
Abstract
Thrombosis is considered as the most typical example of multigenic/multifactorial disorder. The three most common genetic risk factors for thrombotic disorders are the G1691A mutation in factor V gene (FV Leiden), the G20210Α mutation in prothrombin gene (FII), and the C677T mutation in the methylenetetrahydrofolate reductase (MTHFR) gene. An additional panel of biomarkers predisposing for thrombotic events includes the H1299R variant in factor V gene (HR2), A1298C variant in MTHFR gene, the V34L mutation in fibrinogen stabilizing factor XIII (FXIII) gene as well as the 4G/5G polymorphism in plasminogen activator inhibitor type-1 (PAI-1) gene. In this context, we report a novel, rapid and low-cost two-panel diagnostic platform for the simultaneous visual genotyping of the seven mutations (14 alleles). The proposed method comprises the following: (a) a multiplex PCR using genomic DNA isolated from peripheral blood, (b) a multiplex genotyping reaction based on allele-specific primer extension, and (c) visual detection of the genotyping reaction products by means of a multi-allele dipstick-type DNA biosensor, using gold nanoparticles as reporters. The method was applied to 40, previously characterized, and 15 blind clinical samples and the results were 100 % accurate. The proposed assay is simple to perform, requires no specialized and costly equipment, and eliminates multiple pipetting, incubation, and washing steps.

Genotyping platform for visual detection of point mutations using PEXT reaction and multi-allele DNA biosensor
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Thrombophilia or a predisposition for thrombosis can arise from genetic factors, acquired changes in the clotting mechanism, or, more commonly, an interaction between genetic and acquired factors. The genetic factors that can contribute to thromboembolism include mutations at several well-defined loci in genes that code for proteins involved in coagulation, fibrinolysis, and homocysteine metabolism. The three most common genetic risk factors linked with thrombotic events are the G1691A mutation in the gene of coagulation factor V, known as FV Leiden, the G20210A mutation in the FII (prothrombin, PTH) gene, and the C677T mutation in the methylenetetrahydrofolate reductase (MTHFR) gene.
The FV Leiden was first described in 1994 and is the most common inherited risk factor for thrombophilia. It is a point mutation that causes a single amino acid substitution (Gln substitutes Arg in position 506 of the protein) leading to inability of activated protein C to proteolyse and inactivate the coagulation factor V (activated protein C resistance) [1, 2].
The G20210A mutation in the FII gene was first described in 1996 and is a point mutation that occurs in the 3′ untranslated region of the gene, leading to higher levels of normal functional prothrombin. It is the second most common genetic risk factor for thrombotic events [1, 3].
The C677T mutation in the MTHFR gene is the third important genetic risk factor. It is a point mutation that renders the MTHFR enzyme thermo-unstable, reducing its capacity in converting the homocysteine to methionine, in the metabolic pathway of methionine and cysteine. As a consequence, high levels of homocysteine are detected in plasma, a condition known as hyperhomocysteinemia, which is considered as a risk factor for atherosclerosis and thrombosis [4, 5]. The three aforementioned genetic risk factors are part of the screening tests for inherited thrombophilia in patients with thromboembolic episodes.
An additional panel of biomarkers predisposing for thrombotic events includes the H1299R variant in FV gene (known as HR2), the A1298C variant in MTHFR gene, the V34L mutation in fibrinogen stabilizing factor XIII (FXIII) gene as well as the 4G/5G polymorphism in plasminogen activator inhibitor type-1 (PAI-1) gene.
The FV H1299R variant along or in coinheritance with FV Leiden appears to be associated with severe activated protein C (APC) resistance phenotype [6].
The MTHFR A1298C variant reduces the activity of MTHFR enzyme to a lesser extent than in the case of the C677T polymorphism [7]. This reduced enzyme activity leads to hyperhomocysteinemia, which has been associated with several vascular conditions including coronary artery disease and deep vein thrombosis [8].
The factor XIII, or fibrin stabilizing factor, is an enzyme that cross-links fibrin during clot formation. Mutations in factor XIII were found to be associated with protection against some cardiovascular events. The most common V34L sequence variant reduces the clot stability by modifying the structure of the cross-linked fibrin, thus offering protection against thrombotic events. The V34L sequence variant results from a G>T substitution at position 103 in the FXIIIA1 gene, which is located on chromosome 6p24-25 [9].
Furthermore, polymorphisms in the plasminogen activator inhibitor type-1 (PAI-1) gene, including the 4G/5G polymorphism, contribute to the occurrence of thrombotic events by increasing the level of PAI-1 thus leading to excessive build-up of fibrin [10].
Nowadays, most of the hospitals and diagnostic laboratories use the polymerase chain reaction in combination with restriction fragment length polymorphism (PCR-RFLP) methods and allele-specific PCR for the detection of the mutations as they are easy to set up and perform [11–13]. In addition, high-throughput testing platforms, including Luminex xTAG microsphere-based universal array and Sequenome Mass array iPlex Gold genotyping platforms, have been developed for carrying out large-scale analyses in which large numbers of SNPs related to thrombophilia are analyzed in large numbers of patients [14, 15]. The significance of all these tests is high, since they can reveal the patients with an increased probability of recurrence as well as other family members with thrombophilia disorder [16].
The aim of the present work was to develop a simple, rapid, and low-cost two-panel diagnostic platform for visual genotyping (by naked eye) of the above seven mutations, simultaneously, using novel dipstick-type and disposable DNA biosensors. The assay involves a multiplex PCR, a multiplex primer extension reaction, and visual detection of the reaction products by means of single DNA biosensors. Visual detection eliminates the need for specialized equipment and highly qualified technical personnel, and is possible within 30 min after PCR. The biosensor functions in a lateral-flow mode, in which the reagents are deposited in dry-form on the sensor’s strip. Following immersion of the biosensor into the running buffer, the solution migrates along the strip by capillary action, thus redissolving the reagents and enabling capture and detection of the genotyping reaction products without the need for incubation and washing steps. The first biosensor strip (panel 1) has seven spots, i.e., a spot for each of FV Leiden, FII, and MTHFR C677T allele (total of six diagnostic spots) as well as a control spot to confirm the proper function of the sensor. The second biosensor strip (panel 2) has nine spots, i.e., a spot for each of FV H1299R, MTHFR A1298C, FXIII V34L, and PAI-1 4G/5G allele (total eight diagnostic spots) and a control spot. A spot turns red when the corresponding allele is present. Thus, the genotype for each locus is assigned visually by the position of the red spots.
Materials and methods
Instrumentation
PCR and primer extension (PEXT) reaction were performed in the thermal cyclers TaKaRa (Sanyo E&E Europe BV) and MiniCycler PTC-0150 (MJ Research). The electrophoresis apparatus, Sub-Cell GT, was from Biorad (Hercules, CA), and the UV lamp from Vilber Lourmat (Marne la Valee, France). A digital camera, Kodak DC 120, and the Gel Analyzer software for DNA documentation were purchased from Kodak (New York, NY). Microcentrifuge, Mikro-20, was from Hettich (Tuttlingen, Germany). The Guillotine cutting apparatus (Q-Connect) was used for cutting the strips. Images of the strips were captured by scanning the strips, using a simple desktop scanner (ScanJet 4300C, Hewlett-Packard). The intensity of the spots was estimated using the image analysis software (freeware) GelAnalyzer 2010a (www.gelanalyser.com)
Materials and reagents
Kapa 2G Fast multiplex PCR kit was obtained from Kapa Biosystems (London, UK). Vent (exo-) DNA polymerase was purchased from New England Biolabs (Ipswich, MA). Terminal deoxynucleotidyl transferase (TdT) and φΧ174 DNA/BsuRI DNA molecular weight marker were from Fermentas (Vilnius, Lithuania). Ultrapure deoxyribonucleoside triphosphates (dNTPs) were purchased from HT Biotechnology (Cambridge, UK) and biotin-11-dUTPs from Applichem (Darmstadt, Germany). Anti-biotin antibody was from Pierce (ThermoFisher Scientific, Waltham, MA). Gold nanoparticles (40 nm, 9 × 1010 particles/mL) were purchased from British Biocell International (Cardiff, UK), and carboxylated polystyrene microspheres (2 μm) from Polysciences (Warrington, PA, USA). The restriction enzyme Hinf I was obtained from Promega. Immunopore FP membrane (5 μm pore size) was from Whatman (Florham Park, NJ) and the plastic adhesive backing, wicking pad, glassfiber conjugate pad, and absorbent pad were from Schleicher & Schuell (Dassel, Germany). Oligonucleotides used as primers and probes in the course of this study were designed in silico using PrimerPremier 5 and PrimerPlex 2 software (Premier Biosoft International, Palo Alto, CA) and synthesized by the VBC-Biotech (Vienna, Austria). All primers and probes sequences are available on request to the authors. The reagents EDC (C8H17N3-HCl) and MES (C6H13NO4S) were purchased from Applichem. EDTA (C10H16N2O8), Tween-20 (C58H114O26), NaOH, and NaCl were from Fluka. Agarose, borax (Na2B4O7·10 H2O), ethidium bromide (EtBr), glycerol (C3H8O3), sodium citrate (Na3C6H5O7), Tris (C4H11NO3), NH4Cl, and ΚCl were from Sigma. Dimethylsulfoxide (DMSO, C2H6OS) and formamide (CH3NO) were obtained from Roth (Karlsruhe, Germany). Bovine serum albumin (BSA) and SDS (NaC12H25SO4) were from Serva (Heidelberg, Germany). All common reagents were purchased from Sigma (St Louis, MO, USA) or Fluka (Buchs, Switzerland).
Methods
Conjugation of gold nanoparticles with anti-biotin antibody
Conjugation of gold nanoparticles with anti-biotin antibody was performed according to [17] with minor modifications. In brief, a 1-mL aliquot of gold nanoparticles (Au NP, 40 nm, 9 × 1010 particles/mL) (0.15 pmol) was adjusted to pH 9 by adding a 200 mM borax (Na2B4O7·10 H2O) solution. Then, 4 μg of anti-biotin antibody, diluted in 100 μL of 2 mM borax solution, was added to the nanoparticle solution, gradually by stirring. The mixture was incubated in the dark, at ambient temperature, for 45 min, followed by the addition of 100 μL of 100 g/L BSA in 20 mM borax. The mixture was incubated in the dark, at ambient temperature for 10 min, and centrifuged at 4500×g for 10 min. The supernatant was discarded, and the pellet was resuspended in 100 μL wash solution (10 g/L BSA in 2 mM borax) followed by centrifugation at 4500×g for 8 min. The supernatant was discarded, and the pellet was resuspended in 50 μL of reconstitution buffer 1 g/L BSA, 1 g/L NaN3, and 2 mM borax and stored at 4 °C. The functionalized gold nanoparticles were loaded on the conjugate pad at a density of 10 fmol/4 mm and let dry.
Conjugation of polystyrene microspheres with oligonucleotides
Conjugation of polystyrene microspheres with oligonucleotides was performed according to [17] as follows. A 12.8-μL aliquot of a 5.6 × 106/μL suspension of 2-μm white polystyrene microspheres (0.12 fmol) with carboxyl groups on their surface was added in 150 μL of 0.1 M MES, pH 4.5, and centrifuged for 2 min at 15,000×g. The supernatant was discarded and the microspheres were resuspended in 160 μL of 0.1 M MES, pH 4.5. Then, they were mixed with 400 pmol of the oligonucleotide probe that carried an amino group at the 5′ end. A 5-μL aliquot of a freshly prepared 320 g/L EDC solution, in 0.1 M MES, was added and the mixture was incubated under shaking, at ambient temperature for 30 min. The addition of EDC was repeated with a fresh EDC solution (5 μL) and the mixture was incubated for another 30 min, followed by the addition of 2 μL of 100 mL/L Tween-20. The conjugates were pelleted by centrifugation at 15,000×g for 2 min and washed twice with 100 μL of 10 mM Tris–HCl, 1 mM EDTA, 2 mL/L Tween-20, pH 8. Finally, the microspheres were resuspended in 64 μL of 10 mM Tris–HCl, 1 mM EDTA, pH 8. The concentration of microspheres in this suspension is 1.1 × 106/μL, assuming that no loss of beads occurs during the washing steps. The functionalized polystyrene microspheres were loaded on the membrane at a density of 5.6 × 105 particles/spot and let dry.
Tailing of oligonucleotide probes conjugated to the surface of polystyrene microspheres
Oligonucleotide probes (anti-tags) conjugated to the microspheres were tailed with biotin-dUTP using the TdT. The tailing reactions were performed in a total volume of 40 μL containing 0.2 M potassium cacodylate, 25 mM Tris–HCl, pH 7.2, 0.1 mL/L Triton X-100, 1 mM CoCl2, 0.5 mM of each dNTP and 25 μM of biotin-dUTP, 50 U of TdT, and 14.6 × 107 conjugated microspheres. After incubation for 1 h at 37 °C with occasional mixing, the microspheres were separated from the excess of biotin-dUTPs by centrifugation at 15,000×g, for 2 min, washed twice with 100 μL H2O and containing 2 mL/L Tween-20 and, finally, resuspended in 512 μL 10 mM Tris–HCl, 1 mM EDTA, pH 8 (final concentration 2.8 × 105 microspheres/μL).
Construction of dry-reagent dipstick-type DNA biosensor
The dry reagent strip biosensor (4 × 70 mm) was constructed as described previously [12]. It consisted of four parts: an immersion pad, a glass-fiber conjugate pad, a nitrocellulose diagnostic membrane, and an absorbent pad (Fig. 1B). All parts were assembled on a plastic adhesive backing as follows: the membrane (25 mm in length) was first placed on the backing. The absorbent pad (15 mm) was then positioned above the membrane, overlapping it by 2 mm. The conjugate pad (15 mm) was placed below the membrane, overlapping it by 2 mm. The immersion pad (15 mm) was placed below the conjugate pad, with a 2 mm overlapping. A 0.5-μL aliquot of microspheres (5.6 × 105 particles) conjugated with the anti-tag oligos was applied to the appropriate positions of the membrane, to form the test spots. In particular, anti-tag oligos corresponding to the wild-type (normal) alleles were spotted on the left side of the membrane, whereas the oligos that correspond to the variant (mutant) alleles were immobilized on the right side of the membrane. Allelic variants were positioned as follows (from the bottom to the top of the membrane): FV Leiden, FII, and MTHFR C677T for panel 1 and FV H1299R, FXIII, MTHFR A1298C, and PAI-1 for panel 2. A 0.8-μL aliquot of microspheres conjugated with biotinylated oligos was applied to the top of the membrane, to form a control spot. Antibiotin-functionalized gold nanoparticles (3 fmol/μL) were loaded by spotting on the conjugate pad of the strip at a density of 10 fmol/4 mm. Finally, the membrane was air-dried for 5 min. The strips were stored dry (with desiccant) at room temperature.

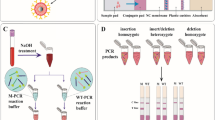
Schematic presentation of the two-panel genotyping platform for visual detection of point mutations related to inherited thrombophilia. (A) Multiplex primer extension genotyping reaction. Panel 1 and panel 2 amplification products are subjected (without prior purification) to a single PEXT reaction each using six or eight allele-specific primers, respectively (two primers for each polymorphic site). Each allele-specific primer comprises a sequence that is adjacent to the polymorphic site, with the 3′-end nucleotide complementary to the allelic variant and a sequence at the 5′-end which is different for each primer and enables capture of the primers by hybridization with the complementary oligonucleotides that are immobilized at the test zones of the sensor. During the 10 cycles of PEXT reaction, biotin-dUTP is incorporated into the extended primer(s) only if the 3′-end of the primer is perfectly complementary with the interrogated DNA sequence. If there is a mismatch at the 3′ end, then extension and biotin incorporation do not occur. (B) Visual detection of genotyping reaction products (by naked eye) by means of multi-allele DNA biosensors (IP—immersion pad; CP—conjugate pad; DM—diagnostic membrane; AP—absorbent pad). Following PEXT reaction, the primers are separated from the interrogated DNA by heat denaturation and the mixture is applied to the sensor. As the developing buffer migrates along the strip, all the allele-specific primers (extended and non-extended) are captured by the immobilized oligos at the test spots of the sensor. The antibiotin-functionalized gold nanoparticles bind only to the extended allele-specific primers. As a result, the test spots are colored red if extension has occurred denoting the presence of the corresponding allele in the original sample. The excess antibiotin-conjugated nanoparticles are captured by immobilized biotinylated oligonucleotides at the control spot of the sensor forming another red zone that indicates the proper performance of the system
Multiplex genotyping based on a dipstick-type DNA biosensor
Multiplex PCR for the FV Leiden-FII-MTHFR C677T mutations (panel 1)
A 181-bp segment in the FV gene, flanking the G1691A mutation, a 295-bp segment in the FII gene, flanking the G20210A mutation, and a 423-bp segment in the MTHFR gene flanking the C677T mutation were amplified, simultaneously, by a triplex PCR. Triplex PCR was performed using the commercially available Kapa 2G Fast multiplex PCR kit with 100 ng of genomic DNA in a final volume of 25 μL. The reaction mixture consisted of 1 U of polymerase, 3 mM MgCl2, 0.2 mΜ of each dNTP, 0.25 μΜ of each of the FV primers and 0.2 μΜ of each of the FII and MTHFR primers. Cycling conditions were set at 95 °C for 3 min followed by 35 cycles at 95 °C for 15 s, 60 °C for 30 s, and 72 °C for 15 s. Negative controls (containing water instead of DNA) were included in each series of PCR to confirm the absence of contamination. The PCR products were visualized by 1.8 % agarose gel electrophoresis and ethidium bromide staining.
Multiplex PCR for the FV H1299R-FXIII-MTHFR A1298C-PAI-1 mutations (panel 2)
A 331-bp segment in the PAI-1 gene, flanking the 4G/5G polymorphism, a 453-bp segment in the MTHFR gene, flanking the A1298C mutation, a 532-bp segment in the FXIII gene flanking the V34L mutation, and a 661-bp segment in the FV gene flanking the H1299R mutation were amplified, simultaneously, by a quadruplex PCR. Quadruplex PCR was performed using the commercially available Kapa 2G Fast Multiplex PCR kit with 100 ng of genomic DNA in a final volume of 25 μL. The reaction mixture consisted of 1 U of polymerase, 3 mM MgCl2, 0.2 mΜ of each dNTP, 0.3 μΜ of each of the FV primers, 0.2 μΜ of each of the FXIII primers, 0.6 μΜ of each of the MTHFR primers, and 0.5 μΜ of each of the PAI-1 primers. Cycling conditions were set at 95 °C for 3 min followed by 35 cycles at 95 °C for 15 s, 65 °C for 30 s, 72 °C for 30 s, and a final extension at 72 °C for 3 min. Negative controls were included in each series of PCR to confirm the absence of contamination.
Multiplex genotyping reaction for panel 1 variants
An aliquot (2–3 μL) of the PCR product (without prior purification) was subjected to a 6-plex PEXT reaction in a final volume of 20 μL containing 1× Vent(exo-) DNA polymerase buffer (20 mM Tris–HCl, 10 mM (NH4)2SO4, 10 mM KCl, 2 mM MgSO4, 0.1 % Triton X-100) supplemented with 0.5 mM MgSO4 (final Mg2+ concentration 2.5 mM), 1 U of Vent(exo-) DNA polymerase, 5 pmol each of the six allele-specific primers, 10 μM each of dATP, dCTP, dGTP, and 5 μΜ each of dTTP and biotin-dUTP. The thermal cycling conditions for the genotyping reaction were as follows: initial denaturation at 95 °C for 5 min, followed by 10 cycles of denaturation at 95 °C for 15 s, primer annealing at 68 °C for 15 s and extension at 72 °C for 15 s.
Multiplex genotyping reaction for panel 2 variants
An 8-plex PEXT reaction was performed in a total volume of 20 μL containing 1× Vent (exo-) DNA polymerase buffer (supplemented with 2.0 mM MgSO4, final Mg2+ concentration 4.0 mM), 1 U Vent (exo-) DNA polymerase, 3 μL of amplified DNA, 2.5 pmol each of the eight allele-specific primers, and 10 μM each of dATP, dCTP, dGTP, 5 μM dTTP, and 5 μΜ biotin-dUTP. The thermal cycling conditions were as follows: initial denaturation at 98 °C for 2 min, followed by 10 cycles of 98 °C for 30 s, 65 °C for 30 s, and 72 °C for 15 s. All PEXT reaction products were subjected to a final denaturation step at 95 °C for 5 min and held at 4 °C until use.
Visual detection of genotyping products by the multi-allele dipstick-type DNA biosensor
A 3-μL aliquot of the denatured PEXT reaction product was mixed with an equal volume of running buffer (2× SSC, pH 7, 5 mL/L Tween-20, 5 g/L SDS, 250 mL/L formamide, 20 mL/L glycerol, 30 g/L BSA). The mixture was then loaded to the upper part of the conjugate pad above the deposited anti-biotin gold nanoparticles (3.5 μL). The immersion pad was then placed into a tube containing 300 μL of running buffer. Visual detection of the PEXT reaction products was completed within 15–20 min. A red spot indicated the presence of the corresponding allele (normal or mutant).
Results and discussion
Assay principle
In total, seven genomic DNA fragments that span the mutations of interest are first amplified by two multiplex PCRs, a 3-plex and a 4-plex PCR for panel 1 and panel 2, respectively, using “Kapa 2G Fast Multiplex PCR kit”. The 3-plex PCR includes fragments flanking the three most common genetic risk factors linked with thrombotic events (FV Leiden, FII, and MTHFR C677T mutations), whereas the second 4-plex PCR includes fragments flanking the four additional biomarkers predisposing for thrombotic events (FV H1299R, MTHFR A1298C, FXIII V34L, and PAI-1 4G/5G allele) (see Electronic Supplementary Material (ESM) Fig. S1A). Subsequently, each of the multiplex PCR products is subjected to a 6-plex and 8-plex PEXT reaction for panel 1 and panel 2, respectively.
Schematics of the proposed multi-allele genotyping platform are shown in Fig. 1. Non purified PCR amplified products of either panel 1 and panel 2 gene fragments are subjected to few (10) cycles of multiplex PEXT reaction in the presence of allele-specific primers (two primers per mutation, one for the wild type, and the second for the mutant allele) and biotin-modified nucleotide that is incorporated in the extended product. For each locus, the allele-specific primers differed only in the last nucleotide, with the normal primer having a 3′ end nucleotide complementary to the normal allelic variant and the mutant primer having a 3′ end nucleotide complementary to the mutant allele. Each genotyping primer contained, at the 5′ end, a characteristic oligonucleotide sequence (tag). Due to the high accuracy of nucleotide insertion by Vent (exo-) DNA polymerase, the extension occurred only under conditions of absolute complementation between the allele-specific primer and the corresponding target sequence. The extended products were labeled with biotin through the incorporation of biotin-dUTP (Fig. 1A).
Visual detection of the genotyping reaction products is performed by means of multi-allele DNA biosensors (Fig. 1B). Besides the single “control spot” that was positioned at the top center of the sensor’s membrane, the six or eight diagnostic spots are arranged in a 3 × 2 or 4 × 2 matrix, in which each row represented a polymorphic locus, i.e., from bottom to top: FV Leiden, FII, and MTHFR for panel 1, and FV H1299R, FXIII, MTHFR A1298C, and PAI-1 for panel 2. The left and right spots of each row corresponded to the normal and mutant alleles, respectively. This positional encoding was enabled by the deposition, on each spot, of microspheres conjugated to specific anti-tag oligonucleotides that hybridize only with the complementary tag sequences at the 5′ end of each allele-specific primer.
An aliquot of the PEXT reaction product (heat-denatured to separate extended genotyping primers from any extended PCR primers and thus ensuring that both the tag sequence and the biotin moieties are located in the same strand) was applied to the biosensor and the lower part of the strip was immersed into the running buffer. Under the effect of capillary action, the solution moved along the diagnostic membrane and the extended primers were captured on the appropriate diagnostic test spot, thus turning the test spot visibly red due to the binding and accumulation of the gold nanoparticles. Non-extended primers were also captured through their 5′ sequence tag but not detected because they lacked biotin. Consequently, the appearance of red diagnostic spots denoted the presence of the corresponding alleles in the sample. The excess of gold nanoparticles was captured from the immobilized biotinylated oligos at the control spot to confirm the proper running of the sample. The genotype was assigned by observing each pair of spots. For any particular mutation, a normal individual would give a red color at the left spot only, whereas a variant homozygote would only give color at the right spot of the pair. Both spots would be red if the sample came from a heterozygote for this polymorphic site.
It should be emphasized that the present assay configuration is radically different from the previously reported panel of three DNA biosensors for genotyping FV Leiden, FII, and MTHFR thrombophilia-associated mutations [18] and offers significant advantages: (A) Three biosensors were necessary for the genotyping of each sample in ref. [18], i.e., one biosensor per mutation. On the contrary, the present paper uses only one multi-allele biosensor for the three most common mutations. (B) In the previous report, each sample was subjected to three separate PEXT reactions, one reaction per mutation, instead of a multiplex genotyping reaction performed in the present work. (C) The capture moieties (tags) of the allele-specific primers, in ref. [18], providing spatial resolution of the extended primers on the diagnostic membrane, are haptens, instead of oligonucleotide tags used in the present work. Consequently, in ref. [18], the diagnostic membrane contained zones of immobilized antibodies, a configuration that limits the multiplicity of the assay because of the commercially limited availability (about four) of antibody–hapten combinations. On the contrary, here the membrane contains microspheres conjugated with oligonucleotide anti-tags thereby providing a means for enhancing the multiplicity of the assay. (D) The detection of the extended allele-specific primers, in ref. [18], is achieved by means of another oligonucleotide probe which was designed to be complementary to the extended segment of the PEXT primers and carried a poly(dA) tail at the 3′-end. In this work, both recognition and detection elements constitute parts of the allele-specific primer, i.e., oligonucleotide tag at the 5′end for the spatial resolution on the membrane and incorporated biotin moieties for the detection when primer extension takes place. As far as both methods used gold nanoparticles for visual detection, in the previous report nanoparticles were conjugated with oligo(dT), whereas the antibiotin antibody is used in the present work for functionalization of nanoparticles.
Optimization studies
Parameters that affect the performance of each step of the proposed genotyping platform were investigated in order to achieve both multiplicity and efficiency of the assay in allele discrimination for all mutations.
Optimization studies of the multiplex PEXT reactions were performed using, as template, PCR-amplified products from clinical samples of various genotypes. We studied the effect of the type of DNA polymerase, the annealing temperature, the amount of allele-specific primers, and the concentration of Mg2+ and dNTPs. Characteristic results are presented in Fig. 2.

Optimization studies of the two-panel multi-allele PEXT-dipstick genotyping platform. Effect of (A, A′) annealing temperature in 6-plex and 8-plex PEXT reactions; (B, B′) amount of allele-specific primers; (C, C′) Mg2+ concentration; (D, D′) dNTPs concentration; (E, E′) formamide concentration in developing solution. For the position of each mutation-specific spot, see cartoon on the left side of the figure. All temperatures, concentrations, and amounts are indicated above the strips. Samples: (A–E) “normal”; (A′) PAI-1 “heterozygote”; (B′) PAI-1 “mutant,” MTHFR A1298C and FV H1299R “heterozygote”; (C′) PAI-1 “mutant” and MTHFR A1298C “heterozygote”; (D′) PAI-1 “heterozygote”; (E′) MTHFR A1298C “mutant,” FXIII “heterozygote”
The primer extension reaction requires the use of a DNA polymerase that lacks the repairing activity. Of the DNA polymerases studied, the Vent(exo-) Taq DNA polymerase, without any 3′→5′ and 5′→3′ exonuclease activity, was found to fulfill parameters such as ability to use biotin-modified dNTPs as substrates, to discriminate non-complementarity at the 3′end of the primer as well as high reaction yield and, therefore, was selected for our studies. The annealing temperature, representing one of the critical parameters for the specificity of the extension reaction, was studied in the range 55–68 °C (Fig. 2A, A′). We chose temperatures between 65 and 68 °C to ensure higher specificity without affecting the reaction yield. The effect of the amount of the six allele-specific primers (panel 1) and that of eight allele-specific primers (panel 2) was studied in the range of 0.5–5 and 1–10 pmol of each primer, respectively. The spot intensity for the panel 1 PEXT products is not practically affected by increasing primer amount up to 5 pmol. This level was thus chosen (Fig. 2B). On the contrary, the spot intensity for the panel 2 PEXT products is practically constant up to the 2.5 pmol but is decreased at higher primer amount (Fig. 2B′). This could be attributed to the lower yield of the panel 2 PEXT products and the competition between extended and non-extended primers for hybridization to the immobilized capture probe. A 2.5-pmol level was chosen for the panel 2 PEXT reaction. The Mg2+ concentration affects the kinetics and the specificity of the extension reaction as it is a cofactor of the DNA polymerase. In the range of 2–6 mM of Mg2+, there were no nonspecific red spots on the strip and the intensity of the specific spots increased with increasing Mg2+ concentration (Fig. 2C, C′). The dNTP concentration was optimized in the range of 5–20 μM of each. The intensity of the test spots increased with the dNTP concentration up to 10 μM. At higher concentrations, the color intensity remained the same; however, some nonspecific signals appeared (Fig. 2D, D′). For these reasons, we choose 10 μM of dNTPs.
Finally, we optimized the formamide concentration in the running buffer in order to eliminate any nonspecific signals. The effect of formamide concentration was studied in the range of 0–250 mL/L (Fig. 2E, E′). At formamide levels greater than 150 mL/L, the nonspecific signals are eliminated with a minimum decrease of the specific red spots. A 250 mL/L formamide concentration was chosen as optimum.
Evaluation of the proposed method
Studies were performed to assess the sensitivity, specificity, reproducibility, and the accuracy of the proposed method. The analytical sensitivity of the genotyping assay was assessed by performing a series of PEXT reactions containing different amount of PCR product in the range 0.05 to 3.0 μL. The PEXT reaction products were analyzed by the dipstick assay and the results are presented in Fig. 3A. As low as 0.2 μL of the PCR product was found to be sufficient to generate detectable spots for correct genotyping. The sensitivity of the dipstick assay expressed as the lowest amount of PCR product providing correct genotype was found to be 3 fmol (10−15 mol) considering the concentration of the amplified product (∼100 fmol/μL), the minimum volume of the PCR product used in PEXT reaction (0.2 μL), the volume of the PEXT reaction mixture (20 μL), and the volume of the PEXT reaction product used for the dipstick assay (3 μL).

Sensitivity (A), specificity (B), and reproducibility (C) of the multi-allele PEXT-dipstick genotyping platform. (A) Effect of the amount of PCR product in the PEXT reaction. (B) A sample normal for all three mutations (1) and a sample heterozygote for all three mutations (2) were subjected to three primer extension reactions. In the first extension reaction, only the three normal primers were added but no mutant primers. The second reaction was performed with the three mutant primers only and the third reaction in the presence of all six primers. The synthetic sample was a mixture of equal amounts of PCR products from samples that contain at least one of the three mutations. Therefore, the synthetic sample represented a heterozygous template for all primers. Normal primers are extended in the presence of all three samples whereas mutant primers are extended and give signals only in the presence of sample carrying the mutation. (C) Overall reproducibility of the multi-allele genotyping assay (including PEXT reaction and the dipstick assay) using two clinical samples: one normal for all three mutations and one with variant genotype (heterozygote for FV Leiden, normal for FII, and homozygote mutant for MTHFR). The detection by the multi-allele biosensor was carried out in quadruplicate and the multiplex primer extension reaction was performed in duplicate for each sample
The specificity of the PEXT reaction and the dipstick assay was assessed by using two samples: one normal for FV Leiden, FII, and MTHFR mutations and the second synthetic heterozygous for the mutations. The synthetic sample was prepared by mixing equimolar amounts of PCR products from samples normal and mutant for the mutations and thus representing a template heterozygous for all primers. PCR products from each sample were subjected to two PEXT reactions. The first reaction was performed in the presence of only the three normal primers, whereas the second one in the presence of only the three mutant primers. The reaction products were visualized by the dipstick biosensor and the results are presented in Fig. 3B. In order for a red spot to be formed, specifically, at the appropriate position of the membrane, two requirements must be fulfilled: (i) Only the allele-specific primers are extended during the extension reaction and (ii) There is no cross-reactivity of the tag sequences of the primers with the anti-tag oligonucleotides attached to the deposited microspheres. Indeed, in Fig. 3B, we observe that the extension reaction of the normal alleles with normal primers gives three red spots only at the positions of the normal alleles. No red spots are observed when the normal alleles were extended with the three mutant primers. Furthermore, the extension products of the mutant alleles with the mutant primers give red spots at the expected locations of the membrane. Consequently, primer extension reaction gave products only with the allele-specific primers and, also, the extension products were captured only at the appropriate positions of the membrane due to the specificity of the tag/antitag hybridization. Figure 3B shows that six red spots were formed when the heterozygote sample was subjected to a PEXT reaction in which all six primers were included.
Data pertaining to the overall reproducibility of the method (including both the genotyping reaction and the dipstick assay) are presented in Fig. 3C. The study was based on two clinical samples: one normal for all three mutations and one with variant genotype (heterozygote for FV Leiden, normal for FII, and homozygote mutant for MTHFR). The multiplex primer extension reaction was performed in duplicate for each sample and the detection by the multi-allele biosensor was carried out in quadruplicate. The results show highly reproducible genotypes for both samples.
The accuracy of the proposed method was assessed by analyzing 40 samples of known genotype and 15 blind samples. Genomic DNA samples were obtained from the Hematology Laboratory-Molecular Biology Unit of Ioannina University Hospital. These clinical samples were already characterized for all seven mutations by using the commercially available genotyping kit “FV-PTH-MTHFR StripAssay” and “CVD StripAssay” of ViennaLab Diagnostics (Vienna, Austria). The principle of this kit comprises a multiplex PCR followed by hybridization (reverse dot-blot) of biotinylated PCR products with allele-specific oligonucleotide probes immobilized on a strip. Detection of the hybrids is performed by means of alkaline phosphatase-conjugated streptavidin in combination with a chromogenic substrate. This is not a lateral flow assay and involves several incubation and washing steps. The genotyping results obtained by the proposed method were in full concordance with those obtained by the kit for 52 out of 55 samples. Characteristic examples of the genotyping assay of 19 samples covering all mutations are shown in Fig. 4A, where N/N refers to normal sample, M/N refers to a heterozygous or double heterozygous sample, and M/M refers to a homozygous mutant sample.

(A) Typical genotyping results from a series of samples using two multiplex PCR reactions, two multiplex primer extension reactions, and two strips. For the position of each mutation-specific probe set in all subsequent samples, see cartoon on the left side of the sample pictures. The spots on the left side correspond to the normal alleles. The spots on the right side correspond to the mutant alleles. The single spot at the top centre of the strip is a control spot that indicates the proper performance of the strip. The genotype is assigned by observing each pair of spots. For any particular mutation, a wild-type individual will give red color at the left spot only, whereas a mutant homozygote will only give color at the right spot of the pair. Both spots will appear red if the sample comes from a heterozygote for this mutation. (B) Quantitative information on genotype of the samples as a plot of allelic fraction vs the number of the sample. Each column of the plot consisting of three or four bars (panel 1 and panel 2, respectively) represents the genotype of each sample. Each bar corresponds to the allelic fraction of each locus as depicted on the right site of the plot. A few homologous mutant loci with allelic fractions (AFwt) = 0 are not seen in the plot
Besides the visual genotyping, the proposed DNA biosensor could also provide quantitative information on sample genotype. For this purpose, the strips were scanned using a common desktop scanner and the images were analyzed using GelAnalyzer 100a software. The allelic fraction for the wild-type allele (AFwt) is calculated from the equation: AFwt = wt/(wt + mut), where wild type (wt) and mutant (mut) correspond to the densitometric signals obtained from spots for N (left) and M (right) allele, respectively [19]. Heterozygous samples are expected to have allelic fractions near 0.5 (a 1:1 ratio of the two alleles) whereas homozygotes for wt and mut are expected to give AFwt values that are higher than 0.8 and lower than 0.2, respectively. Quantitative information on genotype of the 19 characteristic samples (133 mutations or 266 alleles) are also presented as a plot of allelic fraction vs the sample number (Fig. 4B). The allelic fractions for the three genotypes (AF±SD), including all seven loci, where found to be 0.98 ± 0.03, 0.54 ± 0.04, and 0.02 ± 0.03 for wt homozygotes (normal samples), heterozygotes, and mut homozygotes (mutant samples), respectively.
The dispute in three samples concerned the genotype of MTHFR C677T variant. These three samples were further investigated by a PCR-RFLP method [20]. Briefly, the DNA fragment flanking the MTHFR mutation was amplified by PCR using only the MTHFR primers. The amplification products were digested by Hinf I at 37 °C for 1 h. The digestion reaction was performed in a final volume of 20 μL containing 10 U of the enzyme, 1× restriction enzyme buffer, 0.1 g/L BSA, and 200 ng of amplified DNA. The digestion products were separated on agarose gel electrophoresis (1.8 %) and stained ethidium bromide. The amplified DNA contains a Hinf I restriction site, irrelevant of the polymorphic locus, thus producing a main fragment of 389 bp, and a 34-bp fragment. In a normal sample, the 389-bp band is observed. Because the MTHFR mutation creates an additional Hinf I site, a homozygote mutant sample produces a 224- and a 165-bp fragment. In a heterozygote sample, the digestion produces 389, 224, and 165 bp fragments. The digestion products were separated by agarose gel electrophoresis (1.8 %) and the genotype was assigned by comparison with two control samples (a normal and a homozygote for the mutation). The results (see ESM Fig. S1B) proved that all three questionable samples were genotyped accurately by the present method.
Conclusions
The two-panel genotyping platform enables (i) the simultaneous detection of three point mutations (six alleles in total) that constitute the most common and important genetic risk factors related to thrombophilia: the FV Leiden G1691A, the FII G20210A, and the MTHFR C677T mutations, and (ii) the simultaneous detection of four additional variants predisposing for thrombotic events: the FV H1299R, the MTHFR A1298C, the V34L, and the PAI-1 4G/5G polymorphism.
The architecture of the proposed genotyping method comprising (i) multiplex PCR followed by (ii) multiplex primer extension reactions and (iii) detection of the primer extension products is similar with those of the high-throughput Luminex xTAG microsphere-based universal array and Sequenome Massarray iPlex Gold platforms. Even though the proposed method is not high-throughput, it is advantageous in terms of simplicity, rapidity, and cost-effectiveness. More specifically, (i) PCR products in both high-throughput platforms should be treated (before proceeding to primer extension reaction) in order to remove remaining non-incorporated dNTPs and PCR primers. In the present method, no cleanup of the PCR product is required; (ii) 40 cycles of primer extension reaction is required for the xTAG and iPlex platforms. Only few (10) cycles is required for the proposed method; (iii) the primer extension products for xTAG and iPlex platforms should be treated before loading on the readers. In the present method, the primer extension product is loaded onto the strip (reader) without any treatment; (iv) for the detection of the primer extension products, both platforms are using specialized instrumentation along with a software to interpret the results. In the present method, the detection of the products and interpretation of the results are performed visually by naked eye (without instrumentation) using disposable dry-reagent dipstick tests and the total time required for the accumulation of a genotyping result, following all the three steps of the method, takes, approximately, only 3 h.
In recent years, several dipstick-type assays have been developed for the detection of nucleic acids, e.g., detection of characteristic sequences of various pathogens in a variety of samples [21–24]. Contrary to the above methods that simply detect amplicons, the proposed dipstick assay enables the accurate genotyping of single nucleotide variations (polymorphisms or mutations) in the target sequence. The use of spots of oligonucleotide-conjugated microspheres instead of the direct immobilization of the oligonucleotides on the membrane [25] offers the advantage of higher assay multiplexing.
The exponential amplification of the target sequences, through PCR, constitutes an integral part of the whole assay since it allows genotyping of minute amounts of sample and, at the same time, diminishes the interferences from other DNA sequences. However, the method could be simplified by performing the PCR using whole blood, thus avoiding the DNA isolation step, and also using low-cost portable PCR cyclers.
To conclude, the proposed method is a diagnostic tool that combines high specificity, reproducibility, and sensitivity with the additional advantages of simplicity, rapidity, and low cost, and fulfills all the necessary criteria in order to be introduced to the daily routine of small- and medium-sized modern laboratory of Molecular Diagnostics. In addition, the proposed method represents a potentially useful tool for centers with limited resources or for point-of-care testing.
References
Dahlbäck B. Blood. 2008;112:19–27.
Bertina RM. Clin Chem. 1997;43:1678–83.
Poort SR, Rosendaal FR, Reitsma PH, Bertina RM. Blood. 1996;88:3698–703.
Frosst P, Blom HJ, Milos R, Goyette P, Sheppard CA, Matthews RG, et al. Nat Genet. 1995;10:111–3.
Schwahn B, Rozen R. Am J Pharm. 2001;1:189–201.
Otrock ZK, Taher AT, Shamseddeen WA, Zaatari G, Bazarbachi A, Mahfouz RA. Ann Hematol. 2008;87:1013–6.
van der Put NM, Gabreëls F, Stevens EM, Smeitink JA, Trijbels FJ, Eskes TK, et al. Am J Hum Genet. 1998;62:1044–51.
Hanson NQ, Aras O, Yang F, Tsai MY. Clin Chem. 2001;47:661–6.
Ariens RAS, Lai TS, Weisel JW, Greenberg CS, Grant PJ. Blood. 2002;100:743–54.
Glueck CJ, Phillips H, Cameron D, Wang P, Fontaine RN, Moore SK, et al. Metabolism. 2000;49:845–52.
Torabi R, Zarei S, Zeraati H, Zarnani AH, Akhondi MM, Hadavi R, et al. J Reprod Infertil. 2012;13:89–94.
Gaudet M, Fara A, Beritognolo I, Maurizio Sabatti M. Methods Mol Biol. 2009;578:415–24.
Bagheri M, Rad I. Maedica. 2011;6:3–9.
Bortolin S. Methods Mol Biol. 2009;496:59–72.
Gabriel S, Ziaugra L, Tabbaa D. Curr Protoc Hum Genet 2.12.1-2.12.18. 2009.
De Stefano V, Rossi E, Paciaroni K, Leone G. Haematologica. 2002;87:1095–108.
Elenis DS, Ioannou PC, Christopoulos TK. Nanotechnology. 2011;22:155501.
Vlachou MA, Glynou KM, Ioannou PC, Christopoulos TK, Vartholomatos G. Biosens Bioelectron. 2010;26:228–34.
Toubanaki DK, Christopoulos TK, Ioannou PC, Gravanis A. Hum Mutat. 2008;29:1071–8.
Deeparani T, Radhakrishna PM, Elavazhagan T. Middle-East J Sci Res. 2009;2:127–32.
Kalogianni DP, Goura S, Aletras AJ, Christopoulos TK, Chanos MG, Christofidou M, et al. Anal Biochem. 2007;361:169–75.
Deng J, Pei J, Gou H, Ye Z, Liu C, Chen J. J Virol Methods. 2015;213:98–105.
Ang GY, Yu CY, Chan KG, Singh KK, Chan Yean Y. J Microbiol Methods. 2015;118:99–105.
Xu Y, Liu Y, Wu Y, Xia X, Liao Y, Li Q. Anal Chem. 2014;86:5611–4.
Litos IK, Ioannou PC, Christopoulos TK, Traeger-Synodinos J, Kanavakis E. Biosens Bioelectron. 2009;24:3135–9.
Acknowledgments
We would like to thank Dr. Georgios Vartholomatos from the Hematology Laboratory-Molecular Biology Unit of the Ioannina University Hospital, Greece, for providing the clinical samples.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest
Additional information
Nikolaos Fountoglou and Margarita Petropoulou contributed equally to this work.
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM 1
(PDF 339 kb)
Rights and permissions
About this article

Cite this article
Fountoglou, N., Petropoulou, M., Iliadi, A. et al. Τwo-panel molecular testing for genetic predisposition for thrombosis using multi-allele visual biosensors. Anal Bioanal Chem 408, 1943–1952 (2016). https://doi.org/10.1007/s00216-016-9310-9
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00216-016-9310-9