
Abstract
Cocaine is still one of the most abused drugs worldwide and, as such, it is often screened for in driving-under-the-influence or workplace drug - testing scenarios. A large number of samples have usually to be processed in those situations, and this requires fast and simple extraction procedures for the detection and quantification of the drugs. The present work describes an ultrafast and fully validated procedure for the simultaneous detection and quantification of cocaine and its two main metabolites, ecgonine methyl ester and benzoylecgonine, in urine using microextraction by packed sorbent and GC-MS. A small sample volume (200 μL) was used, and a fast extraction procedure together with a microwave-assisted derivatization (800 W, 2 min) allowed the quantification of all analytes in a range of 25 to 1000 ng/mL (r 2 > 0.99). Inter-day precision revealed coefficients of variation (CVs) lower than 10% for all analytes at the tested concentration levels, with an accuracy within a ±7% interval, with the exception of EME’s lowest calibrator (±17%). Intra-day CVs were lower than 15% at the studied concentration levels, with a mean relative error within a ±13% interval. Recoveries ranged from 14.5 to 37.2% (EME), 67.0 to 83.3% (cocaine), and 24.6 to 43.5% (BEG), allowing the limits of detection and quantification to be set at 25 ng/mL for all compounds.

Schematized analysis of cocaine and metabolites in urine by MEPS- GC/MS
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Cocaine is the most commonly used illicit stimulant drug in Europe, and due to the popularity gained in recent years, it is no longer considered an “elite drug.” Cocaine consumption is associated to numerous health problems, such as cardiovascular disorders, neurological impairment, as well as social problems and death [1, 2].
For human performance forensic toxicology purposes in drivers, as well as in workplace drug testing schemes, urine samples are frequently analyzed [3]. Its non-invasive collection procedure and detectable levels of the drugs over an extended period (and at much higher concentrations than those measured in blood), make urine the preferred specimen for regular drug testing analyses. Also, the higher levels of drug metabolites can provide further evidence of drug use. One has to consider, though, that most drug levels in urine do not correlate well with levels in blood, and the relatively short windows of detection (from 1 to 3 days for cocaine) [4].
Although proteins and cellular material are not present in urine at high levels, allowing simple analysis, a sample pre-treatment approach is usually deemed necessary and is considered the limiting step for a fast identification of the target analytes. The described sample preparation methods for cocaine identification and quantification in urine specimens include mainly the widely used solid-phase (SPE) [5–9] and liquid-liquid extraction (LLE) [10–13] procedures, while molecularly imprinted polymers (MIPs) have also been used [14] more recently. Miniaturization is the new trend on sample preparation, and this may be due to the great amounts of solvents and samples usually required for more traditional approaches (which are also more time-consuming). Microextraction by packed sorbent (MEPS) is a miniaturized SPE which reduces operating volumes, sample size, sample processing time, and has been successfully applied for the screening of cocaine and metabolites [15, 16], however, using expensive equipment which is not available in most laboratories for routine analysis. This technique has been successfully applied in both clinical and forensic toxicology fields [17]. In fact, our group has dedicated part of its research activity to this miniaturized procedure, as well on its application to determine a number of illicit drugs, such as cannabinoids [18], ketamine [19], piperazine-type stimulants [20], and salvinorin A [21] in biological samples. The different biological samples used to determine the mentioned compounds, as well as the drugs’ chemical properties play an important role in method development, and will influence the choice of the most adequate sorbent for extraction. Moreover, even if the best sorbent for the target analyte is selected, this perhaps will not suffice unless the whole process is previously optimized. In fact, parameters such as conditioning, proper sample dilution in buffer, sorbent wash, and elution have to be carefully studied in order to maximize recovery, which will be determinant in what concerns the method’s sensitivity [22]. The herein presented work is no different regarding the need for this optimization, since this is the first time that this group of illicit drugs is determined in urine by means of this miniaturized procedure.
Other miniaturized SPE approaches include for instance μSPE procedures; although both can be considered as miniaturized forms of the commonly used SPE, presenting as advantages their relatively low cost, minimized usage of solvents and compatibility with different systems of analyte separation and detection, μSPE has a number of drawbacks, namely the limited range of stationary phases selection, and the possibility of carryover. Moreover, MEPS also involves greater reductions in time and sample volume required to pre-concentrate target analytes, but its overwhelming advantage is the possibility of reusing the sorbent several times, significantly minimizing the costs of the whole procedure [23].
Gas chromatography coupled to mass spectrometry (GC-MS) is commonly used in forensic toxicology laboratories for cocaine determination [5–7, 10, 24–27], despite the derivatization process required to analyze its main metabolites, which is usually time-consuming [7]. In order to accelerate this step, microwave-assisted derivatization was studied and applied. LC-MS-based methods have been used as well [14, 28–34].
The present work describes an ultra fast (for which accounts the accelerated microwave-assisted derivatization), sensitive, selective, accurate and fully validated procedure for the simultaneous detection and quantification of cocaine (COC) and its two main metabolites, ecgonine methyl ester (EME) and benzoylecgonine (BEG) in low volume urine samples (200 μL) using MEPS-GC-MS. The described procedure represents a good alternative for forensic toxicology purposes, as well as in workplace drug testing situations, improving laboratory throughput.
Materials and methods
Reagents and standards
The analytical standards of COC, EME, and BEG were purchased from Sigma Aldrich, (St. Louis, MO, USA) as 1 mg/mL solutions. The internal standards (IS) COC-d3, EME-d3 and BEG-d3 were obtained from Sigma-Aldrich (Lisbon, Portugal), also at 1 mg/mL. Methanol (Merck Co, Darmstadt, Germany) and acetonitrile (Fisher Scientific, Leicestershire, UK) were of HPLC grade. Deionized water was obtained from a Milli-Q System (Millipore, Billerica, MA, USA). Ammonium hydroxide was purchased from J.T. Baker (Deventer, Holland), and Formic Acid was obtained from Merck (Darmstadt, Germany). N-methyl-N-(trimethylsilyl) trifluoroacetamide (MSTFA) and trimethylchlorosilane (TMCS) were acquired from Macherey-Nagel (Düren, Germany). A MEPS syringe (250 μL) was used and M1 cartridges (4 mg; 80% C8 and 20% SCX), both from SGE Analytical Science were purchased from ILC (Porto, Portugal); the microwave oven used for the derivatization process was purchased from Samsung (Lisbon, Portugal). Working solutions were prepared by proper dilution of the stock solutions with methanol to the final concentrations of 10 and 2 μg/mL for COC, BEG, and EME, while a working solution of all IS at 5 μg/mL was prepared also in methanol. All those solutions were stored in the absence of light at 4 °C.
Biological specimens
Drug-free urine samples used in all experiments were provided by laboratory staff. Authentic urine samples used for analysis were kindly provided by volunteers, consumers of cocaine, and provided by the emergency services of the Hospital of Cova da Beira, Covilhã, Portugal. These samples were stored frozen at −20 °C until analysis.
Gas chromatographic and mass spectrometric conditions
An HP 7890A gas chromatography system (Agilent Technologies, Waldbronn, Germany), equipped with a model 5975C Inert XL MSD mass selective detector (Agilent Technologies, Waldbronn, Germany), a MPS2 autosampler and a PTV-injector from Gerstel (Mülheim an der Ruhr, Germany) was used for the chromatographic analysis. The Separation of the analytes was achieved using a capillary column (30 m × 0.25-mm I.D., 0.25-μm film thickness) with 5% phenylmethylsiloxane (HP-5 MS), supplied by J & W Scientific (Folsom, CA, USA).
The oven temperature started at 90 °C for 2 min, followed by an increase at the rate of 20 °C/min until 300 °C and held for 3 min. Splitless mode was used and the inlet and ion source temperatures were set at 220 and 280 °C, respectively. Carrier gas (helium) was set at a constant flow rate of 0.8 mL/min and data were acquired in the selected ion monitoring (SIM) mode. The mass spectrometer was operated with a filament current of 35 μA and electron energy of 70 eV in the positive electron ionization mode. The ions were chosen based on selectivity and abundance in order to maximize the signal-to-noise ratio in matrix extracts. The selected ions were the following (quantifying ions are italicized, and the respective retention time is given between brackets): 82, 96, and 271 for EME (8.25); 82, 182, and 303 for COC (11.70); and 82, 240, and 361 for BEG (11.98). For the internal standards, the selected ions were 85, 185, and 243 for EME-d3, COC-d3, and BEG-d3, respectively.
Sample preparation
The extraction procedure was optimized previously (see Results and discussion), and the final conditions were as follows.
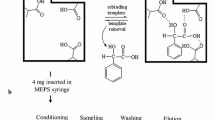
Frozen urine samples were allowed to thaw at room temperature and were afterwards centrifuged at 4500 rpm during 15 min. The supernatant was transferred into 15-mL polypropylene conical tubes. Urine samples (200 μL) were diluted with 100 μL of 0.1 mM potassium phosphate buffer and spiked with 20 μL of the IS working solution. The mixture was homogenized by rotation/inversion movements during 15 min. The M1 MEPS cartridge was conditioned once with 250 μL of methanol and once with 250 μL of 0.1% formic acid (in water). Sample loading was performed with 6 cycles of 150 μL of the diluted urine sample (strokes). Endogenous interferences were removed from the sorbent using 4 cycles of 50 μL of 0.1% formic acid in water followed by a drying step of 4 strokes of air (50 μL) in order to remove all traces of water. The retained analytes were eluted from the sorbent with 4 cycles of 100 μL of 1% ammonium hydroxide in methanol; the extracts were transferred to borosilicate glass tubes and were afterwards evaporated to dryness under a gentle stream of nitrogen. The dry extracts were derivatized with 50 μL of MSTFA with 5% TMCS, the tubes were screw capped and the process was conducted in a microwave oven for 2 min at 800 W. The derivatized extracts were transferred to autosampler vials and a 2 μL aliquot of the resulting solution was injected into the GC-MS system.
After each extraction, the sorbent was washed sequentially with 4 cycles (100 μL) of each of the following solvents: 1% ammonia in methanol:acetonitrile (50:50, v/v) and 1% formic acid in 2-propanol, in order to decrease carryover.
Validation procedure
The described method was fully validated according to the guiding principles of the Food and Drug Administration (FDA) [35], International Conference on Harmonization (ICH) [36] and the Scientific Working Group for Forensic Toxicology (SWGTOX) [37]. A 5-day validation protocol was followed, and the studied parameters included selectivity, linearity and limits, intra- and inter-day precision and accuracy, efficiency, stability, and dilution integrity.
Selectivity
The method’s selectivity was evaluated by analyzing 10 pools of blank urine samples, each pool containing 3 urine samples from different origins, in order to investigate eventual interferences at the retention times and selected ions of the studied compounds. Identification criteria for positivity included an absolute retention time within 2% or ±0.1 min of the retention time of the same analyte in the control sample and the presence of at least three ions per compound. To guarantee a suitable confidence in identification, the maximum allowed tolerances for the relative ion intensities between the three ions (as a percentage of the base peak) were as follows [19]. Considering the relative ion intensity in the control sample, if the relative ion intensity was higher than 50%, then an absolute tolerance of ±10% was accepted; if this value was between 25 and 50%, a relative tolerance of ±20% was allowed; if it was between 5 and 25%, an absolute tolerance of ±5% was accepted and finally, for relative ion intensities of 5% or less, a relative tolerance of ±50% was used [38]. The method would be considered selective if no analyte could be identified in the blank samples by means of those criteria.
Calibration curves and limits
Linearity of the method was established on spiked urine samples prepared and analyzed using the described extraction procedure in the range of 25–1000 ng/mL for all target analytes (five replicates). Calibration curves were obtained by plotting the peak area ratio between each analyte and the IS against analyte concentration. The acceptance criteria included a determination coefficient (R 2) value of at least 0.99 and the calibrators’ accuracy within a ±15% (except at the lower limit of quantification (LLOQ), where ±20% was considered acceptable). Together with each calibration curve, a zero sample (blank sample with IS) and QC samples at 50, 100, 500, and 1000 ng/mL (n = 3) were also analyzed. The LLOQ was defined as the lowest concentration which could be measured with adequate precision and accuracy, i.e., with a coefficient of variation (CV,%) lower than 20% and a relative error (RE,%) within ±20% of the nominal concentration [19].
Intra- and inter-day precision and accuracy
Intra-day precision was evaluated by analyzing in the same day 6 replicates of blank urine samples spiked with the studied analytes at 6 concentration levels. Inter-day precision was evaluated at a minimum of six concentrations within a 5-day period. Intermediate precision was evaluated as well. This precision refers to as run-to-run, analyst-to-analyst, or day-to-day precision. In this work, we have studied day-to-day precision only. This parameter was calculated using the QC samples prepared and analyzed simultaneously with the calibration curves on 5 different days (n = 15). The accuracy of the method was characterized in terms of the mean RE between the concentrations measured using the calibration equation and the spiked concentrations; the accepted limit was 15% for all concentrations, except at the LLOQ, where 20% was accepted.
Extraction efficiency
For the analysis of absolute recovery, two sets of samples (n = 3) were prepared at 4 concentration levels (50, 100, 500, and 1000 ng/mL). Set 1 represented spiked analytes after extraction of a blank sample (representing 100% recovery), and set 2 consisted of spiked analytes in a blank sample before extraction. The IS was added to the two sets of sample only after elution. The recovery results were obtained by comparison of the relative peak areas of sample set 2 with those of the corresponding analytes in sample set 1.
Stability
The stability of COC, BEG, and EME was studied at the abovementioned concentrations applying the MEPS procedure (n = 3) under specific conditions and time intervals (processed samples, short-term and freeze/thaw stability). To study stability in processed samples, the extracts that were previously analyzed were re-analyzed after stored at room temperature in the autosampler for 24 h. This way the target analytes and the internal standard were assessed over the anticipated run time for the batch size, and their concentrations were determined on the basis of the original calibration curve. Short-term stability was evaluated at the same concentration levels (n = 3). Blank samples were spiked and were left at room temperature for 24 h. To study freeze and thaw stability, urine samples were spiked at the previously described concentrations, and were stored at −20 °C for 24 h. After this period, they were thawed unassisted at room temperature, and then refrozen for 12–24 h under the same conditions. This freeze/thaw cycle was repeated twice more, and the samples were analyzed after the third cycle. During the entire stability procedure, the analyzed samples were compared to samples freshly prepared and analyzed in the same day (both sets of samples were quantified in the same calibration curve, the obtained concentrations were compared and the respective RE was calculated relatively to the theoretical concentrations; the CVs between the two sets of samples were calculated as well). The analytes were considered stable if both the CV between the two sets of samples and the calculated REs were in accordance with the abovementioned criteria for precision and accuracy (CVs below 15% and REs within a ±15% interval).
Dilution integrity
This parameter was studied in order to investigate the possibility of sample dilution in the event that the analytes were present at concentrations higher than the method’s upper limit of quantification. The study was performed for all analytes at three levels of concentration (n = 3), which needed three different levels of sample dilution (with blank urine) to fit within the calibration range (1:10, 1:5 and 1:2). Sample dilution would be acceptable if the calculated concentrations after multiplying by the dilution factor were within a ±15% interval from the spiked value.
Results and discussion
Optimization of the sample extraction process
This optimization was performed using spiked urine samples from 5 different origins.
Selection of the extraction procedure
This study was initially carried out in order to find the most suitable extraction procedure that would result in a greater recovery of the target analytes. The most common extraction technique for the analysis of COC and metabolites by GC-MS appears to be SPE using a mixed mode sorbent, and most of the referenced methods using this type of SPE sorbent reported extraction recoveries of at least 70% [39]. For this reason, we have used a mixed mode sorbent (M1) in our 250-μL MEPS syringe. Table 1 resumes five different procedures using this type of sorbent. One can observe that the washing and eluting steps were different for each extraction technique, considering several MEPS protocols parameters [16]. The sorbent conditioning step was the same for all procedures, and consisted on 250 μL of methanol followed by the same volume of 0.1% aqueous formic acid; the sample was aspirated 8 times (150 μL) through the device in all tested procedures [16].
Figure 1 shows the obtained results by application of the five extraction procedures in blank urines previously spiked with COC, EME and BEG (0.5 μg/mL). The respective IS were added after the extraction process. Each extraction procedure (n = 3) was evaluated using one-way ANOVA in order to observe the presence of statistical differences within and between groups. EME is the only analyte revealing no significant differences in the absolute peak area [F(4,10) = 1.20, p < 0.05] when the different procedures were applied; however, one can observe greater responses for techniques 1 and 5. The latter procedures were the only that combined 0.1% formic acid with 1% ammonium hydroxide, and this combination revealed a greater effect on the recovery of EME, still with no significant deviation from that obtained when other techniques were used.

Absolute peak areas and SDs obtained for different extraction procedures
A significant difference in absolute peak areas when the five procedures were performed [F(4,10) = 5.21, p < 0.05] was however observed for COC. Although lower absolute peak areas were observed in general for this analyte, greater recoveries were still observed for techniques 1, 4, and 5 (best for 4 and 5). A significant greater absolute peak area [F(1,4) = 8.18, p < 0.05] as well as a lower coefficient of variation (CV) were observed for technique 5 when compared to technique 4.
BEG reveals similar results to those obtained for COC. The study of the five procedures also resulted in a significant difference [F(4,10) = 64.69, p < 0.05] between them. The absolute peak areas also suggest that techniques 4 and 5 allow better recoveries; however, recovery is significantly higher for technique 5 [F(1,4) = 185.20, p < 0.05], presenting also a lower CV when compared to technique 4. Therefore, technique 5 was considered the most suitable extraction procedure for the target analytes, and hence, it was adopted for further analysis.
Number of strokes
The number of extraction cycles (strokes) during the sample loading step is a parameter that should be optimized, hence leading to higher recoveries of the target analytes [16]. Given the previous extraction procedure evaluation, the selected technique was subjected to a study of the number of strokes, regarding the present method application. The number of extraction cycles was the only parameter which was modified, ranging from 6 to 12 strokes (n = 3), while all the remaining MEPS parameters were kept constant. The IS was added only after the extraction process, in order to use peak areas ratio to compare different method conditions.
Figure 2 shows the obtained results. One can observe that 8 strokes would result in greater recovery, but also in a larger CV for EME. COC and BEG present better recoveries with an acceptable CV (<12%) when 10 extraction cycles were performed. However, no significant statistical difference is observed for EME [F(3,8) = 0.61, p < 0.05], COC [F(3,8) = 0.67, p < 0.05] or BEG [F(3,8) = 1.55, p < 0.05] when the number of extraction cycles varied in the range of 6 to 12.

Absolute peak areas and SDs obtained when different number of strokes were applied on MEPS loading step
One of the main goals of present method development is a fast determination of COC and metabolites with the use of a minimal sample volume and a simple analyte pre concentration. Therefore, 6 cycles of extraction seemed a reasonable option for a simple MEPS procedure, since no statistically significant difference was observed when higher number of strokes was applied.
Derivatization procedure
Most GC-MS methods for the analysis of COC and metabolites require a derivatization process to increase volatility, detectability or stability, and to facilitate chromatographic separation. The most common agents described in the literature for the target analytes are MSTFA [5, 10, 24], N,O-Bis(trimethylsilyl) trifluoroacetamide (BSTFA) [6, 40], pentafluoropropionic anhydride (PFPA) [26, 27], and pentafluoropropanol (PFPOH) [26, 27]. TMCS (1%) can often be combined as a catalyst to increase TMS donor potential [10, 40], and we increased its concentration to 5% combined with MSTFA as a derivatization agent in the present work.
However, derivatization procedures using heating blocks are usually time-consuming [7]. In order to accelerate this analytical step, Brunetto et al. [7] made an extensive study on microwave-assisted derivatization of COC and BEG with BSTFA after SPE in urine samples. The authors reported a higher increase on the signal of BEG derivative after exposure to 700 W up to 90 s. The study of the level of irradiation made by the same authors revealed, however, greater signals for BEG and reproducible measurements when 630 W were applied [7].
In the present work, we have used microwave-assisted derivatization (800 W) with MSTFA and 5% TMCS during 120 s, a period of time considered a critical limit for safety reasons [7]. The greater level of irradiation applied with these agents during this period of exposure gave reproducible measurements for all three analytes, and CVs lower than 15%.
A comparison was made between microwave assisted derivatization applied under our conditions and using a heating block at 85 °C for 45 min. Peak areas for EME were greater when the derivatization reaction was microwave-assisted, revealing an increase of the reaction rate for this analyte. Different results are observed for COC and BEG, as a greater peak area was observed using a heating block. The results obtained for BEG differ from the previously mentioned study [7]. The authors report greater peak areas for BEG when a microwave assisted derivatization is applied comparing this process with a common thermostated bath. The present work uses a different agent (MSTFA) and different microwave conditions (800 W, 120 s) which could have caused these results. Under our conditions, no significant difference is observed for COC peak area, since this compound does not derivatize, and these results are also in agreement with the previous study [7].
Method validation
The present method was fully validated according to the guiding principles of the Food and Drug Administration (FDA) [35], International Conference on Harmonization (ICH) [36] and those of the Scientific Working Group for Forensic Toxicology (SWGTOX) [37]. The parameters included selectivity, efficiency, linearity and calibration model, limits, intra- and interday precision and trueness, stability and dilution integrity. All validation experiments were studied using blank urine samples from 5 different origins (except, selectivity, for which 30 different origins were used).
Selectivity
Selectivity of the methodology was evaluated as previously described, and it was checked for interferences at the retention times and selected ions of the studied analytes. No interferences from endogenous substances were observed at the retention times and selected ions for each analyte (Fig. 3).

Chromatograms of urine sample spiked at the LLOQ (25 ng/mL) and one pool of blank urine in SIM mode
Calibration curves and limits
The present method was considered linear within the adopted calibration ranges for all analytes, yet weighted least squares regressions had to be adopted in order to compensate for heterocedasticity. For each target analyte, six weighting factors were evaluated (1/√x, 1/x, 1/x 2, 1/√y, 1/y, 1/y2), and that revealing the best results was selected, given the data obtained during the assessment of the inter-day precision and accuracy [35, 36]. The choice was made by calculating the mean relative error (RE) for each factor, and summing the absolute value. The factor that revealed the lowest sum of errors and simultaneously presented a mean R2 value of at least 0.99 was chosen. A weighting factor of 1/x 2 was deemed necessary for cocaine analysis. The calibrators’ accuracy [mean relative error (bias) between the measured and spiked concentrations] was within a ±15% interval for all concentrations, except at the LLOQ, for which a ±20% interval was accepted. Table 2 shows calibration data.
The obtained LLOQs were 25 ng/mL for all compounds and can be considered quite satisfactory, especially when compared to analytical methods published by other authors for the same analytes. Among those applied to urine samples, many published methods presented greater LLOQs, namely for EME [9, 15, 27], COC [7, 13, 15, 27, 41, 42] and BEG [5, 10, 14]. Greater LLOQs were also obtained when analysis were carried out using a GC-MS systems [5, 7, 10, 27], similarly to the present work, but also with systems presenting higher selectivity, such as LC-MS [14] or LC-MS/MS [9]. So far, only Jagerdeo and Abdel-Rehim [15] have developed an analytical method utilizing MEPS as a preconcentration procedure with the main goal of screening COC and metabolites in human urine samples, reporting LLOQS of 65 ng/mL (EME), 75 ng/mL (BEG) and 95 ng/mL (COC). However, the present method reaches lower LLOQs (25 ng/mL) with a simple and fast derivatization process, using only 200 μL of sample, making it an excellent option for routine application in a forensic toxicology laboratory. Another application of MEPS has demonstrated its efficiency when applied to pre-concentrate the target analytes of the present work, as well as other illicit drugs, but in oral fluid [33]. The authors of the mentioned work coupled MEPS with a highly sensitive separation and detection equipment, LC-MS/MS, which resulted in lower quantification limits, although with lower recoveries for BEG and EME. The method’s limits of detection were not evaluated systematically, since positive results below the LLOQ are not reported, and therefore were considered to be 25 ng/mL.
Intra- and inter-day precision and accuracy
Intra-day precision and accuracy were evaluated by analyzing 6 replicates of blank urine spiked with COC and metabolites at 6 concentration levels on the same day. The obtained coefficients of variation (CVs) were lower than 15% at the studied concentration levels, with a mean relative error within a ±13% interval (Table 3). The evaluation of the inter-day precision and accuracy was made within a 5-day period at 6 concentration levels. The obtained CVs were lower than 10% for all analytes at the tested concentration levels, with an accuracy within a ±7% interval, with the exception of EME’s lowest calibrator (±17%) (Table 3).
Additionally, intermediate (combined intra- and inter-day) precision and accuracy was also evaluated using the QC samples prepared and analyzed simultaneously with the calibration curves on 5 different days, and CVs lower than 12% and accuracy within ±6% interval were obtained. This evaluation was made by analysis of the quality control samples (4 concentration levels related to the linearity range) which were analyzed in triplicate over the same 5-day period (n = 15) (Table 3).
Stability
The study of stability in processed samples was carried out at the same QC concentration levels (n = 3), in which previously analyzed samples were re-analyzed after 24 h in the autosampler. COC and metabolites concentrations were determined on the basis of the original calibration curve, obtaining CVs lower than 11% and mean relative errors within a ±14% interval. The obtained results demonstrate the possibility of performing sample re-analysis after 24 h in the autosampler with no significant change in the target analytes’ concentration.
Short-term stability evaluation was carried out for blank urine samples spiked with the same concentration levels, also analyzed in triplicate. For this study, samples were spiked and left at room temperature for 24 h. These samples were then compared to freshly prepared ones. The CVs obtained were lower than 12% for all target analytes, while mean relative error was within a ±15% interval.
Freeze/thaw stability was also studied in samples at the same 4 concentration levels (n = 3). This evaluation was made by freezing the spiked samples at −20 °C for 24 h, after which they were thawed unassisted at room temperature. When completely thawed, the samples were re-frozen for 24 h. In total, three cycles of freeze/thaw were performed, after which samples were reanalyzed and subsequently compared to samples prepared and analyzed on the same day. One can also observe that COC and metabolites were also stable after 3 freeze/thaw cycles, resulting in CVs lower than 13% and mean relative errors within ±13%. All stability data is shown in Table 4.
Extraction efficiency
For the study of extraction efficiency, two sets of samples (n = 3) were prepared by spiking blank urine with the target analytes at four concentration levels (50, 100, 500, and 1000 ng/mL).
EME presented the lowest extraction efficiencies, ranging from 14.5 to 37.2%; however, it could be considered adequate when compared to previous publications [12, 14]. BEG extraction efficiencies ranged from 24.6 to 43.5%, also considered appropriate considering other authors’ results using LLE [10, 14] or SPE [5] in urine samples. Greater recoveries were obtained for COC, ranging from 67.0 to 83.3%. Similar extraction efficiencies were reported when LLE [10, 12, 14] or SPE [43] were applied to detect this drug of abuse. According to the present extraction procedure, MEPS can be considered a powerful technique, resulting in a fast and efficient extraction of the target analytes with lower sample and solvent consumption.
Dilution integrity
To evaluate this parameter, urine samples were spiked with analyte concentrations which would need proper dilution in order to fit within the calibration range. Three different concentrations were used (1600, 4000 and 8000 ng/mL) which were diluted respectively by 1:2, 1:5 and 1:10 with blank urine prior to analysis. The calculated concentrations, after multiplied by the dilution factor, were within a ±15% interval from the spiked value. Therefore, too concentrated samples can be adequately analysed after proper dilution.
Method applicability
Subsequent to the analytical method validation, this was successfully applied to a routine analysis of COC and its main metabolites (EME and BEG) in urine samples. Ten authentic urine samples were kindly provided by volunteers, consumers of cocaine, and as well provided by the emergency services of the Hospital of Cova da Beira, Covilhã, Portugal to outwit cocaine consumption in patients. The concentrations of analytes ranged from 113 to 962 ng/mL (EME), 40 to 377 ng/mL (COC), and 530 to 3350 ng/mL (BEG). Figure 4 is an example of a positive result, and the obtained concentrations were 962, 44 and 3350 ng/mL for EME, COC and BEG, respectively. Table 5 shows the ion ratios for each analyte in this sample, and their comparison with the control sample, allowing the positive identification of the compounds.

Chromatogram of an authentic urine sample (EME: 962 ng/mL; COC: 44 ng/mL; BEG: 3350 ng/mL)
Conclusions
An ultrafast (15 min), sensitive, selective, accurate, and fully validated procedure is described for the simultaneous detection and quantification of COC and its two main metabolites (EME and BEG) in urine using MEPS-GC-MS. This analytical method has shown to be linear within the adopted ranges for all target analytes with a LLOQ of 25 ng/mL.
The analyses were carried out using small sample volumes (0.2 mL of urine) and the method was successfully applied to authentic samples, proving its usefulness in routine drug testing.
The microwave-assisted derivatization process (800 W) with MSTFA and 5% TMCS for 120 s was considered a better approach, substituting the commonly used and more time-consuming heating block. The herein presented derivatization procedure takes only 2 min, improving laboratory throughput.
This is the first report on the use MEPS combined with GC-MS for quantification of COC and metabolites in urine samples. MEPS was considered a better alternative to the traditionally used pre-concentration techniques, resulting in a lower sample and solvents consumption. The cartridge was efficiently reutilized for more than 200 extractions in urine samples.
References
Reid MJ, Langford KH, Grung M, Gjerde H, Amundsen EJ, Morland J, et al. Estimation of cocaine consumption in the community: a critical comparison of the results from three complibrsmentary techniques. BMJ Open. 2012; 2(6). doi:10.1136/bmjopen-2012-001637.
Ruiz MJ, Paolieri D, Colzato LS, Bajo MT. Chronic and recreational use of cocaine is associated with a vulnerability to semantic interference. Psychopharmacology. 2015;232(10):1717–26. doi:10.1007/s00213-014-3806-9.
Valente MJ, Carvalho F, Bastos MdL, Carvalho M, Pinho PGd (2012) Chromatographic methodologies for analysis of cocaine and its metabolites in biological matrices. In: DBS, editor. Gas chromatography - biochemicals, narcotics and essential oils. CC By, pp 163–167. doi:10.5772/32225.
Lum G, Mushlin B. Urine drug testing: approaches to screening and confirmation testing. Lab Med. 2004;35(6):368–73. doi:10.1309/QHJCKA4235EGPEGF.
Lerch O, Temme O, Daldrup T. Comprehensive automation of the solid phase extraction gas chromatographic mass spectrometric analysis (SPE-GC/MS) of opioids, cocaine, and metabolites from serum and other matrices. Anal Bioanal Chem. 2014;406(18):4443–51. doi:10.1007/s00216-014-7815-7.
Alvear E, von Baer D, Mardones C, Hitschfeld A. Determination of cocaine and its major metabolite benzoylecgonine in several matrices obtained from deceased individuals with presumed drug consumption prior to death. J Forensic Legal Med. 2011;23:37–43. doi:10.1016/j.jflm.2014.01.003.
Brunetto Mdel R, Delgado Y, Clavijo S, Contreras Y, Torres D, Ayala C, et al. Analysis of cocaine and benzoylecgonine in urine by using multisyringe flow injection analysis-gas chromatography–mass spectrometry system. J Sep Sci. 2010;33(12):1779–86. doi:10.1002/jssc.200900833.
Langman LJ, Bjergum MW, Williamson CL, Crow FW. Sensitive method for detection of cocaine and associated analytes by liquid chromatography-tandem mass spectrometry in urine. J Anal Toxicol. 2009;33(8):447–55.
Jagerdeo E, Montgomery MA, Sibum M, Sasaki TA, LeBeau MA. Rapid analysis of cocaine and metabolites in urine using a completely automated solid-phase extraction-high-performance liquid chromatography-tandem mass spectrometry method. J Anal Toxicol. 2008;32(8):570–6.
Strano-Rossi S, Bermejo AM, de la Torre X, Botre F. Fast GC-MS method for the simultaneous screening of THC-COOH, cocaine, opiates and analogues including buprenorphine and fentanyl, and their metabolites in urine. Anal Bioanal Chem. 2011;399(4):1623–30. doi:10.1007/s00216-010-4471-4.
Dowling G, Regan L, Tierney J, Nangle M. A hybrid liquid chromatography-mass spectrometry strategy in a forensic laboratory for opioid, cocaine and amphetamine classes in human urine using a hybrid linear ion trap-triple quadrupole mass spectrometer. J Chromatogr A. 2010;1217(44):6857–66. doi:10.1016/j.chroma.2010.08.043.
Johansen SS, Bhatia HM. Quantitative analysis of cocaine and its metabolites in whole blood and urine by high-performance liquid chromatography coupled with tandem mass spectrometry. J Chromatogr B Analyt Technol Biomed Life Sci. 2007;852(1–2):338–44. doi:10.1016/j.jchromb.2007.01.033.
Fernandez P, Lago M, Lorenzo RA, Carro AM, Bermejo AM, Tabernero MJ. Microwave assisted extraction of drugs of abuse from human urine. J Appl Toxicol. 2007;27(4):373–9. doi:10.1002/jat.1216.
Thibert V, Legeay P, Chapuis-Hugon F, Pichon V. Molecularly imprinted polymer for the selective extraction of cocaine and its metabolites, benzoylecgonine and ecgonine methyl ester, from biological fluids before LC-MS analysis. J Chromatogr B Analyt Technol Biomed Life Sci. 2014;949–950:16–23. doi:10.1016/j.jchromb.2013.11.051.
Jagerdeo E, Abdel-Rehim M. Screening of cocaine and its metabolites in human urine samples by direct analysis in real-time source coupled to time-of-flight mass spectrometry after online preconcentration utilizing microextraction by packed sorbent. J Am Soc Mass Spectrom. 2009;20(5):891–9. doi:10.1016/j.jasms.2009.01.010.
Abdel-Rehim M. Microextraction by packed sorbent (MEPS): a tutorial. Anal Chim Acta. 2011;701(2):119–28.
Barroso M, Moreno I, da Fonseca B, Queiroz JA, Gallardo E. Role of microextraction sampling procedures in forensic toxicology. Bioanalysis. 2012;4(14):1805–26.
Rosado T, Fernandes L, Barroso M, Gallardo E. Sensitive determination of THC and main metabolites in human plasma by means of microextraction in packed sorbent and gas chromatography-tandem mass spectrometry. J Chromatogr B. 2016. doi:10.1016/j.chromb.2016.09.007.
Moreno I, Barroso M, Martinho A, Cruz A, Gallardo E. Determination of ketamine and its major metabolite, norketamine, in urine and plasma samples using microextraction by packed sorbent and gas chromatography-tandem mass spectrometry. J Chromatogr B. 2015;1004:67–78.
Moreno IED, Da Fonseca BM, Magalhães AR, Geraldes VS, Queiroz JA, Barroso M, et al. Rapid determination of piperazine-type stimulants in human urine by microextraction in packed sorbent after method optimization using a multivariate approach. J Chromatogr A. 2012;1222:116–20.
Moreno I, da Fonseca B, Oppolzer D, Martinho A, Barroso M, Cruz A, et al. Analysis of Salvinorin A in urine using microextraction in packed syringe and GC-MS/MS. Bioanalysis. 2013;5(6):661–8.
Yang L, Said R, Abdel-Rehim M. Sorbent, device, matrix and application in microextraction by packed sorbent (MEPS): a review. J Chromatogr B. 2016. doi:10.1016/j.jchromb.2016.10.044.
Plotka-Wasylka J, Szczepańska N, de la Guardia M, Namieśnik J. Modern trends in solid phase extraction: new sorbent media. TrAC Trends Anal Chem. 2015;77:23–43.
Pelicao FS, Peres MD, Pissinate JF, De Martinis BS. A one-step extraction procedure for the screening of cocaine, amphetamines and cannabinoids in postmortem blood samples. J Anal Toxicol. 2014;38(6):341–8. doi:10.1093/jat/bku039.
Fleming SW, Dasgupta A, Garg U. Quantitation of cocaine, benzoylecgonine, ecgonine methyl ester, and cocaethylene in urine and blood using gas chromatography–mass spectrometry (GC-MS). Methods Mol Biol. 2010;603:145–56. doi:10.1007/978-1-60761-459-3_14.
Romberg RW, Jamerson MH, Klette KL. Rapid analysis of benzoylecgonine in urine by fast gas chromatography–mass spectrometry. J Anal Toxicol. 2006;30(8):554–8.
Saito T, Mase H, Takeichi S, Inokuchi S. Rapid simultaneous determination of ephedrines, amphetamines, cocaine, cocaine metabolites, and opiates in human urine by GC-MS. J Pharm Biomed Anal. 2007;43(1):358–63. doi:10.1016/j.jpba.2006.06.031.
Jagerdeo E, Montgomery MA, LeBeau MA, Sibum M. An automated SPE/LC/MS/MS method for the analysis of cocaine and metabolites in whole blood. J Chromatogr B. 2008;874(1-2):15–20. doi:10.1016/j.jchromb.2008.08.026.
de Jager AD, Bailey NL. Online extraction LC-MS/MS method for the simultaneous quantitative confirmation of urine drugs of abuse and metabolites: amphetamines, opiates, cocaine, cannabis, benzodiazepines and methadone. J Chromatogr B Analyt Technol Biomed Life Sci. 2011;879(25):2642–52. doi:10.1016/j.jchromb.2011.07.029.
Schaefer N, Peters B, Schmidt P, Ewald AH. Development and validation of two LC-MS/MS methods for the detection and quantification of amphetamines, designer amphetamines, benzoylecgonine, benzodiazepines, opiates, and opioids in urine using turbulent flow chromatography. Anal Bioanal Chem. 2013;405(1):247–58. doi:10.1007/s00216-012-6458-9.
Dulaurent S, El Balkhi S, Poncelet L, Gaulier J-M, Marquet P, Saint-Marcoux F. QuEChERS sample preparation prior to LC-MS/MS determination of opiates, amphetamines, and cocaine metabolites in whole blood. Anal Bioanal Chem. 2016;408(5):1467–74.
Ellefsen KN, da Costa JL, Concheiro M, Anizan S, Barnes AJ, Pirard S, et al. Cocaine and metabolite concentrations in DBS and venous blood after controlled intravenous cocaine administration. Bioanalysis. 2015;7(16):2041–56.
Montesano C, Simeoni MC, Curini R, Sergi M, Lo Sterzo C, Compagnone D. Determination of illicit drugs and metabolites in oral fluid by microextraction on packed sorbent coupled with LC-MS/MS. Anal Bioanal Chem. 2015;407(13):3647–58. doi:10.1007/s00216-015-8583-8.
Koster RA, Alffenaar J-WC, Greijdanus B, VanDerNagel JEL, Uges DRA. Application of sweat patch screening for 16 drugs and metabolites using a fast and highly selective LC-MS/MS method. Ther Drug Monit. 2014;36(1):35–45. doi:10.1097/FTD.0b013e3182a04feb.
FDA. Food and Drug Administration, Guidance for Industry: Bioanalytical Method Validation. 2001. http://www.fda.gov/downloads/Drugs/GuidanceCompilanceRegulatoryInformation/ Guidances/ucm070107.pdf.
ICH. International Conference on Harmonization (ICH), Validation of Analytical Procedures: Text and methodology Q2 (R1). 2005. http://www.ich.org/fileadmin/Public_Web_Site/ICH_Products/Guidelines/Quality/Q2_R1/Step4/Q2R1Guideline.pdf.
SWGTOX. Scientific Working Group for Forensic Toxicology (SWGTOX) standard practices for method validation in forensic toxicology. J Anal Toxicol. 2013;37(7):452–74.
WADA. World Anti-doping Agency, International standard for laboratories: identification criteria for qualitative assays incorporating column chromatography and mass spectrometry. 2010. http://www.wada-ama.org/Documents/WorldAnti-DopingProgram/WADP-IS-Laboratories/WADATD2010IDCRv1.0IdentificationCriteriaforQualitativeAssaysMay082010EN.doc.pdf.
Smith R, Bogusz MJ. Handbook of analytical separations: forensic science, vol 6. 2nd edn. Amsterdam: Elsevier; 2011.
Marchei E, Colone P, Nastasi GG, Calabro C, Pellegrini M, Pacifici R, et al. On-site screening and GC-MS analysis of cocaine and heroin metabolites in body-packers urine. J Pharm Biomed Anal. 2008;48(2):383–7. doi:10.1016/j.jpba.2007.11.025.
Znaleziona J, Petr J, Maier V, Knob R, Horakova J, Smetanova D, et al. Capillary electrophoresis as a verification tool for immunochemical drug screening. Biomed Pap Med Fac Univ Palacky Olomouc Czech Repub. 2007;151(1):31–6.
Zaitsu K, Miki A, Katagi M, Tsuchihashi H. Long-term stability of various drugs and metabolites in urine, and preventive measures against their decomposition with special attention to filtration sterilization. Forensic Sci Int. 2008;174(2–3):189–96. doi:10.1016/j.forsciint.2007.04.224.
Virag L, Mets B, Jamdar S. Determination of cocaine, norcocaine, benzoylecgonine and ecgonine methyl ester in rat plasma by high-performance liquid chromatography with ultraviolet detection. J Chromatogr B Biomed Appl. 1996;681(2):263–9.
Acknowledgements
This work is supported by FEDER funds through the POCI—COMPETE 2020—Operational Programme Competitiveness and Internationalisation in Axis I—Strengthening research, technological development and innovation (Project POCI-01-0145-FEDER-007491) and National Funds by FCT—Foundation for Science and Technology (Project UID/Multi /00709/2013).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
All individuals participating in this study have signed an informed consent form. The described study was approved by the appropriate ethics committee and has been conducted according to ethical standards.
Conflict of interest
The authors declare that they have no conflict of interest.
Rights and permissions
About this article

Cite this article
Rosado, T., Gonçalves, A., Margalho, C. et al. Rapid analysis of cocaine and metabolites in urine using microextraction in packed sorbent and GC/MS. Anal Bioanal Chem 409, 2051–2063 (2017). https://doi.org/10.1007/s00216-016-0152-2
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00216-016-0152-2