial fluid, or they may be incorporated into lipoproteins.
In this study, we evaluated the antioxidant activity of coffee with several roasting degrees by using different methods of assessing antioxidant activity, namely in vitro LDL oxidation, the oxygen radical absorbance capacity (ORAC) assay and the 2,2′-azinobis(3-ethylbenzothiazoline-6-sulfonic acid) (ABTS) decolorization assay. In addition, we assessed the incorporation of coffee components into human LDL particles and evaluated whether the incorporated compounds may protect human lipoproteins against oxidation promoted by different pro-oxidants. Plasma was spiked with different volumes of coffee extracts prior to isolation of LDL particles; then LDL oxidation was promoted with different prooxidants. This work builds on our previous research [7, 8] showing that coffee brews, regardless of their roasting degree, are able to exert antioxidant activity.
Materials and methods
Chemical and reagents
All chemicals used throughout the study were obtained from Sigma (Gillingham, U.K.).
Coffee samples
Brazilian Arabica coffee beans were roasted for 180 s at different temperatures: 270, 290 and 300 °C to produce light-, medium- and dark-roasted samples, respectively. Green coffee beans from the same origin were also used. The different beans were supplied by Kraft Foods (Munich, Germany).
Preparation of coffee extracts
Green and roasted beans were ground for 1 min in a Moulinex (Paris, France) coffee grinder model Super Junior “S”. Coffee extracts were prepared using ground beans (15 g) and hot water (250 ml at 90 °C) in a commercial automatic coffee brewing machine. Coffee extracts prepared in triplicate were stored for less than one month at −20 °C before analysis.
Capillary zone electrophoresis (CZE) analysis of coffee samples
The method was based on that of Ames et al. [14]. Separations used a Hewlett-Packard (subsequently Agilent, Bracknell, U.K.) 3D capillary electrophoresis instrument equipped with Chemstation software. The capillary was 48.5 cm long (40 cm to the detector) with an internal diameter of 50 μm and a ×3 bubble cell. Other conditions of analysis were as follows: buffer, 50 mM borate (pH 9.5); voltage, 20 kV; temperature of analysis, 25 °C; injection, 50 mbar for 5 s; electroosmotic flow (EOF) marker, benzyl alcohol. Electropherograms (e-grams) were monitored at 200 and 420 nm, and spectra were collected from 190 to 600 nm. The capillary was conditioned after each sample run by flushing for 3 min with 0.1 M NaOH and for 3 min with buffer. Samples were analyzed in duplicate.
ABTS•+ decolorization assay
The assay was carried out according to Re et al. [15]. The ABTS•+ chromophore was produced by the oxidation of 7 mM ABTS with potassium persulfate (2.45 mM final concentration) in water. Before performing experiments, the radical was diluted with phosphate buffered saline (PBS) at pH 7.4 to give an absorbance of 0.7 ± 0.02 at 734 nm. Tenfold diluted coffee extracts (20 μL) were combined with 2 mL of ABTS•+ reagent and the absorbance monitored for 10 min at 30 °C using a Lambda Bio 20 UV/Vis spectrophotometer (Perkin-Elmer, Cambridge, U. K.). Readings taken after 5 min were used to calculate antioxidant activity. Trolox, the water-soluble analog of vitamin E, was used as a reference standard. A standard curve was prepared by measuring the percent inhibition values at different concentrations of Trolox. Inhibition values were calculated as follows:
The Trolox equivalent antioxidant capacity (TEAC) of each coffee extract represents the concentration of Trolox with the same antioxidant capacity as the extract. Triplicate coffee extracts of each sample were prepared and the activity of each was measured in duplicate.
ORAC assay
The method of Ou et al. [16] was used with minor modifications as described below. The reaction was carried out in 75 mM phosphate buffer (pH 7.4) and the final reaction volume was 3 mL. Into a cuvette was placed 2.25 mL of fluorescein (100 μM in 75 mM phosphate buffer) and 375 μL of coffee extract. The mixture of sample and fluorescein was incubated at 37 °C for 10 min before adding 375 μL of AAPH (150 in 75 mM phosphate buffer). The fluorescence was recorded every 5 min until the final value was less than 10% of the initial fluorescence value. During the experiments, solutions were kept at 37 °C in a water bath and only removed to perform the measurements. Phosphate buffer (75 mM) was used as a blank. To calculate ORAC values, the area under the fluorescence decay curve (AUC) was determined for Trolox at different concentrations to create a calibration curve. The AUC values were calculated as follows:
where f 0 is the initial fluorescence reading at 0 min and f i is the fluorescence reading at time i. For each batch of samples, a standard of Trolox (30 μM) was analyzed as a quality control. The relative ORAC activity (Trolox equivalents) of each coffee sample was calculated as:
Three different coffee extracts of each sample were prepared and their activity was determined in duplicate.
Rapid LDL isolation
LDL (1.019–1.063 g/mL) was isolated by sequential density ultracentrifugation from normal human blood in the presence of EDTA [17]. Briefly, after obtaining the plasma by centrifugation (800g for 30 min), the density was adjusted to 1.21 g/mL by the addition of KBr and overlaid with a solution of density 1.006 g/mL. A first ultracentrifugation step was carried out using a Beckman NVT 65-near vertical rotor at 402,000 g for 50 min at 4 °C. The orange band was collected and the density adjusted to 1.15 g/mL with high-density solution (2.62 M NaCl, 2.98 M KBr and 297 μM EDTA). After overlaying this band with a solution of 1.063 g/mL, a second ultracentrifugation step was carried out at 40,200 g for 3 h at 4 °C. The aims of this second step were to remove contaminating albumin and to concentrate the LDL. Finally, the LDL was collected from the top of the tubes and dialyzed overnight against phosphate buffer (NaCl 140 mM, Na2HPO4 8.1 mM, NaH2PO4 1.9 mM and EDTA 100 μM) to remove KBr. LDL protein was measured according to the method of Lowry et al. [18] using bovine serum albumin as standard.
LDL oxidation
LDL (50 μg protein/mL) was oxidized in PBS (Gibco, Paisley, U. K.) at 37 °C either with CuSO4 (usually 5 μM net above the concentration of EDTA carried over from the storage buffer, which was below 1 μM) or AAPH (final concentration 1 mM). The oxidation of LDL was performed in the absence (control) or presence of the different coffee extracts tested in this study. Coffee extracts were diluted in PBS (1/10) and 10 μL was placed in a cuvette together with copper/AAPH and LDL (final volume of 2 mL). The kinetics of LDL oxidation was determined by monitoring the change of absorbance at 234 nm with a Lambda Bio 20 UV/Vis (Perkin-Elmer, Cambridge, U. K.) spectrophotometer equipped with an eight position automated sample changer. Three different coffee extracts of each sample were prepared and their activity was determined in duplicate.
The time needed to reach 50% of the maximal amount of conjugated dienes was used.
Plasma incubation
Pooled plasma (5 mL) from healthy individuals was incubated at 37 °C for 3 h with the green coffee and dark coffee extracts. The incubations were planned to obtain a final concentration of 1% and 10 % (v/v) of coffee extracts in plasma. Experiments were performed in duplicate with PBS as control. LDL was isolated and oxidized by AAPH or CuSO4 at 37 °C, as described above.
Statistical analysis
Statistical analysis was performed by one-way analysis of variance (ANOVA) to compare means of more than two groups using Statgraphics plus for Windows, version 5.1. (Manugistics, Maryland, USA).
Results and discussion
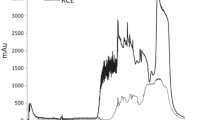
The different coffee extracts were analyzed by CZE and the e-grams obtained at 200 nm are shown in Fig. 1. Five different peaks possessed a UV-diode array spectrum matching that of CGA. Based on the migration time and the spectrum of the available standard, the main isomer of CGA was identified as 3-caffeoylquinic acid with a migration time of around ten minutes. Vanillic and ferulic acids were also detected but their peaks were much smaller than those for CGA isomers. As the roasting process intensified, CGA isomer peaks decreased significantly. In the dark coffee extracts, the total phenolic compound area was only 2.5% of that for the green coffee extracts. This agrees with Borrelli et al. [19] who also reported that phenolic compounds were almost absent from dark-roasted coffees. At the same time, the increase of the roasting temperature caused a remarkable generation of compounds absorbing at 420 nm. This absorbance is characteristic of the development of brown compounds resulting from the Maillard reaction, especially melanoidins. This increase of brown compounds was directly related to the decrease of phenolic compounds (Fig. 2), in line with previous reports [3, 7].

E-grams (200 nm) of coffee extracts. CGA Chlorogenic acid isomers, FA ferulic acid, VA vanillic acid

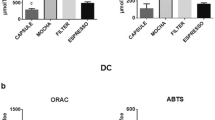
Area at 200 nm (phenolic compounds) and at 420 nm (brown pigments) in the different coffee extracts analyzed by CZE
The antioxidant activity of the coffee extracts determined by ABTS•+ and ORAC assays is given in Table 1. No significant differences were found among the aqueous coffee extracts by the ABTS•+ assay. These results differ from those previously reported by ourselves and other laboratories using the same method [7, 20]. The different findings may be caused by differences in the origin of the beans together with the subsequent roasting process and coffee preparation [21, 22]. For instance, beans from Brazil were used in this study, Del Castillo et al. [7] used Colombian beans and Delgado-Andrade et al. [20] studied instant coffee.
In contrast with the ABTS•+ assay, the ORAC assay showed that dark coffee extracts possessed significantly higher antioxidant activity than the rest of the coffee extracts (Table 1). The ORAC method provides information on the ability of the different samples to scavenge peroxyl radicals, one of the most harmful radicals that can be generated inside the organism. As it can be observed in Fig. 2, brown compounds were completely absent from green coffee extracts while phenolic compounds were barely detected in dark coffee extracts. It seems that brown compounds present in dark coffee extracts are significantly more effective at scavenging peroxyl radicals than phenolic compounds present in green coffee extracts. Among the different brown compounds detected in coffee beverages, melanoidins are the most abundant, accounting for up to 25% of the dry matter [23]. Antioxidant activity of melanoidins has been well characterized using different methods, including those involving scavenging of peroxyl radicals [5, 9, 19, 24]. Other MRPs such as heterocyclic compounds (thiophenes, thiazole, oxazoles, pyrroles, pyrazines, imidazoles and furans) have been identified in coffee [25]. Although these compounds were shown to exhibit antioxidant activity, their levels in brewed coffee range from only micrograms to milligrams per kilogram [26, 27]. Therefore, melanoidins might be considered the main responsible of the peroxyl radical scavenging activity found in the dark coffee extracts.
The differing results obtained by means of ORAC and ABTS•+ methods show that the antioxidant activity cannot be evaluated satisfactorily by a single antioxidant test, especially when a mixture of different antioxidant compounds is present, as with coffee. To obtain more physiologically relevant information concerning the antioxidant effect of the coffee extracts and their possible protection against atherosclerosis, their ability to protect LDL oxidation by copper or AAPH was assessed.
Data showing the ability of the coffee extracts to protect LDL against oxidation are shown in Table 2. LDL particles were more sensitive to oxidation when the pro-oxidant was copper than when AAPH was used. For copper-mediated oxidation, green coffee exhibited significantly higher antioxidant activity than the other coffees. Green coffee was able to prolong the lag phase by almost 1,000 min. Most of the antioxidant activity of green coffee is attributed to the phenolic compounds present in this beverage, especially CGA. Experiments carried out in our laboratory (unpublished results) and those reported in the literature show the protective effect of CGA against LDL oxidation [28]. When the roasting process was applied, the decrease in phenolic compounds was accompanied by a reduction of antioxidant activity. This observation is in line with results from Richelle et al. [10] who analyzed coffee with different degrees of roasting using similar conditions to those employed in the current study.
AAPH-mediated oxidation of LDL provided further evidence of the important role of MRPs as antioxidants. Green coffee extracts were again the most active samples, but this time followed by dark and medium coffee extracts. Light and medium coffee extracts showed no significant differences among them (Table 2). In this case the disappearance of phenolics during the roasting process did not imply as important loss of activity as was the case for copper-mediated oxidation. MRPs generated during the roasting process seem to have the ability to minimize the impact provoked by loss of phenolic compounds and protect LDL from oxidation when the peroxyl radical is the pro-oxidant. The explanation for this dissimilar behavior when LDL oxidation is promoted by different pro-oxidants is based on the different mechanisms involving each experiment. When AAPH is used, the inhibition of LDL oxidation is due almost exclusively to the quenching of peroxyl radicals. MRPs present in dark coffee exhibit a high capacity to scavenge peroxyl radicals (Table 1). Therefore, despite the decrease of phenolic compounds in roasted coffees, an effective protective effect against LDL oxidation can be expected due to the generation of MRPs in these samples.
When the oxidation is mediated by copper, the disappearance of phenolic compounds (mainly CGA) upon roasting seems to be the most important factor in explaining the loss of antioxidant activity for LDL (Table 2). Copper has been suggested to decompose preexisting lipid hydroperoxides in the LDL particle to generate peroxyl, alkoxyl or hydroxyl radicals which oxidize the LDL [29, 30]. Different mechanisms may be involved in the defense of LDL against oxidation by copper compared to AAPH, as regards the capacity to chelate copper to the capacity to scavenge various radicals [31].
Phenolic compounds bearing ortho-dihydroxyl groups have been reported to possess a protective effect against LDL oxidation [28]. CGA and caffeic acid share these groups, and the latter compound has been shown to act not only as a metal chelator but also as a peroxyl and alkoxyl scavenger. Furthermore, it can spare and protect the α-tocopherol and β-carotene present in the LDL particles [31, 32]. All these properties may also be shown by CGA. In fact, when copper was used in great excess (100 μM) the extension of the lag phase was the same for all the coffee extracts (results not shown). This demonstrates that CGA is able to inhibit LDL oxidation by mechanisms other than copper chelation.
Having established the importance of MRPs as in vitro antioxidant components of roasted coffee and their ability to protect against LDL oxidation, we tried to assess the ability of coffee components to be incorporated into LDL particles. Based on the CZE results, green and dark coffee extracts were selected as representatives of phenolic- and MRP-rich coffees, respectively. Plasma was incubated with both 1 and 10% (v/v) of these two coffee extracts. Although 10% (v/v) of coffee might seem at first sight a high concentration, it should be borne in mind that plasma circulating through the gastrointestinal tract may well encounter a higher local concentration of absorbed coffee components than elsewhere in the body and might bind some of these components before entering the general circulation.
The incubation of the two coffee extracts with plasma (coffee extract:plasma = 1:99, v:v) and the subsequent isolation of the LDL particles did not significantly affect the lag time of the LDL oxidation mediated either by copper or AAPH. However, the result was different after the incubation of the dark roasted coffee extracts with plasma at a ratio of 10:90 (v:v). The lag time of the oxidation of the isolated LDL particles was extended by 25 ± 2 min (n = 4) comparing with the control when copper was used as pro-oxidant (Fig. 3a). The incubation of green coffee with plasma at a ratio of 10:90 (v:v) did not have any effect in the same experiment (results not shown). This is clear evidence that some compounds from the dark coffee bind to the lipoproteins, and remain bound during LDL isolation and act as antioxidants. Taking into account the composition of the coffee extracts shown by CZE it may be concluded that the coffee compounds associated with the LDL particles are formed as a result of the roasting process.

Effect of pre-incubation of plasma with dark coffee extract on the LDL oxidation by copper (a) or AAPH (b). LDL was isolated after the incubation of plasma with 10% (v/v) of dark coffee extract or with 10% (v/v) PBS (control) and oxidized by 5 μM CuSO4 or 1 mM AAPH
Since melanoidins are known to possess antioxidant activity and they are the most abundant MRPs detected in coffee beverages [23], they are prime candidates for incorporation into the LDL particles. As said above, other MRPs such as heterocyclic compounds are detected in too small amounts to consider them crucial in the antioxidant activity of dark coffee. To our knowledge, this is the first time that the incorporation of MRPs into human lipoproteins has been reported. In addition, when AAPH was used as pro-oxidant, the time to reach 50% of the maximum production of conjugated dienes increased 45 ± 11 min (n = 4) comparing to the control (Fig. 3b).
It is well known that the generation of MRPs involves the condensation of the carbonyl group of reducing sugars with the free amino group of amino acids and proteins. In addition, some phenolic compounds also participate in the Maillard reaction and become part of the MRPs, including the melanoidins [33]. However, knowledge about the chemical structure and composition of coffee melanoidins is still in its infancy and no information is available on the nature of the phenolic compounds incorporated, although for coffee the main candidate is CGA.
The mechanisms by which melanoidins might interact with human lipoproteins are also unclear as well as the fractions that are mainly responsible for the antioxidant activity. Melanoidins with different molecular masses are detected in coffee [7, 19, 20]. The low water-solubility of high molecular weight melanoidins (>100 kDa) could contribute to the incorporation of this fraction into the lipids of lipoproteins. On the other hand, the interaction of smaller melanoidins with the protein moiety of the lipoproteins, especially with apo B-100, cannot be ruled out. Phenolic compounds have been reported to bind to lipoproteins by ionic interactions involving charged residues on the surface of the particles [34]. The incorporation of phenolic moieties in the melanoidin structure could promote this kind of interaction between melanoidins and lipoproteins. In addition, these phenolics seem to have a key role in the antioxidant activity of melanoidins. Delgado-Andrade et al. [20] detected a high antioxidant activity in coffee melanoidins by using different methods. However, when the low molecular weight compounds that were non-covalently bound to the melanoidin fraction were removed, the antioxidant activity of the melanoidins decreased significantly.
In conclusion, coffee brews possessed strong antioxidant activity at all degrees of roasting. Data reported here demonstrate the importance of coffee MRPs as antioxidant compounds in dark coffee extracts by several in vitro methods including those with human lipoproteins. Although other MRPs such as heterocyclic compounds could be also involved, melanoidins are the compounds most likely to be responsible for this activity. In particular, data are presented for the first time showing that MRPs present in dark coffee are incorporated into LDL and provide protection against oxidation. Regarding green coffee, the relevance of the plasma incubation based model could be of course limited as the main phenolic compound in green coffee (i.e. CGA) is very poorly absorbed. However, several studies show that low-molecular-weight MRPs including melanoidins are absorbed to a certain extent (for a review see Somoza, [35]). Therefore, the results obtained for the incubation of plasma with dark coffee extracts might be used as the starting point for further research. Future work should use dietary intervention studies to investigate if any components of coffee are incorporated into LDL in vivo and to find out which coffee antioxidants formed during roasting are absorbed and metabolized and, thus, likely to cause health effects.
References
Parliment TH, Ho CT, Schieberle P (2000) In ACS Symposium Series 754, American Chemical Society, Washington DC, pp 188–201
Czerny M, Mayer F, Grosch W (1999) J Agric Food Chem 47:695–699
Friedman M (1996) J Agric Food Chem 44:631–653
Higdon JV, Frei B (2006) Crit Rev Food Sci Nutr 46:101–123
Nicoli MC, Anese M, Manzocco L, Lerici CR (1997) Lebensm Wiss Technol 30:292–297
Daglia M, Papetti A, Gregotti C, Berté F, Gazzani G (2000) J Agric Food Chem 48:1449–1454
Del Castillo MD, Ames JM, Gordon MH (2002) J Agric Food Chem 50:3698–3703
Del Castillo MD, Ames JM, Gordon MH (2005) Eur Food Res Technol 221:471–477
Delgado-Andrade C, Morales FJ (2005) J Agric Food Chem 53:1403–1407
Richelle M, Tavazzi I, Offord E (2001) J Agric Food Chem 49:3438–3442
Vinson JA, Jang J, Yang J, Dabbagh Y, Liang X, Serry M, Proch J, Cai S (1999) J Agric Food Chem 47:2502–2504
Baynes JW, Dominiczak MH (1999) In: Baynes JW (eds) Medical Biochemistry. Mosby, London, pp 139–155
Chisolm GM, Steinberg D (2000) Free Radical Biol Med 28:1815–1826
Ames JM, Royle L, Bradbury GW (2000) In ACS Symposium Series 754, American Chemical Society, Washington DC, pp 364–373
Re R, Pellegrini N, Proteggente A, Pannala A, Yang M, Rice-Evans C (1999) Free Radical Biol Med 26:1231–1237
Ou B, Hampsch-Woodill M, Prior RL (2001) J Agric Food Chem 49:4619–4626
Vieria OV, Laranjinha JAN, Madeira VMC, Almeida LM (1996) J Lipid Res 37:2715–2721
Lowry OH, Rosenbergh NJ, Farr AL, Randall LJ (1951) J Biol Chem 193:265–275
Borrelli RS, Visconti A, Mennella C, Anese M, Fogliano V (2002) J Agric Food Chem 50:6527–6533
Delgado-Andrade C, Rufián-Henares JA, Morales FJ (2005) J Agric Food Chem 53:7832–7836
Sánchez-Gónzalez I, Jiménez-Escrig A, Saura-Calixto F (2005) Food Chem 90:133–139
Parras P, Martínez-Tomé M, Jiménez AM, Murcia MA (2007) Food Chem 102:582–592
Belitz DH, Grosch W (1999) Food Chemistry. Springer, Berlin
Morales FJ, Jiménez-Pérez S (2004) Eur Food Res Technol 218:515–520
Flament I, Chevalier C (1988) Chem Ind:592–596
Fuster MD, Mitchell AE, Ochi H, Shibamoto T (2000) J Agric Food Chem 48:5600–5603
Yanagimoto K, Lee KG, Ochi H, Shibamoto T (2002) J Agric Food Chem 50:5480–5484
Cheng C-J, Dai F, Zhou B, Yang L, Liu Z-L (2007) Food Chem 104:132–139
Esterbauer H, Gebiki J, Puhl H, Jurgens G (1992) Free Radical Biol Med 13:341–390
Burkitt MJ (2001) Arch Biochem Biophys 394:117–135
Chimi H, Cillard J, Cillard P, Rahmani M (1991) J Am Oil Chem Soc 68:307–312
Nardini M, D’Aquino M, Tomassi G, Gentili V, Di Felice M, Scaccini C (1995) Free Radic Biol Med 19:541–552
Nunes MF, Coimbra MA (2001) J Agric Food Chem 49:1773–1782
Manach C, Scalbert A, Morand C, Rémésy C, Jiménez L (2004) Am J Clin Nutr 79:727–747
Somoza V (2005) Mol Nutr Food Res 49:663–672
Acknowledgments
J. A. Gómez-Ruiz thanks the Ministerio de Educación, Cultura y Deporte for a scholarship. The authors would like to thank Alan Bradbury (Kraft Foods, Munich, Germany) for providing coffee samples and Dr. A. B. Gerry (Reading University) for advice and helpful suggestions on measuring LDL oxidation.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Gómez-Ruiz, J.Á., Ames, J.M. & Leake, D.S. Antioxidant activity and protective effects of green and dark coffee components against human low density lipoprotein oxidation. Eur Food Res Technol 227, 1017–1024 (2008). https://doi.org/10.1007/s00217-007-0815-5
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00217-007-0815-5