
Abstract
The interest in metabolomic studies has rapidly increased over the past few years. Changes of endogenous compounds are typically detected in plasma or urine. However, the use of hair allows for long-term monitoring of metabolomic changes and has recently started being applied to metabolomic studies. Within the proposed study, we aimed for a systematical investigation of different pre-analytical parameters on detected metabolites from different chemical classes in hair. For this purpose, three different parameters were varied: (1) multi-step decontamination (dichloromethane (DCM), acetone, H2O, acetone; H2O, acetone, DCM, acetone; and H2O, methanol/acetone), (2) homogenization (pulverization vs. cutting into snippets), and (3) extraction (acetonitrile (ACN)/buffer pH 4 vs. ACN/H2O vs. ACN/buffer pH 8.5). To include as many metabolites as possible, samples were analyzed by high-resolution time of flight mass spectrometry coupled to liquid chromatography (HPLC-HRMS) and additionally by gas chromatography high-resolution mass spectrometry (GC-HRMS) followed by untargeted-like data processing, respectively. The application of different decontamination procedures yielded similar results, although pointing to a trend towards increased washing-out effects if protic solvents were used as a first washing step. Pulverization of hair samples was favorable in terms of detected and tentatively identified metabolites. Evaluation of extraction solvents showed differences in extraction yield for the majority of investigated metabolites, yet, a prediction of metabolite extraction according to their pKa values was not possible. Overall, successive decontamination with DCM, acetone, H2O, and acetone; homogenization by pulverization; and extraction with ACN/H2O produced reliable results.

Graphical abstract
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Global metabolomics aims to analyze the entirety of small endogenous biomolecules (< 1000 Da). As a phenotype signature, it has proven to be a powerful tool to measure metabolome changes in order to search for biomarkers, typically related to diseases [1]. Modern techniques such as liquid chromatography (LC) or gas chromatography (GC) coupled to high-resolution (HR) mass spectrometry (MS) theoretically allow for simultaneous screening of all endogenous metabolites of interest and are currently state-of-the-art for metabolomic profiling [2,3,4]. Until now, such metabolomic studies are mainly performed in classic and common matrices such as blood (plasma, serum) or urine. Yet, the metabolome of those body fluids is highly dynamic, resulting in an extremely variable composition in dependence on daily activities, dietary variations, stress, or circadian variations. Those fluctuations currently present one of the biggest challenges in metabolomics [5]. In addition, small influences on the metabolome are often conserved just for a very short time period [6], limiting its use for a robust biomarker detection. In contrast to these conventional matrices, hair as a complementary matrix generally allows for a long-term and retrospective detection of analytes. Substances of both, endo- and exogenous origin, are constantly incorporated into the growing hair shaft [7, 8]. Knowing that the hair growth rate is approximately 1 cm/month, segmentation of hair allows the detection of cumulative analyte levels consistent to the corresponding hair growth period [9]. Its application for retrospective consumption monitoring (e.g., drugs of abuse, alcohol markers) is therefore already widely applied to, e.g., abstinence controls in the process of regaining the driver’s license, workplace drug testing, or doping controls [7]. Furthermore, the stable accumulation of substances over time allows searching for robust and long-term biomarkers that are less influenced by short-term fluctuations, e.g., circadian variations [10]. In addition, hair samples bear several advantages allowing non-invasive sampling and uncritical storage at room temperature [7]. Considering this, hair appears as a new, promising matrix for metabolomic studies.
So far, studies for endogenous compounds in hair mainly focused in a targeted way on specific chemical compounds or groups such as cortisol, amino acids, or hair lipids [9, 11, 12], but an extended global picture of the hair metabolome composition is still missing. Only recently, first reports were published on the topic of untargeted analysis of metabolomic changes in human hair under certain conditions [13,14,15,16]. For example, the identification of potential biomarkers for fetal compromise or gestational diabetes using hair metabolomics was reported by Sulek et al. [15] and He et al. [16], respectively. Delplancke et al. investigated the segmental analysis of the maternal hair metabolome depending on the different pregnancy trimesters in healthy pregnancies [13]. Combining LC-MS/MS and GC-MS, 980 metabolites were identified. Of those, 78 were considered as significantly altered either between first and second trimester or second and third trimester. However, the process of metabolite identification was not completely reproducible, e.g., confidence of identification as proposed by the metabolite standard initiative (MSI) [17] is missing for LC-MS/MS data. This inconclusive assignment of metabolite identifications prevented a reasonable elucidation of the hair metabolome composition. That is why, despite these recent findings, data about the composition of the hair metabolome is still scarce, highlighting the need for further systematic studies in this research field.
One key step that will have a major impact on the obtained metabolic profile [18] in metabolomic studies particularly for hair analysis is the sample preparation. Variances originating from the sample preparation should be kept to a minimum to avoid mis- or over-interpretation of results. In the past years, many studies have been published about the impact of different sample pre-treatment parameters on the quality of metabolomics data [19, 20] in blood while data on hair sample preparation is still missing. The well-established workflow for hair sample preparation for analysis of xenobiotics comprises segmentation, decontamination, pulverization/cutting, extraction, cleanup (optional), and analysis [7, 21]. Each step represents a potential source of error and variation that needs to be kept as small as possible. Recommendation for sample decontamination from the Society of Hair Testing (SoHT) is an initial washing step with an organic solvent, to remove oils, followed by aqueous washes [22]. Specific washing protocols used for hair analysis of endogenous compounds include, among others, a brief wash with dichloromethane (DCM) [23], H2O and methanol (MeOH) [16], H2O and acetone [9], MeOH, H2O and MeOH [24], acetone, 0.1% sodium dodecyl sulfate (SDS), and H2O and acetone, respectively [25]. Sample homogenization by pulverization or cutting is usually applied after suitability evaluation of each method in regard to the targeted analyte. Extraction of the target analyte from the solid hair matrix into solution presents the key part of the sample preparation. Many experimental parameters can potentially affect the extraction efficiency: particle size, extraction time, energy (temperature/shaking/ultra-sonication), type of solvent, solvent volume, and frequency of extraction steps [26]. Similar to the decontamination procedures, extraction protocols differ widely, ranging from alkaline hydrolysis [15] over acidic hydrolysis [11] to simple methanolic extraction [9].
Within this study, we aimed to systematically analyze the impact of different hair sample pre-treatment parameters namely decontamination, homogenization, and extraction, and their effects on a selection of identified metabolites. Analysis of the metabolites was performed with LC-MS/MS and GC-MS, two commonly employed techniques in metabolomic studies [27].
Materials and methods
Chemicals and reagents
The following analytical standards and chemicals were purchased from Sigma-Aldrich (Buchs, Switzerland): 1-methylhistidine, adenine, adenosine, adipic acid, ammonium formate, ammonium bicarbonate, arginine, aspartic acid, azelaic acid, butyrylcarnitine (C4), caffeine, L-carnitine (C0), chenodeoxycholic acid, cholic acid, choline, citrulline, cortisol, cortisone, creatinine, decanoic acid (C10:0), decanoylcarnitine (C10), deoxycholic acid, dodecanoic acid (C12:0), formic acid, glutamic acid, glutaric acid, glycocholic acid, glycine, hippuric acid, histidine, hypoxanthine, inosine, isoleucine, leucine, methionine, methyladenosine, methylguanosine, methylmalonic acid, mevalonolactone, myristic acid (C14:0), N,N-dimethylglycine, N-acetylneuraminic acid, nicotinamide, nicotinic acid, norvaline, octanoylcarnitine (C8), p-aminobenzoic acid, pentadecanoic acid (C15:0), phenylalanine, proline, propionylcarnitine (C3), pyroglutamic acid, raffinose, riboflavin, serine, suberic acid, taurine, taurocholic acid, tridecanoic acid (C13:0), triethanolamine, trimethyllysine, theobromine, threonine, tryptophan, tyrosine, uracil, uric acid, urocanic acid, and valine. Deuterated and heavy-labeled internal standards (IS) arginine-13C6, alanine-D4, hippuric acid 15N, and testosterone-D2 were sourced from Cambridge Isotope Laboratories, Inc. (Andover, MA, USA) and delivered by ReseaChem Life Science (Burgdorf, Switzerland) or Sigma-Aldrich (Buchs, Switzerland). The derivatization reagents methoxyamine hydrochloride and 1 mL solutions of N-methyl-N-trimethylsilyl trifluoroacetamide (MSTFA) were purchased from Sigma-Aldrich (Steinheim, Germany) and Macherey-Nagel (Düren, Germany), respectively. Water was filtered with a Purelab Ultra Millipore filtration unit (Labtech, Villmergen, Switzerland). Acetonitrile (ACN), DCM and MeOH of HPLC grade were purchased from Fluka (Buchs, Switzerland). All other chemicals used were from Merck (Zug, Switzerland) and of the highest grade available.
Hair samples
Cosmetically untreated hair samples were collected from two healthy volunteers who provided written informed consent. According to Swissethics (Humanforschungsgesetz), no further ethical approval from the cantonal ethic commission is necessary if the research is not aiming to investigate diseases or functions of the human body as is the case in the current study. Four-centimeter-long hair was sampled approximately 1 cm away from the scalp. The hair samples were stored in aluminum foil at room temperature until further analysis. For the evaluation of decontamination and extraction procedures, hair samples that have not yet been homogenized were sieved prior to decontamination (Sieve shaker AS 200 control, Retsch, Haan, Germany).
Evaluation of sample pre-treatment parameters
In the following, different sample pre-treatment parameters were investigated. Namely, decontamination, homogenization, and extraction were varied. When one parameter was modified as described in detail in the following chapters, all other parameters were kept constant. An IS solution (alanine-D4 (250 μM), arginine-13C6 (100 μg/mL), hippuric acid 15N (200 μg/mL), norvaline (100 μM), and testosterone-D2 (20 μg/mL)) was used in all protocols.
Evaluation of decontamination procedures
Three different multi-step washing protocols were tested for their efficiency to remove metabolites from the hair in comparison with unwashed hair samples. For this purpose, hair samples were weighed into a 30-mL Sarstedt tube and washed with the following washing protocols: wash protocol A (DCM; acetone; H2O; acetone), wash protocol B (H2O; acetone; DCM; acetone), and wash protocol C (H2O; MeOH/acetone (1:1, v/v)), respectively. Washing volumes were 10 mL for DCM, H2O, and MeOH/acetone steps and 5 mL for acetone steps, which were added in order to adjust solvent polarity and improve cleaning efficacy. Each washing step was conducted during 2 min of vigorously shaking followed by cautious decantation of the washing solution. Finally, the hair samples were dried overnight at room temperature. Wash solutions of DCM, H2O, and MeOH/acetone steps were kept, spiked with 20 μL IS mix, evaporated under nitrogen (N2), and reconstituted in 500 μL of ACN/H2O (2:8, v/v). Further analysis was performed as discussed under the “Analytical methods” section.
Evaluation of homogenization procedures
For the evaluation of homogenization procedures, an average of 30 mg dried hair (decontaminated according to wash protocol A) was either pulverized or cut into snippets. For the pulverization, hair samples were weighed into a 2-mL Eppendorf tube containing one tungsten carbide ball (weight approx. 3 g, diameter 7 mm) and homogenized for 10 min at 30 Hz using a bench-top mill (Mixer Mill MM 400, Retsch, Haan, Germany). Snipped hair samples were obtained by manually cutting the hair samples into millimeter segments with a pair of scissors. Extraction was carried out using ACN/H2O (2:8, v/v) (see “Evaluation of extraction protocols”).
Evaluation of extraction protocols
In each case, hair samples examined here were decontaminated according to wash protocols A to C and homogenized by pulverization. To test the impact of different extraction protocols and a possible pH-dependent extraction effect, the following extraction mixtures were tested: ACN/ammonium formate buffer 0.5 M (2:8, v/v, pH 4, pH adjusted with formic acid), ACN/H2O (2:8, v/v, pH 6), and ACN/ammonium bicarbonate buffer 0.5 M (2:8, v/v, pH 8.5, pH adjusted with ammonium hydroxide).
Sample preparation workflow
Apart from decontamination, homogenization, and extraction (as described in detail above), all samples were prepared in the same way. In brief, an average of 30 mg of decontaminated and homogenized hair was weighed into a 2-mL Eppendorf tube. After spiking with 20 μL IS solution, extraction was performed by addition of 1 mL extraction mixture followed by vortexing (15 s) and incubation in an ultrasonic bath for 16 h. After centrifugation (5 min, 9′000 rpm), 900 μL of the supernatant was transferred into a pill glass and dried under a gentle stream of N2 at 35 °C. The residue was reconstituted in 500 μL of ACN/H2O (2:8, v/v) by vortexing (15 s) followed by combined filtration and centrifugation (5 min, 9′000 rpm, VWR centrifugal filter, 0.45 μM pore size, VWR, Dietikon, Switzerland) as described by Steuer et al. [28]. Seventy microliters of the filtrate was transferred into a 250-μL insert and analyzed by LC-MS/MS as described under “Analytical methods.” Pooled quality control (QC) samples were prepared from 30 μL of the filtrates, aliquoted into 26 vials whereof half was analyzed with LC-MS/MS and half with GC-MS (see “Analytical methods”).
The remaining filtrate and aliquoted QC samples were again evaporated to prepare the samples for derivatization. For this purpose, a 2-step derivatization was performed: dried residues were reconstituted with 50 μL freshly prepared methoxyamine hydrochloride in pyridine (20 mg/mL), vortexed (15 s), and transferred into a 250-μL insert followed by incubation at 80 °C for 15 min. After cooldown of the samples, they were evaporated again under N2 at 40 °C. Following the methoximation, dried samples were crimp-capped and the second derivatization step was performed shortly before the injection on a sample preparation robot (GERSTEL Multi-Purpose Sampler (MPS), Gerstel, Mülheim, Germany) controlled by the integrated GERSTEL MAESTRO software (version 1.4.40.1; Gerstel, Mülheim, Germany) by automatically adding 50 μL MSTFA. This automatization permitted derivatization of each sample under the exact same conditions. After addition of MSTFA, samples were vortexed (30 s) and silylation was performed by incubation at 80 °C for 15 min under continuous shaking. Finally, after a cooldown time of 5 min, samples were injected onto the GC-MS system (see “Analytical methods”).
In addition, the same sample preparation was conducted for solvent blanks of wash solutions and extraction solvents (without hair matrix) spiked with IS as negative controls to eliminate false-positive peaks.
Analytical methods
All samples were analyzed in triplicates and in randomized order throughout the batch. Comparison of results was only performed within one batch. Pooled QC samples, as a measure for repeatability [27], were injected every five samples, except for the evaluation of homogenization procedures due to the small sample size.
HPLC-HRMS analysis
Samples were analyzed by LC-MS/MS according to Boxler et al. [29] with minor changes. In brief, analysis was performed on a Thermo Fischer Ultimate 3000 UHPLC system (Thermo Fischer Scientific, San Jose, CA) coupled to a HR time-of-flight (TOF) instrument system (TripleTOF 6600, Sciex, Concord, Ontario, Canada). The chromatographic separation was carried out using a Waters (Baden-Daettwil, Switzerland) XSelect HSST RP-C18 column (150 mm × 2.1 mm i.d.; 2.5 μM particle size) within a total run time of 20 min. Mobile phases were 10 mM ammonium formate containing 0.1% (v/v) formic acid in water (eluent A) and 0.1% (v/v) formic acid in MeOH (eluent B). An LC gradient elution was performed as follows: 1 min 100% eluent A, increase from 0 to 100% eluent B during 1–15 min, 3 min held at 100% eluent B before decreasing to 0% eluent B at 18.01 min, and re-equilibration for 2 min. The flow rate was programmed to 0.5 mL/min, increased to 0.7 mL/min after 15 min, and held at 0.7 mL/min for 5 min. The column oven temperature was set to 40 °C, the autosampler to 7 °C. For all samples, injection volume was 5 μL.
The HRMS analysis was conducted using electrospray ionization (ESI) in positive ionization mode with a DuoSpray ion source at a resolving power (full width at half-maximum (fwhm) at m/z 400) of 30,000 in MS and 30,000 in MS2 (HR mode) or 15,000 (high-sensitivity mode). An automatic calibration was done using an atmospheric-pressure chemical ionization (APCI) positive calibration solution (Sciex) every 5 samples.
A full-scan TOF-MS acquisition was performed over a mass range from m/z 50 to m/z 1000 (accumulation time 100 msec, collision energy (CE) of 5 eV). In addition, for one out of three replicates, an information-dependent acquisition (IDA) scan was performed as an MS/MS experiment for compound identification over a mass range of m/z 50 to m/z 1000 (accumulation time 100 msec, CE 35 eV with a CE spread of 15 eV in high-sensitivity mode). Further IDA criteria were set as follows: dynamic background subtraction on the 4 most intense ions, intensity threshold above 100 cps, and exclusion time of 5 s (half peak width) after two occurrences. The data acquisition was controlled by Analyst TF software (version 1.7, Sciex, Concord, Ontario, Canada).
At the beginning, at the end, and repeatedly in between the samples, a system suitability test (SST) was measured and checked for reproducibility of the data and retention time (RT) shifts. The SST contained the following analytes (concentration 10 μg/mL each): 1-methylhistidine, adenine, adenosine, arginine, azelaic acid, chenodeoxycholic acid, cholic acid, citrulline, cortisol, cortisone, creatinine, deoxycholic acid, glutaric acid, glycocholic acid, hippuric acid, inosine, isoleucine, leucine, methionine, methylmalonic acid, mevalonolactone, N,N-dimethylglycine, nicotinic acid, p-aminobenzoic acid, phenylalanine, proline, pyroglutamic acid, raffinose, riboflavin, taurine, taurocholic acid, tryptophan, and uracil. The relative abundance of the analytes was determined by integration of the respective peak area using MultiQuant V 2.1 (Sciex).
GC-HRMS analysis
Derivatized samples were analyzed on a TRACE 1300 GC system (Thermo Scientific, Bremen, Germany) coupled to a HR Orbitrap MS (Thermo Scientific, Bremen, Germany) as recently published elsewhere [30]. In brief, sample injection was performed by the sample preparation robot GERSTEL MPS. Chromatographic separation was achieved using a TraceGOLD TG-5SilMS (30 m × 0.25 mm I.D. × 0.25 μM) film capillary column (Thermo Scientific, Bremen, Germany) and a constant helium gas flow of 1 mL/min. GC settings were as follows: inlet temperature 250 °C; gradient elution with initial column temperature set to 70 °C with a hold time of 4 min, increasing to a final temperature of 320 °C in steps of 20 °C/min which was hold for 8 min, resulting in a total run time of 24.50 min. Sample injection volume was 1 μL with a split ratio of 5:1. The HRMS was operated with electron ionization (EI) at 70 eV energy in positive mode at an ion source temperature of 230 °C. Full scan acquisition was carried out over a mass range of m/z 50 to m/z 650 and HR EI fragment spectra were acquired at a resolving power of 60,000 (fwhm at m/z 200) and started after a filament delay of 5.3 min. The temperature of the transfer line was set to 250 °C. To maintain mass accuracy, internal lockmass-correction was performed with masses m/z 207.03240, m/z 225.04290, and m/z 381.05110. All MS parameters were controlled by Xcalibur software (Version 4.0; Thermo Scientific, Bremen, Germany).
Data evaluation
Data evaluation for HPLC-HRMS analysis
On the one hand, untargeted, database-assisted identification was conducted using Progenesis QI (Waters Corp, Milford, USA) after alignment of all samples measured within one batch, peak-picking, and deconvolution by searching against different online databases: National Institute of Standards and Technologies (NIST) [31], METLIN [32], Lipidblast [33], and the Human Metabolome Database (HMDB, V4.0) [34]. Peak-picking settings in Progenesis QI were set on default: automatic sensitivity method, no minimum peak width, no RT limits. Data files from IDA scans were integrated into the software in order to have MS/MS spectra for improved identification of the most abundant compounds. On the other hand, further identification of compounds was performed in PeakView V 2.2 (Sciex) by searching against an in-house library. Identification of analytes was done based on the library match, exact masses on the MS and MS/MS level, and RT, if available. The list of (tentatively) identified metabolites was manually reviewed and compared to negative controls to remove false positives. MultiQuant V 2.1 was used for peak integration.
Data evaluation for GC-HRMS analysis
TraceFinder 4.1 software (Thermo Scientific, Bremen, Germany) was used for GC-MS data processing and deconvolution using the incorporated deconvolution plugin. Data was retention time aligned by exclusion across all samples within one batch and subsequently screened against the NIST 17 spectral library (containing EI MS data of 267,376 compounds; National Institute of Standards and Technology, Gaithersburg, MD, USA) and a HR metabolomics library (containing HR EI MS data of > 800 metabolites; Thermo Scientific, Bremen, Germany) for compound identification. The obtained peak list with tentatively identified metabolites was manually reviewed. Hits were considered positive based on the search index score, the HR filtering value, and manual assessment by comparison of the library spectrum with the acquired spectrum. RT of tentatively identified metabolites were confirmed with reference standards, if available. Afterwards, a compound database (CD) was created in TraceFinder 4.1 containing information of the exact mass of the most abundant and specific fragment in combination with one or more additional specific fragments (for confirmation purposes) and the respective RT. Data was screened against the CD and peak areas were integrated therein.
Statistics
Statistical analyses were done using GraphPad Prism 7 (GraphPad Software, CA, USA). Comparisons of extraction results from pulverized and snipped hair samples as well as for comparison of wash water sums were performed with a two-tailed t test and a confidence interval of 95%. An ordinary one-way ANOVA was applied for the comparison of not decontaminated and decontaminated hair samples and for the evaluation of extraction tendencies. Statistical significance was considered as follows: p ≤ 0.05 (*): significant; p ≤ 0.01 (**): very significant; p ≤ 0.001 (***) and p ≤ 0.0001 (****): extremely significant. Normalization of peak area values through the respective sample weight was done for all hair samples.
Results and discussion
Analytical methods and compound identification
To investigate the effect of different sample pre-treatment parameters on the detection of endogenous hair metabolites, a broad spectrum of endogenous compound groups should be covered. Since information about the exact composition of the human hair metabolome is very limited, an untargeted approach for metabolite identification was applied, without prior selection of a set of metabolites. In fact, the chemical diversity of the metabolome is challenging regarding the sample analysis. Using several analysis methods for a broader coverage of the metabolome is generally suggested and accepted [27]. Especially the use of instruments with high resolution and high mass accuracy are preferred. To that end, each analytical method, LC-MS/MS, and GC-MS, used in combination within the present study, contributes additional strong aspects: easy sample preparation and handling of non-volatile and heat-sensitive analytes for LC-MS/MS and high chromatographic resolution and peak capacity for GC-MS. Due to limited availability of hair samples from one person, hair samples of two individuals were used to conduct different parts of the study. They were not compared for biological variances as this was not aimed for within this study.
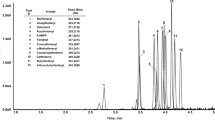
Final identification of compounds measured with LC-MS/MS was based on exact mass, fragmentation spectra, and RT (if available) by screening against an in-house library in PeakView and different online databases in Progenesis QI. HR EI spectra obtained by GC-MS analysis were screened against the NIST 17 spectral library and a high-resolution metabolomics library using TraceFinder 4.1. Identification results were grouped in different levels according to the classification suggested by the MSI [17]: level 1 identifications are identified compounds confirmed by means of a chemical reference standard using two or more orthogonal parameters such as RT and mass spectrum and analyzed under the same analytical conditions as the respective compound. Level 2 identifications have not been verified with a reference standard, are based on physicochemical properties, and/or require spectral similarities to publically available databases. They are so-called “putatively annotated compounds.” “Putatively characterized compound classes” are level 3 identifications that assign characteristic physicochemical properties and/or mass spectral data and exact mass information to chemical compound classes. Unknown compounds are assigned to level 4. After peak-picking (considering only adducts of [M+H]+, [M+Na]+, [M+NH4]+, and [M+H-H2O]+) and deconvolution, across all samples, LC-MS/MS analysis initially lead to the detection of around 6500 features with MS/MS spectra, whereas GC-MS generated a maximum of around 590 EI spectra. As the identification of unknown spectra was out of scope of this study, all features without available MS/MS spectra from IDA experiments and without a library hit were filtered out. Subsequently, compound identifications without reasonable RT, with very low abundances, missing occurrences in one of the triplicates, and false positives from comparison with negative controls were also neglected. Finally, to follow-up the aim of this project, only metabolites with an identification level of 1 or 2 were further investigated under the different conditions. These restrictions resulted in the identification of 51 metabolites by combined use of LC-MS/MS and GC-MS, ranging over different chemical classes (i.e., amino acids and derivatives thereof, carnitines, saturated fatty acids). The majority of metabolites could be detected and identified via LC-MS/MS analysis (38 metabolites) compared to 18 metabolites identified by GC-MS analysis. Five metabolites were identified via both analytical techniques, indicating that the use of two different analytical techniques is beneficial as they complement each other. An overview of identified metabolites and their identification level according to the suggestions of the MSI [17] is given in Table S1 of the Electronic Supplementary Material (ESM). The total ion chromatograms of LC-MS and GC-MS including some selected main metabolites and results of QC-pools are given in Fig. 1 and Table S2 of the ESM, respectively.

Extraction results (mean of three each ± standard deviation) of ACN/H2O (2:8) for extracts of pulverized hair samples without decontamination and with decontamination according to wash protocols A to C exemplified for a nicotinamide and b arginine. Statistical analysis with ordinary one-way ANOVA: p > 0.05 (not significant, without indication), p ≤ 0.001 (***), and p ≤ 0.0001(****)
Besides the identified compound classes, other compound classes expected to be present in hair samples and measurable with the applied procedures, e.g., phospholipids, were not identified. This was due to an insufficient differentiation of isomers that would lead to an identification level 3 according to MSI guidelines which was considered insufficient for the purpose of our study.
Also, the number of identified features in the present study appears rather small compared to a recent study by Delplancke et al., who described the identification of 980 metabolites in human hair by combined use of GC- and LC-MS [13]. However, identification in that study was not based on MSI guidelines which prevents direct comparison of the numbers. Indeed, it seems that automatic identification by the software was accepted without further verification considering adduct forms and putative identifications or any other criteria.
Furthermore, for unambiguous identification using LC-MS, MS/MS data in addition to accurate precursor ions is required. However, a lack of IDA data especially for low abundant compounds prevented clear identification of further metabolites. Sequential window acquisition of all theoretical fragment-ion spectra (SWATH) as a data-independent acquisition (DIA) method produces MS/MS data of all ions across the selected mass range of interest by recording fragmentation data of precursor ion windows (usually 20 to 35 Da wide) [35]. Though, the (current) incompatibility with common metabolomics software tools such as Progenesis QI limits its application for untargeted metabolomic data evaluation [29].
Further, it is known that long-term weathering effects (influence of sunshine, rain, or wind) damage the hair shaft which provokes increased wash-out of drugs or endogenous compounds, such as steroids, along the hair shaft [7, 36]. In this study, hair samples were cut 1 cm away from the scalp. Missing analysis of the first proximal centimeter might have prevented the detection of higher metabolite concentrations and therefore the identification of further metabolites.
As indicated in Table S1 and Fig. S1 of the ESM, many of the identified metabolites analyzed by LC-MS eluted rather early (< 3 min) on the used reversed phase column which is considered as the area particularly prone to matrix effects. In the present study, the main focus was on the influence of different pre-analytical parameters in only two hair samples. Therefore, the influence of matrix effects—which particularly plays a role once different samples from different subjects are analyzed—should be negligible. Common metabolomics studies apply different systems (e.g., reversed phase and hydrophilic interaction liquid chromatography (HILIC), capillary electrophoresis and ion-pairing reversed-phase chromatography) to improve retention and separation of polar metabolites [37]. For the purpose of the present study and taking into consideration the already large amount of data when dealing with GC- and LC-MS, we decided to reduce the analysis to a single chromatographic column for LC-MS in order to limit the data handling to reasonable measures.
However, implementation of analysis in negative ionization mode as well as the use of additional chromatographic columns, such as HILIC, could improve the separation of polar compounds and thus even more increase the identification rate in future studies.
Obviously, metabolites investigated here only represent a small selection of the metabolome, yet, compound classes mainly identified as significantly altered in previous studies were included (amino acids and derivatives, alcohols, organic acids, and fatty acids [13, 15, 16]) and were considered as sufficient for the intended purpose of the study.
Evaluation of decontamination procedures
Decontamination is a necessary step, as hair samples might be contaminated with sweat, sebum, or dust that deposited on the hair shaft, possibly leading to misinterpretation of results or interfering with analytical measurements. To evaluate the suitability of different decontamination procedures, hair samples were washed applying three different washing protocols and extracts were analyzed, along with genuine (not decontaminated) hair samples. Washing solutions were kept and analyzed to monitor metabolites that had been removed during the different washing steps. Subsequently, the relative amounts of metabolites found in the wash solutions were compared. Decontamination solvents used in this study were chosen according to SoHT guidelines where a washing procedure with an aqueous and organic washing step is recommended [38] and based on the method development and routine experience in our laboratory. The influence of the first washing step, either using a protic solvent which swells the hair matrix or an aprotic solvent that only removes greasy deposits from the hair shaft without hair swelling properties, was investigated.
Comparing extracts of genuine and decontaminated hair samples in general, different results were obtained as depicted exemplarily for nicotinamide and arginine in Fig. 1. For nicotinamide, dexpanthenol, and triethanolamine, a significant difference between genuine and decontaminated samples could be observed, whereas other metabolites such as arginine did not show any significant differences. Overall, the abundances of nicotinamide, dexpanthenol, and triethanolamine were relatively high suggesting a high contamination from the outside. Nicotinamide, dexpanthenol, and triethanolamine are common ingredients in cosmetic hair care products [39,40,41] and triethanolamine is described to be detected in sweat [42]. The impact of external contamination cannot be estimated in advance and should be kept to a minimum to allow correct interpretation of biological variances. These results therefore strongly support the need for a decontamination step of hair samples prior to metabolomic analysis as it has already been applied in current hair metabolomics literature. Furthermore, a standardization of wash protocol procedures would be desirable in order to ensure a better comparability of results.
Besides these examples where an external contamination is very likely, other metabolites did not show any significant difference between genuine and decontaminated hair even though they were detected in wash water solutions (see Figs. 1 and 2). This leads to the hypothesis that some metabolites are most likely mainly incorporated in inner hair compartments and are hardly deposited on the hair surface. Protic solvents, such as H2O, swell the hair matrix and therefore entail extraction already during the washing step, which could explain these differences.

Heatmaps of analytes found in wash solutions of wash protocols A (1. DCM, 2. H2O) and B (1. H2O, 2. DCM) with LC-MS/MS analysis. a Black (higher quantity found in relation to corresponding washing step), gray (lower quantity), black check (equal quantity), white dotted (not detected). b Sum of analytes: black: higher quantity than gray, same color: equal sum (not significant)
A more detailed distribution of metabolites in different wash water fractions of wash protocols A and B being directly comparable, analyzed with LC-MS/MS, is given in Fig. 2. Generally, higher quantities were detected in the aqueous phases compared to the DCM phases for polar metabolites. Despite the different amounts found in the isolated wash waters, the total sum of metabolites removed during the decontamination steps did not differ between wash protocols A and B for the majority of the investigated metabolites (see Fig. 2b). The higher recovery of polar metabolites in aqueous wash waters is not surprising, since theoretically, a washing step consisting of H2O should remove hydrophilic compounds to a larger extent than DCM due to better dissolving properties and vice versa. For wash protocol C, the total quantity of removed metabolites was slightly higher for some metabolites. As the acetone washing steps from wash protocols A and B were not analyzed and wash protocol C did not include an additional acetone washing step, the difference reflects metabolites in the acetone washing step as no differences in extraction yields could be observed. Hence, for polar compounds, the order of aqueous and organic washing steps does not seem to affect the total amount of polar metabolites being removed.
When looking at the results obtained from GC-MS analysis as given in Fig. 3, a different pattern is observed for rather hydrophobic compounds: the total amount of removed saturated fatty acids or squalene hardly varied in the first step, regardless of the washing solvent used. In contrast, if H2O was used as the first washing step (wash B and C), an increased removal in the second step could be seen compared to wash A. These results could be reproduced by repetition of the initial experiment. The fact that similar quantities of saturated fatty acids and squalene were observed in the first washing steps, regardless of the washing solvent used, leads to the hypothesis that those analytes had presumably been deposited by sebum and were removed mechanically trough the shaking process [43]. In addition, further washing out of hydrophobic compounds observed during the second washing step when H2O was used as the initial wash (wash B and C) suggests an incipient extraction process. As widely described, non-protic solvents (e.g., DCM) do not swell the hair and are believed to remove contaminants from the surface of the hair shaft only, in contrast to protic solvents (e.g., H2O) that cause swelling of the hair matrix [7]. As a consequence, washing the hair with a solvent that possesses good swelling properties might facilitate extraction of analytes in the subsequent washing steps.

Decontamination findings (sample weight-corrected peak areas, mean of three each ± standard deviation) for C12:0, C14:0, C15:0, and squalene found in wash solutions of wash protocols A (1. DCM, 2. H2O), B (1. H2O, 2. DCM) and C (1. H2O, 2. MeOH/acetone 1:1), analyzed with GC-MS. Black: first washing step, light gray: second washing step
Though, we believe that an aqueous washing step is necessary as a non-protic solvent such as DCM is not suitable to remove polar metabolites that originate from, e.g., sweat. The impact of a possible external contamination should be kept to a minimum to allow correct interpretation of analysis results. Moreover, the aqueous washing step is not enough to achieve a full extraction and presents just a small percentage of the amount which is detected through extraction.
Finally, a two-step washing protocol that removes the greasy layer on the hair shaft with a non-protic solvent (e.g., DCM) followed by a second, H2O-only step to avoid early extraction of analytes, is favorable. Therefore, wash protocol A presented a good compromise and was consequently used for the following experiments.
Evaluation of homogenization procedures
After the decontamination step, hair samples are usually reduced in size to promote better extraction by increasing the sample surface. In theory, the higher and more consistent the surface, the higher the extraction rate and the better the reproducibility should become. To investigate the impact of different homogenization procedures on the detection of metabolites, hair was either pulverized using a ball mill or cut into millimeter segments using a pair of scissors (see “Materials and methods”).
An initial comparison of pulverized hair vs. snippets was performed on the number of features detected for each homogenization procedure. After a search against the databases METLIN, NIST, HMDB, and Lipidblast, a total of 23,036 features could be detected for pulverized hair samples and 17,335 for snipped samples for LC-MS/MS analysis (see Table 1). Thereof, 18,505 features for pulverized and 13,530 for snipped samples could be tentatively identified. For GC-MS analysis, a total of 287 peaks for pulverized and 276 peaks for snipped samples could be detected.
In detail, significantly higher extraction results (p < 0.05) were obtained for trimethyllysine, N-acetylneuraminic acid, and riboflavin after LC-MS/MS analysis, if samples were pulverized. Six out of 35 identified metabolites showed significantly higher peak area ratios for snipped hair samples (choline, histidine, C10, adenosine, inosine, and methylguanosine). Extraction results for all other metabolites were not significantly different. For GC-MS analysis, tyrosine showed a significantly higher peak area ratio in pulverized samples, whereas peak area ratios of myristic acid and pentadecanoic acid were significantly higher for snipped samples. Again, all other metabolites did not show significant differences. Destruction of the hair structure and increasing the hair surface by pulverization has been shown to effectively improve the extraction of drugs of abuse [44], ethyl glucuronide (EtG) [45, 46], and steroid esters [47]. In the present study, pulverization yielded a higher number of detected and tentatively identified metabolites, which raises the likelihood to detect metabolome changes.
On the other hand, an enormous increase of the hair surface by pulverization might lead to an adsorption of analytes on the hair debris and higher matrix effects potentially leading to ion suppression or increased background noise when analyzed with LC-MS/MS. This might explain the comparably higher extraction yields of some single analytes in snipped samples.
Whenever possible, sample preparation steps should be standardized as much as possible. Relative standard deviations (rSD) ranged from 1.7 to 55% for pulverized and from 3.7 to 68% for snipped samples as summarized in Table 2. However, for 14 analytes from snipped samples, the rSD was over 30%. In contrast, for pulverized samples, rSD was over 30% for only 6 analytes, demonstrating a better homogeneity for pulverized samples. Manually cutting hair samples into snippets automatically leads to a greater variation of hair segment lengths. Mechanical pulverization of hair samples results in more homogenous samples and is advantageous for the reproducibility of the method as observed with the smaller rSD values. Those results are in line with findings observed by Aqai et al. [47] who found higher standard deviations for unground hair samples and Becker et al. who reported smaller interlaboratory variances in case of pulverization [48]. In conclusion, to maintain reproducible results with sufficient recovery, pulverization was chosen as homogenization procedure.
Evaluation of extraction protocols
As the metabolome is chemically very diverse, the sample extraction from the solid hair matrix into solution for further MS analysis is a challenging task. The detection of all metabolites within one method combined with sufficient sensitivity is not possible to date. Hence, expanding the knowledge about the extraction of endogenous compounds from hair could be of great benefit, especially if metabolites of interest are known, e.g., for the evaluation of metabolic pathways. Beforehand, different solvent mixture ratios for ACN/H2O were tested for extraction efficiency in terms of quantity and number of detected metabolites. A solvent mixture of ACN/H2O (2:8, v/v) provided the best extraction efficiencies overall (data not shown). Based on these preliminary results, the extraction of metabolites with ACN/ammonium formate buffer 0.5 M (2:8, v/v, pH 4), ACN/H2O (2:8, v/v, pH 6), and ACN/ammonium bicarbonate buffer 0.5 M (2:8, v/v, pH 8.5) was compared in this study. As depicted in Fig. 4, for most analytes, a difference in extraction tendencies could be seen for the three different pH conditions. With ACN/H2O pH 6, best extraction results were obtained for amino acids, citrulline, trimethyllysine, riboflavin, and triethanolamine. Glycerophosphocholine was equally well extracted with ACN/H2O pH 6 and ACN/buffer pH 8.5, and tyrosine with ACN/H2O pH 6 and ACN/buffer pH 4. In general, extraction with ACN/buffer pH 8.5 was favorable for the detection of carnitines and saturated fatty acids as well as several single metabolites (methyladenosine, methylguanosine, or pantothenic acid). Urocanic acid, nicotinamide, nicotine, uric acid, and hypoxanthine were best extracted with ACN/buffer pH 4. However, for metabolites out of almost every compound class, no clear extraction tendency towards one pH could be observed. In theory, basic substances should be effectively extracted in acidic conditions due to protonation of present nitrogen atoms which increases their aqueous solubility [21]. This is true for nicotine (Fig. 5c) and nicotinamide that were effectively extracted using ACN/buffer pH 4. On the other hand, acidic molecules should be best extracted under basic conditions due to deprotonation of the carboxylic acid group, again increasing their water solubility. This was shown to be the case for saturated fatty acids that were best extracted with ACN/buffer pH 8.5. Despite these plausible examples, for some metabolites, a different extraction pattern would have been expected if underlying the hypothesis that metabolites would be extracted according to the pKa only. As an example, phenylalanine (Fig. 5a) has pKa values of 2.47 (strongest acid) and 9.45 (strongest basic) and an isoelectric point (pI) of 6. Considering the (de)protonation stages, the least protonation would be expected around pH 6 where phenylalanine is suspected to be uncharged, therefore having the least water solubility at that pH. Increasing the pH to 8.5 would increase the deprotonation of the carboxylic acid group, resulting in higher extraction yields for pH 8.5 compared to pH 6. However, extraction with ACN/buffer pH 8.5 resulted in lowest extraction of phenylalanine. These results are in line with studies from Madry et al. and Chata et al. who concluded that the pKa is not necessarily a prediction marker for the extraction yield [26, 49].

Heatmap of pH extraction tendencies. Extraction of pulverized hair samples decontaminated according to wash protocol A, a analysis with LC-MS/MS, b analysis with GC-MS. Black: highest extraction, gray: lowest extraction, black check: medium extraction, white dotted: no tendency towards one pH, cross: not detected

Exemplary extraction tendencies (mean of three each ± standard deviation) for extraction with ACN/H2O (2:8) of pulverized hair samples, decontaminated according to wash protocol A and analyzed with LC-MS/MS: a phenylalanine, b guanosine, and c nicotine. Statistical analysis with ordinary one-way ANOVA: p > 0.05 (not significant, without indication), p ≤ 0.05 (*), and p ≤ 0.01 (**)
Surprisingly, exceptional poor extraction results were obtained for the extraction with ACN/buffer pH 4 and subsequent analysis with GC-MS (Fig. 4b). To exclude a possible interference of the buffer mixture with the GC-MS system, another additional experiment was performed. Three hair samples were decontaminated with wash protocol A, pulverized, and extracted with ACN/H2O as described above. Extracts were pooled, separated into three aliquots, and evaporated to dryness followed by reconstitution in ACN/buffer pH 4, ACN/H2O pH 6, and ACN/buffer pH 8.5 respectively. Except for pyroglutamic acid with a very low abundance, no analytes were detected in the sample that was reconstituted in ACN/buffer pH 4, whereas abundances for metabolites in samples of ACN/H2O pH 6 and ACN/buffer pH 8.5 were comparable. Hence, the poor extraction results were not due to an extraction effect and sample preparation with ACN/buffer pH 4 for GC-MS analysis was considered unsuitable.
Taken together, a combination of different extraction methods would be the best to cover as many metabolites as possible. Nevertheless, this is connected with a large amount of time spent on the sample preparation and the subsequent data processing. Therefore, extraction with ACN/H2O was considered the best compromise as previous studies on the hair metabolome mostly identified amino acids, amino acid derivatives, organic acids, and fatty acids as significantly altered [13, 15, 16].
Conclusion
Using a combination of the two complementary analytical techniques, LC-MS/MS and GC-MS, the reliable detection of a multitude of metabolites from different chemical classes in hair was possible as well as the successful study of decontamination, homogenization, and extraction parameters. A multi-step decontamination with DCM, acetone, H2O, acetone, and homogenization by pulverization can be suggested. In any case, to maintain a high reproducibility, a standardization of sample preparation procedures for hair metabolomics is highly recommended. However, we are aware that the metabolites investigated within this study present only a small selection out of the (hair-) metabolome. However, our current studies now provide the basis for further studies on the longitudinal composition of the hair metabolome (accounting for influences of weathering effects), the analysis of biological variances between individuals and a quantitative determination to report endogenous levels of metabolites under different conditions (e.g., drug consumption).
References
Patti GJ, Yanes O, Innovation SG. Metabolomics: the apogee of the omics trilogy. Nat Rev Mol Cell Biol. 2012;13(4):263–9.
Dinis-Oliveira RJ. Metabolomics of drugs of abuse: a more realistic view of the toxicological complexity. Bioanalysis. 2014;6(23):3155–9.
Dettmer K, Aronov PA, Hammock BD. Mass spectrometry-based metabolomics. Mass Spectrom Rev. 2007;26(1):51–78. https://doi.org/10.1002/mas.20108.
Nicholson JK, Lindon JC, Holmes E. ‘Metabonomics’: understanding the metabolic responses of living systems to pathophysiological stimuli via multivariate statistical analysis of biological NMR spectroscopic data. Xenobiotica. 1999;29(11):1181–9.
Monteiro MS, Carvalho M, Bastos ML, Guedes de Pinho P. Metabolomics analysis for biomarker discovery: advances and challenges. Curr Med Chem. 2013;20(2):257–71.
Boxler MI, Liechti ME, Schmid Y, Kraemer T, Steuer AE. First time view on human metabolome changes after a single intake of 3,4-methylenedioxymethamphetamine in healthy placebo-controlled subjects. J Proteome Res. 2017;16(9):3310–20.
Pragst F, Balikova MA. State of the art in hair analysis for detection of drug and alcohol abuse. Clin Chim Acta. 2006;370(1–2):17–49.
Binz TM, Rietschel L, Streit F, Hofmann M, Gehrke J, Herdener M, et al. Endogenous cortisol in keratinized matrices: systematic determination of baseline cortisol levels in hair and the influence of sex, age and hair color. Forensic Sci Int. 2018;284:33–8.
Binz TM, Braun U, Baumgartner MR, Kraemer T. Development of an LC-MS/MS method for the determination of endogenous cortisol in hair using (13)C3-labeled cortisol as surrogate analyte. J Chromatogr B Anal Technol Biomed Life Sci. 2016;1033-1034:65–72.
Greff MJE, Levine JM, Abuzgaia AM, Elzagallaai AA, Rieder MJ, van Uum SHM. Hair cortisol analysis: an update on methodological considerations and clinical applications. Clin Biochem. 2019;63:1–9.
Rashaid AH, Harrington Pde B, Jackson GP. Profiling amino acids of Jordanian scalp hair as a tool for diabetes mellitus diagnosis: a pilot study. Anal Chem. 2015;87(14):7078–84.
Masukawa Y, Narita H, Imokawa G. Characterization of the lipid composition at the proximal root regions of human hair. J Cosmet Sci. 2005;56(1):1–16.
Delplancke TDJ, de Seymour JV, Tong C, Sulek K, Xia Y, Zhang H, et al. Analysis of sequential hair segments reflects changes in the metabolome across the trimesters of pregnancy. Sci Rep. 2018;8(1):36.
Jones B, Han TL, Delplancke T, McKenzie EJ, de Seymour JV, Chua MC, et al. Association between maternal exposure to phthalates and lower language ability in offspring derived from hair metabolome analysis. Sci Rep. 2018;8(1):6745.
Sulek K, Han TL, Villas-Boas SG, Wishart DS, Soh SE, Kwek K, et al. Hair metabolomics: identification of fetal compromise provides proof of concept for biomarker discovery. Theranostics. 2014;4(9):953–9.
He X, de Seymour JV, Sulek K, Qi H, Zhang H, Han TL, et al. Maternal hair metabolome analysis identifies a potential marker of lipid peroxidation in gestational diabetes mellitus. Acta Diabetol. 2016;53(1):119–22.
Sumner LW, Amberg A, Barrett D, Beale MH, Beger R, Daykin CA, et al. Proposed minimum reporting standards for chemical analysis chemical analysis working group (CAWG) metabolomics standards initiative (MSI). Metabolomics. 2007;3(3):211–21.
Deda O, Chatziioannou AC, Fasoula S, Palachanis D, Raikos N, Theodoridis GA, et al. Sample preparation optimization in fecal metabolic profiling. J Chromatogr B Anal Technol Biomed Life Sci. 2017;1047:115–23.
Raterink RJ, Lindenburg PW, Vreeken RJ, Ramautar R, Hankemeier T. Recent developments in sample-pretreatment techniques for mass spectrometry-based metabolomics. TrAC Trends Anal Chem. 2014;61:157–67.
Yin P, Lehmann R, Xu G. Effects of pre-analytical processes on blood samples used in metabolomics studies. Anal Bioanal Chem. 2015;407(17):4879–92.
Vogliardi S, Tucci M, Stocchero G, Ferrara SD, Favretto D. Sample preparation methods for determination of drugs of abuse in hair samples: a review. Anal Chim Acta. 2015;857:1–27.
Society of Hair Testing. Recommendations for hair testing in forensic cases. Forensic Sci Int. 2004;145(2–3):83–4.
Kintz P, Cirimele V, Jamey C, Ludes B. Testing for GHB in hair by GC/MS/MS after a single exposure. Application to document sexual assault. J Forensic Sci. 2003;48(1):195–200.
Choi B, Kim SP, Hwang S, Hwang J, Yang CH, Lee S. Metabolic characterization in urine and hair from a rat model of methamphetamine self- administration using LC- QTOF-MS-based metabolomics. Metabolomics. 2017. https://doi.org/10.1007/s11306-017-1257-0.
Xie P, Wang TJ, Yin G, Yan Y, Xiao LH, Li Q, et al. Metabonomic study of biochemical changes in human hair of heroin abusers by liquid chromatography coupled with ion trap-time of flight mass spectrometry. J Mol Neurosci. 2016;58(1):93–101.
Madry MM, Kraemer T, Baumgartner MR. Systematic assessment of different solvents for the extraction of drugs of abuse and pharmaceuticals from an authentic hair pool. Forensic Sci Int. 2018;282:137–43.
Dunn WB, Broadhurst D, Begley P, Zelena E, Francis-McIntyre S, Anderson N, et al. Procedures for large-scale metabolic profiling of serum and plasma using gas chromatography and liquid chromatography coupled to mass spectrometry. Nat Protoc. 2011;6(7):1060–83.
Steuer AE, Arnold K, Kamber D, Kraemer T. Suitability evaluation of new endogenous biomarkers for the identification of nitrite-based urine adulteration in mass spectrometry methods. Drug Test Anal. 2018. https://doi.org/10.1002/dta.2481.
Boxler MI, Schneider TD, Kraemer T, Steuer AE. Analytical considerations for (un)-targeted metabolomic studies with special focus on forensic applications. Drug Test Anal. 2018. https://doi.org/10.1002/dta.2540.
Brockbals L, Habicht M, Hajdas I, Galassi FM, Ruhli FJ, Kraemer T. Untargeted metabolomics-like screening approach for chemical characterization and differentiation of canopic jar and mummy samples from Ancient Egypt using GC-high resolution MS. Analyst. 2018;143(18):4503–12.
Linstrom PJ, Mallard WG. NIST Chemistry WebBook. NIST Standard Reference Database Number 69, 2001.
Guijas C, Montenegro-Burke JR, Domingo-Almenara X, Palermo A, Warth B, Hermann G, et al. METLIN: a technology platform for identifying knowns and unknowns. Anal Chem. 2018;90(5):3156–64.
Kind T, Liu KH, Lee DY, DeFelice B, Meissen JK, Fiehn O. LipidBlast in silico tandem mass spectrometry database for lipid identification. Nat Methods. 2013;10(8):755–8.
Wishart DS, Tzur D, Knox C, Eisner R, Guo AC, Young N, et al. HMDB: the human metabolome database. Nucleic Acids Res. 2007. https://doi.org/10.1093/nar/gkl923.
Zhu X, Chen Y, Subramanian R. Comparison of information-dependent acquisition, SWATH, and MS(All) techniques in metabolite identification study employing ultrahigh-performance liquid chromatography-quadrupole time-of-flight mass spectrometry. Anal Chem. 2014;86(2):1202–9.
Noppe G, de Rijke YB, Dorst K, van den Akker EL, van Rossum EF. LC-MS/MS-based method for long-term steroid profiling in human scalp hair. Clin Endocrinol. 2015;83(2):162–6.
Cuykx M, Rodrigues RM, Laukens K, Vanhaecke T, Covaci A. In vitro assessment of hepatotoxicity by metabolomics: a review. Arch Toxicol. 2018;92(10):3007–29.
Cooper GA, Kronstrand R, Kintz P, Society of Hair Testing. Society of Hair Testing guidelines for drug testing in hair. Forensic Sci Int. 2012;218(1–3):20–4.
De AK, Chowdhury PP, Chattapadhyay S. Simultaneous quantification of dexpanthenol and resorcinol from hair care formulation using liquid chromatography: method development and validation. Scientifica (Cairo). 2016;2016:1537952.
Cosmetic Ingredient Review Expert P. Final report of the safety assessment of niacinamide and niacin. Int J Toxicol. 2005;24(Suppl 5):1–31.
Fiume MM, Heldreth B, Bergfeld WF, Belsito DV, Hill RA, Klaassen CD, et al. Safety assessment of triethanolamine and triethanolamine-containing ingredients as used in cosmetics. Int J Toxicol. 2013;32(3 Suppl):59S–83S.
Delgado-Povedano MM, Calderon-Santiago M, Priego-Capote F, Luque de Castro MD. Development of a method for enhancing metabolomics coverage of human sweat by gas chromatography-mass spectrometry in high resolution mode. Anal Chim Acta. 2016;905:115–25.
Camera E, Ludovici M, Galante M, Sinagra JL, Picardo M. Comprehensive analysis of the major lipid classes in sebum by rapid resolution high-performance liquid chromatography and electrospray mass spectrometry. J Lipid Res. 2010;51(11):3377–88.
Eser HP, Potsch L, Skopp G, Moeller MR. Influence of sample preparation on analytical results: drug analysis [GC/MS] on hair snippets versus hair powder using various extraction methods. Forensic Sci Int. 1997;84(1–3):271–9.
Salomone A, Baumgartner MR, Lombardo T, Alladio E, Di Corcia D, Vincenti M. Effects of various sample pretreatment procedures on ethyl glucuronide quantification in hair samples: comparison of positivity rates and appraisal of cut-off values. Forensic Sci Int. 2016;267:60–5.
Monch B, Becker R, Nehls I. Quantification of ethyl glucuronide in hair: effect of milling on extraction efficiency. Alcohol Alcohol. 2013;48(5):558–63.
Aqai P, Stolker AA, Lasaroms JJ. Effect of sample pre-treatment on the determination of steroid esters in hair of bovine calves. J Chromatogr A. 2009;1216(46):8233–9.
Becker R, Lo I, Sporkert F, Baumgartner M. The determination of ethyl glucuronide in hair: experiences from nine consecutive interlaboratory comparison rounds. Forensic Sci Int. 2018;288:67–71.
Chata C, E MH, Grova N, Appenzeller BM. Influence of pesticide physicochemical properties on the association between plasma and hair concentration. Anal Bioanal Chem. 2016;408(13):3601–12.
Acknowledgements
The authors would like to express their gratitude to Emma Louise Kessler, MD, for her generous legacy; she donated to the Institute of Forensic Medicine at the University of Zurich, Switzerland, for research purposes.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Hair samples were collected from two healthy volunteers who provided written informed consent. According to Swissethics (Humanforschungsgesetz), no further ethical approval from the cantonal ethic commission is necessary if the research is not aiming to investigate diseases or functions of the human body as is the case in the current study.
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
ESM 1
(PDF 231 kb)
Rights and permissions
About this article

Cite this article
Eisenbeiss, L., Steuer, A.E., Binz, T.M. et al. (Un)targeted hair metabolomics: first considerations and systematic evaluation on the impact of sample preparation. Anal Bioanal Chem 411, 3963–3977 (2019). https://doi.org/10.1007/s00216-019-01873-4
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00216-019-01873-4