
Abstract
A duplex PCR method was developed to simultaneously detect the presence of hazelnut and peanut in raw materials and commercial products. It was found to be able to specifically detect traces of the investigated products down to 50 pg of their target DNA.
A PNA array device has been designed and implemented to be used in combination with the duplex PCR in order to investigate the presence of traces of potentially allergenic nuts in foodstuffs. A PNA probe for each target amplified by the duplex PCR was designed, synthesized and characterized. The PNA probes were then deposited on commercial slides in order to build a PNA array to be used for recognizing the PCR products; the concentration of the probes as function of the concentration of the target DNA, together with the specificity of the probes were investigated.
The conditions optimized during the setting of the experiment were used to obtain the final version of the PNA array which was then used to test several commercially available foodstuffs claiming to contain or not to contain the targeted nuts.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
During the recent years food allergy prevalence has emerged as a serious public health issue affecting around 1% of the world's adult population and up to 2.5% of the children population, respectively [1]. Food allergens are proteins or glycoproteins with molecular weight ranging from 10 to 70 kDa, and although any protein is potentially allergenic, the most severe adverse reactions to food are associated with the consumption of a small number of products classified into eight main food groups responsible for about 90% of all the IgE-mediated food allergies: cow's milk, eggs, fish, crustaceans, peanuts, soybeans, tree nuts and wheat. Since anaphylactic reactions are reported to take place even after consumption of very low amounts of allergenic proteins [2], the presence of hidden allergens in food products, not listed on the label, is a great problem which causes concern in sensitive people. To face the problem, avoidance of the allergenic food or component is at present the only practical way for allergic patients; nevertheless hidden allergens cause serious concern even after this precaution. In this context the new directive of the European parliament (Dir. 2003/89/EC) regarding the comprehensive labelling of allergenic food ingredients is of great importance: the declaration of all the major allergenic ingredients (Annex IIIbis: cow's milk, eggs, fish, crustaceans, peanuts, soybeans, tree nuts, wheat, celery, mustard, sesame seeds and sulphites), regardless of the total amount in the final product, is made compulsory. The problem of threshold levels of the allergens still persists, i.e. the minimum quantity of a protein which elicits an allergic reaction.
Among the categories of allergenic foods, peanuts and tree nuts are probably the ones most likely to be responsible for the contamination of other materials during food manufacturing on account of their widespread use as ingredients in many food products, and for this reason they should be considered as potential sources of hidden food allergens. Moreover, peanuts and tree nuts are not only responsible for 1.1% of all the allergic reactions in the USA [3], but are becoming an emerging problem also in Europe [4] probably on account of the increased consumption.
Although the determination of allergens in food [5, 6] is mainly based on immunological methods via the use of either specific IgE from human serum or mono and polyclonal antibodies of animal origin, a sensitive indirect analysis of the presence of allergenic proteins can be performed targeting their specific cDNA via PCR methods. This approach has already been used for the indirect detection of specific food allergens in various matrices such as wheat [7], olive [8], apple [9] and hazelnut [10, 11] and seems to be suitable in particular when applied to matrices in which protein concentration is low (e.g. hidden allergens) or proteins are partially degraded by processing. However, PCR systems can sometimes lead to ambiguous interpretations on account of the low specificity of the priming sequences or of “carry-over” contaminations producing false positives [12]. Although the latter can be avoided by applying good laboratory practices, in all cases of ambiguous interpretations it is advisable to confirm the identity of the PCR amplicons. This may be done in different ways including sequencing of the amplicons, using nested PCR or using probes specifically hybridizing target DNA sequences. Among these, particularly useful turned out to be peptide nucleic acids (PNAs), oligonucleotide analogues in which the sugar–phosphate backbone has been replaced by a pseudopeptide chain of N-aminoethylglycine monomers, endowed of a higher sequence specificity [13–15].
In two recent works we successfully applied PNA probes to the post-PCR detection of a specific DNA sequence identifying the presence of hazelnut in foodstuffs via HPLC [16], and of GM soybean via an array device [17].
In the present work we report the implementation of a PNA array for the detection of hazelnut and peanut in various foodstuffs after the simultaneous amplification of their DNA by a duplex PCR designed and developed for this purpose. The method here proposed allows to detect the presence of DNA coming from two potential sources of hidden allergens (i.e. hazelnut and peanut) and confirming simultaneously their identity via hybridization with PNA probes.
Materials and methods
Sample preparation
Commercial samples
A set of 10 commercial foodstuffs, including breakfast cereals, snacks, biscuits and chocolate, were purchased from randomly chosen food stores.
DNA extraction
DNA extraction was performed using the commercial kit Wizard ® Plus Minipreps DNA purification system (Promega), opportunely modified as described in a previous work [16]. The modifications can be summarized as follows: once the DNA was extracted and purified following the instructions provided in the commercial kit, it underwent a further purification step by addition of 0.1 volumes of 3 M NaOAc (pH 5.2) and two volumes of cold 95% ethanol, followed by incubation at −70°C for 30 min. After precipitation the DNA was then pelleted by centrifugation at 12000 rpm for 15 min at 4°C. The DNA pellet was washed with 80% ethanol and incubated at room temperature for 10 min, then centrifuged for 5 min at 12000 rpm; the DNA pellet was finally dried and then dissolved in 100 μl of water. The DNA concentration was evaluated by UV absorption at 260 nm; 30 ng/μl stock solutions were prepared by dilution with bidistilled water.
Duplex PCR
A duplex PCR system simultaneously targeting a DNA sequence characterizing the Cor a 1.0301 isoform of the hazelnut allergen (GenBank acc. number Z72440.1) and one characterizing the Ara h 2 peanut allergen (GenBank acc. number L77197.1) was designed, developed and validated. The primer sequences chosen for the analyses are reported in Table 1.
The PCR primers to be incorporated in the single DNA strand to be hybridized with the PNA array (Cor a1_74 and Ara h2_455) were labelled with a Cy5 fluorophore at their 5′ end.
The samples for PCR screening were amplified using the method described later, whereas all samples to be used on the array platform underwent a double amplification: the first step was used to amplify the target sequences, while the second step was an unbalanced PCR used to selectively amplify the target DNA single strands to be hybridized on the array.
Polymerase chain reactions were performed in a final volume of 50 μl. All the reagents were supplied by Euroclone and the primers purchased from Thermoelectron. The concentration of the reagents in the first PCR were: 1× reaction buffer, 3 mM MgCl2, 0.2 mM dNTPs, 0.4 μM of each primer, 0.1 u/μl of hot start Blue Taq, 150 ng of DNA. The concentrations for the second step of the unbalanced PCR were the same, apart from the primer concentrations brought to 0.2 μM for the oligonucleotide priming the non-target DNA strand, and 2.0 μM for the oligonucleotide priming the target DNA strand (Table 1). The PCR was carried out in a PCR-sprint thermal cycler (Thermohybaid) using the following conditions: one cycle of DNA denaturation and Blue Taq activation at 95°C for 5 min; 40 cycles consisting of DNA denaturation at 95°C for 50 s, primer annealing at 60°C for 50 s and elongation at 72°C for 50 s; one step of final elongation at 72°C for 5 min.
PNA array preparation
PNA design
The PNA sequences were first checked to minimize any secondary structure which would result in a loss of hybridization efficiency, using the online available program Mfold (version 3.1) [18]. The sequence specificity of the probes was first evaluated using the BLAST homology search system from DDBJ (http://www.ddbj.nig.ac.jp/E-mail/homology.html) and then both probes were checked in order to avoid hybridization with any other non-target region among those amplified by the duplex PCR, by aligning the PNA sequences to the DNA sequences of the other amplified products. The sequences chosen for the analyses are reported in Table 1.
PNA synthesis
The PNA oligomers were synthesized by solid-phase synthesis on a 433A Peptide Synthesizer (Applied Biosystems) using the BOC strategy and HBTU/DIEA coupling, as described in a previous work [19]. Swelling, downloading and cleavage of the PNAs from the resin were done manually. Two 2-(2-aminoethoxy)ethoxyacetic acid spacers were added at the N-terminus of the PNA to link it to the solid surface of the array. The crude products were purified by reversed phase HPLC using a Phenomenex C18 peptide column (3 μm, 250 mm × 10 mm) with a binary gradient (flow rate: 4 ml/min); eluent A: water/TFA = 100:0.2; eluent B: water/acetonitrile/TFA = 60:40:0.2; detector UV (260 nm). The purified products were characterized by Electrospray Ionization-mass spectrometry (ESI-MS).
Array preparation
“CodeLink™ Activated Slides” (Amersham Biosciences) were used as solid supports to which the amino-terminal group of the PNA probes were covalently linked. The deposition of the probes was carried out using a GMS 417 Arrayer (Genetic Microsystem) with a pin-and-ring deposition system. The manufacturer's instructions for the deposition protocol were slightly changed in order to comply with the special requirement of the chemical structures of PNAs: in particular a 100 mM carbonate buffer (pH 9.0) containing 10% acetonitrile and 0.001% sodium dodecyl sulphate (SDS) was used as deposition buffer. Moreover, after every deposition, the pin-and-ring system was purged with water for 10 s and further washed with acetonitrile/water (1:1), in order to avoid dragging of the probes in subsequent depositions. The probes were coupled to the surface and the remaining reactive sites were blocked by leaving the slides in a humid chamber (relative humidity 75%) at room temperature for 12 h, followed by immersion in a glass rack containing a 50 mM solution of ethanolamine, 0.1 M TRIS, pH 9, prewarmed at 50°C, for 30 min. The slides were washed twice with bidistilled water at room temperature and then slowly shaken for 30 min in plastic tubes containing a 4× saline/sodium citrate (SSC) solution and a 0.1% SDS buffer prewarmed at 50°C. Each slide was then washed with bidistilled water at room temperature and centrifuged in a plastic tube at 800 rpm for 3 min. Slides were then ready to undergo the hybridization protocol or could be stored in a desiccated chamber for future use. It has to be remarked that, since a fluorescent control probe (CP sequence: NH2–(AC)11–Cy5) was deposited to check the efficiency of the deposition step, all the previously described operations were carried out away from direct light in order to prevent degradation of the Cy5 fluorophore.
Array analysis
Sample hybridization
DNA samples to be tested were prepared by diluting 50 μl of the PCR product to a final volume of 65 μl and a final concentration of 4× SSC and 0.1% SDS buffer. Hybridization was performed by loading the samples to “in situ frame” chambers (Eppendorf, 0030 127.510) and leaving the slides under slow shaking for 2 h at 40°C. After the hybridization step all the slides were treated individually to prevent cross contamination. The slides were washed under slow shaking for 5 min at 40°C with a 2× SSC, 0.1% SDS buffer prewarmed at 40°C, followed by treatment for 1 min with 0.2× SSC and for 1 min with 0.1× SSC at room temperature. The slides were then spin-dried at 1000 rpm for 5 min. It has to be noticed that all post-hybridization steps were performed in a dark environment to prevent degradation of the Cy5 fluorophore used to label the target sequences.
Image acquisition
The fluorescent signal deriving from the hybridization was acquired using a GMS 418 Array Scanner (Genetic Microsystem) at λ ex=646 nm and λ em=664 nm. In order to correctly compare the hybridization data, all the images reported were acquired with laser power = 100 and photomultiplier gain = 40.
Results
The primer pairs chosen for the development of the duplex PCR, assessed for their specificity during the design step by alignment in the online available databases, were individually tested with real standard samples in order to verify their ability to amplify only their specific targets. For this reason the primer pairs designed for the detection of hazelnuts and peanuts were individually tested with a panel of 15 in-house standard food matrices composed by nuts (brazilian nut, almond, hazelnut, pistachio and walnut), legumes (lupine, chickpea, peanut, pea and soybean) and cereals (barley, maize, wheat, oat and rice), using the PCR method developed, as described in the “Materials and methods” section. In order to avoid false negative results and to confirm the presence of vegetable amplifiable DNA, all the standards, once extracted, were tested using a protocol for the amplification of different non-coding regions of chloroplast DNA [20]. The results (data not shown) indicated that a positive amplification could be observed only when the designed primer pairs were applied to their specific targets and no amplification was observed for non-target matrices. The length of the amplicons obtained was found to be consistent with the design of the single PCRs: 156 bp for hazelnut and 201 bp for peanut.
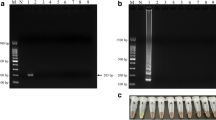
The limits of detection for the single and duplex PCR designed were then evaluated using subsequent dilutions of stock solutions of their target DNA ranging from 150 ng to 1 pg. Detection limits for the single PCRs were found to be 5 pg (data not shown), while the duplex PCR system, in agreement with what is already known in the literature [21], was found to have a lower sensitivity, being able to amplify the target sequences down to 50 pg of target DNA (Fig. 1).

Limits of detection of the duplex PCR for the detection of hazelnut and peanut. Hazelnut amplicon size: 156 bp; peanut amplicon size: 201 bp. Samples were prepared with concentration of the target DNAs corresponding to: 150 ng (1), 50 ng (2), 10 ng (3), 5 ng (4), 1 ng (5), 500 pg (6), 100 pg (7), 50 pg (8), 10 pg (9), 5 pg (10), 1 pg (11). M: 100 bp size marker. N: negative control
The limit of detection for the PNA probes was assessed by hybridization of the PNA designed to target hazelnut with a synthetic 15mer oligonucleotide complementary to the probe and labelled with a Cy5 fluorescent group. The same set of probe concentrations (50, 40, 30, 20 and 10 μM) was spotted on 10 slides which were then individually hybridized with concentrations of the target oligonucleotide corresponding to 0.8 μM, 0.4 μM, 0.2 μM, 0.1 μM, 50 nM, 25 nM, 12.5 nM, 6.75 nM, 1 nM, 0.01 nM. The best hybridization signal (data not shown) in the whole range of target concentrations was observed for the 30 μM PNA, while no significant improvement was observed by increasing the PNA concentration. A statistically significative enhancement of the fluorescent signal (more than ten times the signal/noise ratio) was detected down to 1 nM, which can then be assumed as limit of detection of the array system. Although the oligonucleotide cannot completely mime the behaviour of a longer single stranded DNA, such as those resulting from amplification, these results were taken as indicative for choosing the concentration of the PNA probes to be spotted.
The array device was prepared by depositing nine replicates for each probe to be coupled in three parallel lines corresponding to: H—30 μM solution of the PNA for the detection of hazelnut; P—30 μM solution of the PNA for the detection of peanut; CP—30 μM Cy5 labelled control probe to evaluate the efficiency of the deposition step.
In order to test the selectivity of hybridization, the PNA array was tested with two samples amplified from hazelnut and peanut standards, respectively. The samples were prepared by double amplification as described in the “Materials and methods” section, in order to produce a consistent amount of the target single strands. The samples were individually hybridized on two different arrays according to the protocol and then their images were acquired via a fluorescent reader. The results show that the PNA probes were able to selectively recognize the presence of hazelnut (Fig. 2a) and peanut (Fig. 2b), respectively.

Evaluation of the PNA specificity. The arrays were tested with the PCR product deriving from amplification of DNA extracted from hazelnut (a) and peanut (b) H: hazelnut PNA; P: peanut PNA; B: blank lane; CP: Cy5 labelled control probe
In order to evaluate the applicability of the PNA array method to the detection of traces of hazelnut and peanut in foodstuffs, a panel of commercial food products, purchased from randomly chosen food stores, was chosen in order to test their content of hazelnut and/or peanut as claimed on the label, or for their possible presence as a result of accidental contamination during manufacturing (Table 2). Out of 10 selected products, the presence of hazelnut as ingredient was declared in five samples and of peanut in one sample, while the possible presence of traces of tree nuts and peanuts as a result of manufacturing carryover was declared in three samples.
All samples were preliminarily tested with the duplex PCR method in order to evaluate their composition and to compare the results with what observed with the subsequent PNA array test. The results (Fig. 3) show that almost all the selected samples, apart from sample 6, contained hazelnut at various levels, while only sample 3 contained both hazelnut and peanut. This means that traces of hazelnut were found in four foods which did not report hazelnut among the ingredients (samples 1, 3, 5, 9), two of which did not declare it as possible contaminant (samples 3 and 5); no contamination was found for peanut.

Duplex PCR for the detection of hazelnut and peanut applied to commercial foodstuffs, as reported in Table 2. Hazelnut amplicon size: 156 bp; peanut amplicon size: 201 bp. M: 50 bp size marker. N: negative control

PNA array analyses for the detection of hazelnut and peanut applied to commercial foodstuffs (the numbers refer to the products reported in Table 2): sample 1 (a) and sample 3 (b). H: hazelnut PNA; P: peanut PNA; B: blank lane; CP: Cy5 labelled control probe
Samples 1 and 3 were then selected to be tested on the PNA array as representative for the analyses of hidden allergens. In fact sample 1, in which the presence of hazelnut was declared as possible contaminant, was found to contain hazelnut; sample 3, in which the presence of peanut as ingredient was declared but not the presence of hazelnut either as ingredient or as possible contaminant, was found to contain both peanut and hazelnut.
The samples selected were doubly amplified, and then hybridized as described in the “Materials and methods” section. The results obtained with the PNA array device were in agreement with those previously observed with the duplex PCR: sample 1 gave raise to a strong hybridization signal only on the first lane, indicating the presence of hazelnut in the tested product (Fig. 4a), whereas in sample 3 a strong fluorescent signal was observed on the second lane and a weaker signal was also observed for lane one, indicating the simultaneous presence of peanut and hazelnut in the tested product (Fig. 4b).
Conclusions
In this paper a new duplex PCR method for the detection of hidden allergens in foodstuffs was developed and tested. Its sensitivity was found to be comparable with similar methods published for other applications and was evaluated to be 50 pg of target DNA when using standards. Its applicability to commercial samples for screening purposes was also assessed by testing a number of selected products. It has to be noticed that the limit of detection of the PCR method proposed here might differ when applied to food samples on account of the processing they underwent. Moreover, commercial samples different from those tested might need specific extraction methods to obtain a pure DNA extract.
The availability of the duplex PCR method constituted the basis for the development of a PNA array device which allowed detecting simultaneously the presence of DNA from hazelnut and peanut, two ingredients which are possible sources of hidden allergens in food products. The results presented here show that this combined approach can constitute a fast and reliable method for the indirect unambiguous determination of hazelnut and peanut in foodstuffs. Furthermore, when compared to conventional electrophoresis methods, this platform has the advantage to perform in one step the simultaneous detection of the presence of target organisms (i.e. hazelnut and peanut) and to confirm the identity of the amplified products via specific sequence recognition. On account of its sensitivity the method proposed is particularly suitable for detecting hidden allergens in food.
The model proposed here can be easily furtherly extended with new target DNA sequences and new PNA probes, in order to build up a complete array device for the detection of many potential sources of food allergens in foodstuffs.
References
Jackson WF (2003) Food allergy. Concise monograph series. International Life Sciences Institute Europe, Belgium
Yunginger JW, Sweeney KG, Sturner WQ, Giannandrea LA, Teigland JD, Bray M, Benson PA, et al. (1988) JAMA 260:1450–1452
Sicherer SH, Munoz-Furlong A, Burks AW, Sampson HA (1999) J Allergy Clin Immunol 103:559–562
Ewan P (1996) Br Med J 312:1074–1078
Besler M (2001) Trends Anal Chem 20 (11):662–672
Poms RE, Klein CL, Anklam E (2004) Food Additives Contam 21(1):1–31
Köppel E, Stadler M, Lüthy J, Hübner P (1998) Lebensm Unters Forsch A 206:399–403
Aché JD, Castro AJ, Rodriguez-Garcia MI (2002) Micron 33(1):33–37
Son DY, Scheurer S, Hoffmann A, Haustein D, Vieths S (1999) Eur J Nutr 38(4):201–215
Holzhauser T, Wangorsch A, Vieths S (2000) Eur Food Res Technol 211:360–365
Herman L, De Block J, Viane R (2003) Int J Food Sci Technol 38:633–640
Quist D, Chapela IH (2001) Nature 414:541; Metz M, Futterer J (2002) Nature 416:10; Mann CC (2002) Science 295:1617; Quist D, Chapela IH (2002) Nature 416:11
Egholm M, Behrens C, Christensen L, Berg RH, Nielsen PE, Buchardt O (1993) J Chem Soc Chem Commun, 800–801
Jensen KK, Orum H, Nielsen PE, Nordén B (1997) Biochemistry 36:5072–5077
Dueholm KL, Nielsen PE (1997) New J Chem 21:19–31
Germini A, Scaravelli S, Lesignoli F, Sforza S, Corradni R, Marchelli R (2005) Eur Food Res Technol (in press)
Germini A, Mezzelani A, Lesignoli F, Corradini R, Marchelli R, Bordoni R, Consolandi C, De Bellis G (2004) J Agric Food Chem 52(14): 4535–4540
Zuker M (2003) Nucleic Acids Res 31(13):1–10
Lesignoli F, Germini A, Corradini R, Sforza S, Galaverna G, Dossena A, Marchelli R (2001) J Chromatogr A 922:177–185
Taberlet P, Gjelly L, Pautou G, Buovet J (1991) Plant Mol Biol 17:1105–1109
Matsuoka T, Kawashima Y, Akiyama H, Miura H, Goda Y, Kusakabe Y, Isshiki K, Toyoda M, Hino A (2000) J Food Hyg Soc Japan 41:137–143
Acknowledgements
This work was part of the project “PNA-based nanotechnology and microsystems for food diagnostics” financed by MIUR—Programmi di Ricerca Scientifica di Rilevante Interesse Nazionale
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Rossi, S., Scaravelli, E., Germini, A. et al. A PNA-array platform for the detection of hidden allergens in foodstuffs. Eur Food Res Technol 223, 1–6 (2006). https://doi.org/10.1007/s00217-005-0034-x
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00217-005-0034-x