
Abstract
Mandelic acid (MA) is a major metabolite of ethylbenzene and styrene. For the first time, a selective, fast, and easy-to-use procedure was developed for the determination of MA in urine samples. The new procedure is based on MIMEPS, the combination of a molecularly imprinted polymer (MIP) and microextraction by packed sorbent (MEPS). High-performance liquid chromatography with ultraviolet detection (HPLC–UV) was used for the separation and determination of MA. The bulk polymerization method was used to synthesize the MIP, and the MIP and non-imprinted polymer (NIP) were characterized by Fourier transform infrared spectroscopy. The selectivity of the MIP was investigated in the presence of interferents. In addition, we investigated the various parameters that affect the performance of the MEPS, including the pH of the sample, the number of extraction cycles, sample volume, and the types and volumes of the washing and elution solvents. A six-point calibration curve was obtained in the range of 0.2–20 µg/mL (R 2 = 0.9994). The extraction recovery was more than 88.8%. The limit of detection and the limit of quantitation were 0.06 and 0.2 µg/mL, respectively. The intra- and inter-day precisions were in the range of 3.6–4.7% and 3.8–5.1%, respectively. The accuracy was −8.4 to −11.1%. The optimized procedure was selective, sensitive, and rapid, and it was both user friendly and environmentally friendly. The sample preparation process took only about 5 min, so the MIMEPS–HPLC–UV procedure is recommended as an alternative for the biomonitoring of workers exposed to ethylbenzene and/or styrene.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Mandelic acid (MA) is a metabolite of ethyl benzene (EB) and styrene. EB is primarily used for the production of styrene, a monomeric unit for polystyrene materials, and it is ubiquitous in ambient air. Styrene is used extensively in plastics, latex paints and coatings, synthetic rubber, and polyester resins (Collins and Richey 1992; ATSDR 2010a). Some people are exposed to EB and styrene in their occupations, and many people are exposed to these chemicals every day through routine activities, such as the use of automobiles, boats, aircraft, and gasoline-powered tools and equipment. Paints, varnishes, and solvents release EB into the environment (Bahrami 2001; ATSDR 2010a, b). Both EB and styrene have been classified as group 2B carcinogens (possible carcinogens) by the International Agency for Research on Cancer (IARC 2002, 2006). The main route of exposure to EB and styrene is inhalation. Once inside the human body, these organic compounds are metabolized to yield the main urinary metabolites, MA and phenylglyoxylic acid, which are used as biomarkers of exposures to EB and styrene (Šperlingová et al. 2004; ACGIH 2016).
Various analytical methods are used to analyze MA in urine samples, including conventional solid phase extraction (SPE) (Lee et al. 2010; Mao et al. 1996), hollow-fiber liquid-phase microextraction (Bahrami et al. 2017), and liquid–liquid extraction (LLE) (Inoue et al. 1995; Ohashi et al. 2006; Wang et al. 2006). Also, the “dilute-and-shoot” approach, which does not require a sample preparation step, is used (Manini et al. 2002; Šperlingová et al. 2004; Paci et al. 2013). In addition, to the best of our knowledge, there has been only one study regarding the use of MIP as the stationary phase in high-performance liquid chromatography (HPLC) column to chiral separation of diastereoisomers of MA (Hung et al. 2005). However, the sample preparation using SPE and LLE methods is a bottleneck of these analytical methods (including the derivatization step in GC (Ohashi et al. 2006; Lee et al. 2010) and some HPLC analytical methods (Wang et al. 2006), since it is time and labor consuming). When conventional SPE columns were introduced to the market, they were well accepted because of their superiority over LLE methods. However, the lack of selectivity, which is the main drawback of the conventional SPE sorbents (e.g., C2, C8, C18, and SAX), led researchers to use selective sorbent materials, such as molecularly imprinted polymers (MIPs). MIPs are smart sorbents that have a defined selectivity for a given analyte or structurally related compounds (Chen et al. 2016). The main advantages of MIPs are their high selectivity, low cost, robustness, resistance to a wide range of pH values and temperatures, and their high mechanical strength (Qiao et al. 2006; Dopico-Garcio et al. 2011).
On the other hand, the removal of matrix interferences, cleanup and preconcentration, and providing a robust and reproducible method are the main objectives of sample preparation. In addition, recent objectives such as miniaturization, automation, high-throughput performance, online coupling with analytical instruments, smaller initial volumes of samples and solvents, minimizing the amount of glassware used, and reducing the time required (Smith 2003; Abdel-Rehim 2011) have led researchers to develop more advanced extraction techniques. From this perspective, microextraction by packed sorbent (MEPS), which was introduced by Abdel-Rehim, has emerged as a powerful sample preparation technique with an increasing range of applications (Du et al. 2014; Moein et al. 2015a; Soleimani et al. 2017).
In MEPS, about 2–4 mg of the sorbent are packed inside the barrel of a syringe as a plug or between the barrel and the needle as a cartridge, and thus the solvent consumption is significantly reduced as compared to conventional SPE columns. In MEPS, the sample is passed directly through the sorbent with no need for a separate robot, as that noted in conventional SPE columns. In addition, in contrast to single-use conventional SPE columns, MEPS can be used about 100 times for plasma or urine samples. Working with much smaller volumes to fully elute the sorbent is the most important benefit of MEPS. Therefore, there is no need for the concentration steps that usually result in losses of the analyte (Abdel-Rehim 2011; Pereira et al. 2014; Moein et al. 2015a). In general terms, compared to SPE or LLE methods, MEPS reduces the sample preparation time and solvent consumption. In recent years, according to the selectivity and low cost of MIPs and the advantages of MEPS over conventional SPE outlined above, the combination of MIPs and MEPS (MIMEPS) has been welcomed as a simple, rapid, selective, sensitive, and both user-friendly and environmentally friendly method for sample cleanup and preconcentration of analytes in environmental (Prieto et al. 2011a, b), biological (Asgari et al. 2017; Daryanavard et al. 2013; Moein et al. 2014, 2015b; Soleimani et al. 2017), and food samples (Du et al. 2014; Fumes et al. 2016).
In this study, for the first time, we developed a novel approach based on MIMEPS for the determination of MA in urine samples. Fourier transform infrared (FTIR) spectra were used to characterize the polymers. The imprinting effect and selectivity of the synthesized MIP were also evaluated. We optimized the important factors that affected the performance of MIMEPS, e.g., extraction cycles, sample volume, type and volume of the washing solution, the type and volume of the elution solution, and the pH of the sample. Also, we compared the MIMEPS technique with other existing methods for the determination of MA in urine samples.
Materials and methods
Reagents and materials
All chemicals were of analytical reagent grade. MA, dimethyl sulfoxide (DMSO), ethyleneglycol dimethacrylate (EGDMA), 2,2ʹ-azobisisobutyronitrile (AIBN), methacrylic acid (MAA), hippuric acid (HA), and trans,trans-muconic acid (t,t-MA) were all supplied by Sigma-Aldrich (Darmstadt or Hohenbrunn, Germany). Methanol, ethanol, acetic acid, hydrochloric acid (HCl), and sodium hydroxide were purchased from Merck (Darmstadt, Germany). Water was obtained from an ultra-water system (TKA, Niederelbert, Germany). A 250-µL gastight syringe (Hamilton, Nevada, USA) was used as the MEPS syringe. Stock solutions (1000 μg/mL) of MA, HA, and tt-MA were prepared in the methanol–water (8:2, v/v) mixture. A working standard solution was prepared daily by diluting the stock solution with water. Blank urine samples were taken from non-smoker subjects who did not drink alcoholic beverages.
Instrumentation
The chromatographic system was a Knauer HPLC system (Berlin, Germany) equipped with a K-2600 ultraviolet detector, a K-1001 pump, a D-14163 solvent degasser, and a 3950 autosampler with a sample loop of 100 µL. A reversed-phase C18 column (250 mm × 4.6 mm i.d., 5 µm diameter particles) maintained at room temperature (25 °C) was used to separate MA. A water–methanol–acetic acid (69:30:1, v/v/v) solution was used as isocratic mobile phase at a flow rate of 1.0 mL/min. The injection volume was 10 µL and the detector was set at 247 nm. To record the FTIR spectra of the polymers, prepared by the KBr pellet conventional method, a Perkin Elmer Spectrum 65 FTIR spectrophotometer was used (4000–500 cm−1).
MIP and non-imprinted polymer (NIP) preparation
The bulk polymerization method was used to synthesize MIP according to the optimized mole ratio (Table 1). Figure 1a shows a schematic of the MIP preparation procedure. In the first step, 0.11 mmol of MA (0.017 g) was dissolved in 2 mL of DMSO, and then 0.8 mmol of MAA (67 µL) was added. The solution was ultrasonicated for 15 min. Subsequently, 2.2 mmol of EGDMA (415 µL) and 0.25 mmol of AIBN (0.041 g) were added. The solution was sonicated for an additional 20 min and then purged with nitrogen for 5 min to eliminate oxygen. The resulting solution was placed in an oil bath at 60 °C for 24 h. After cooling to room temperature, the polymer was ground and sieved to obtain particles that were <40 μm in size. To remove the template molecule, the particles were washed in a methanol–acetic acid solution (9:1, v/v) on a shaker for 8 h (8 washing cycles). The process of eliminating the template was monitored by UV–Vis spectrophotometry. The particles were dried and stored at room temperature for later use. The corresponding NIP was prepared in the same manner, but without the template molecule.

Schematic illustration of a the MIP preparation procedure and b the MIMEPS procedure
Study of the imprinting effect and selectivity of the MIP
We investigated the selectivity of the MIP for MA in the presence of HA and tt-MA as interfering compounds. These acids were chosen because they are metabolites of toluene and benzene, respectively, chemicals that workers are exposed to concurrently in workplaces (Online Resource 1). For the experiment, 50 mg of MIP or NIP was added to 20 mL of the binary mixtures of MA/HA and MA/tt-MA (1 µg/mL each) prepared in water at a pH of 2.0 (see the section of Optimization of MEPS procedure—pH of the sample). Each mixture was stirred for 20 min at room temperature and then centrifuged at 4000 rpm for 5 min. The concentrations of the free analyte and interferents in the supernatant were measured at 247, 264, and 225 nm for MA, tt-MA, and HA, respectively. The distribution coefficient (K d), selectivity coefficient (k), and relative selectivity coefficient (k′) (Tarley et al. 2011; Andrade et al. 2014) were calculated according to the following equations:
where C i and C f are the initial and final concentrations of the analyte, and V s is the volume of the solution.
In addition, the selectivity of MIP was assessed by analyzing spiked urine samples with 2 µg/mL of each analyte (MA, HA, and tt-MA) using MIMEPS under the same conditions. The pH of the spiked samples was adjusted to the optimized value of 2.0. The sample was percolated through 4 mg of MIP with 8 × 100-µL extraction cycles. Then, the analytes were eluted using a methanol–acetic acid solution (8:2, v/v). The eluent was injected into the HPLC–UV.
MEPS procedure
Figure 1b shows the MIMEPS procedure. The extraction was performed manually by packing approximately 4 mg of MIP between two polyethylene frits inside the barrel of a 250-µL Hamilton syringe. After conditioning the MIP with 3 × 100 µL of methanol, followed by 3 × 100 µL of water, the urine sample was percolated through the MIP sorbent eight times (8 × 100 µL) at the speed of 10 µL/s (extract–discard mode). Then, the sorbent bed was washed once with 100 µL of water. In the next step, the analyte was eluted by 2 × 100 µL of methanol–acetic acid (8:2, v/v) solution. An aliquot of 10 µL was injected into the HPLC system. To avoid carryover, the MIMEPS bed was cleaned with 4 × 150 µL of the elution solution, followed by 4 × 150 µL of the washing solution between extractions. The same MIP was used about 70 times without the loss of extraction efficiency and/or the MIP clogging.
Validation of the method
Method validation was performed according to the Food and Drug Administration guidelines for bioanalytical method validation (FDA 2001). The selectivity of the method was investigated to ensure that no interfering peaks occurred by comparing the chromatograms of five different blank urine samples with the chromatograms of urine samples spiked with MA. For the determination of linearity, a six-point calibration curve was established using spiked urine samples that contained 0.2–20 µg/mL of MA (five replicates of each, n = 30). The calibration curve was prepared by plotting the MA peak areas against MA concentrations. Intra-day precision [expressed as percent relative standard deviation (%RSD)] and accuracy [expressed as the degree of deviation of the determined value from the nominal value] were obtained on the same day by the determination of five replicates of each quality control (QC) sample [0.2 µg/mL (low QC), 1 µg/mL (medium QC), and 10 µg/mL (high QC)]. To calculate inter-day precision and accuracy, five replicates of the QC samples were investigated over three consecutive days.
Limit of detection (LOD) and limit of quantitation (LOQ) were estimated based on signal-to-noise ratios of 3:1 and 10:1, respectively. The extraction recoveries of MA in the three QC levels were obtained by measuring the concentration after the MIMEPS procedure and dividing it by the concentration before the MIMEPS procedure multiplied by 100 (after MIMEPS/before MIMEPS × 100).
Collection and analyses of urine samples
The subjects signed an informed consent form before the collection of their urine samples. In addition, the protocol of the study was approved by the university’s Ethics Committee (Approval No. IR.UMSHA.REC.1394.371). End-of-shift urine samples were obtained from eight workers who had been exposed to EB in a petrochemical plant. The urine samples were collected in polypropylene bottles and stored at −20 °C until they were analyzed. Aliquots of the urine samples were acidified and adjusted to pH 2.0, as described in the section of Optimization of MEPS procedure—pH of the sample. The levels of MA in the urine samples were determined by the MIMEPS–HPLC–UV method that we developed.
Results and discussion
MIP characterization, the study of imprinting effect, and selectivity of the MIP
The FTIR spectra of the MIP (with template removal), MIP* (without template removal), and NIP are provided in Online Resource 2. An apparent broad O–H stretching vibration peak was observed for both MIP and NIP at 3454 cm−1, indicating the presence of hydroxyl (O–H) bonds that are attributed to the MAA carboxylic group (COOH). The stretching peaks at 2995 and 2953 cm−1 for both MIP and NIP are related to the stretching vibration of the C–H bonds in the methyl groups. The carbonyl group C=O stretching peak was observed at 1728 cm−1 for both NIP and MIP, and this peak might have originated from MAA and EGDMA.
Both MIP and MIP* had similar typical peaks, which indicated that they had similar backbone structures. As a result of the formation of hydrogen-binding bonds, the electric cloud density of OH and C=O decreased, leading to decreasing the frequency of vibration. Therefore, the C=O stretching and the O–H stretching peaks at 1730 and 3460 cm−1, respectively, in MIP* (without template removal) were shifted to 1728 and 3454 cm−1 in the related MIP (with template removal), respectively. In addition, in the MIP*, there was one band with the high relative intensity at 1466 cm−1 that was observed at 1459 cm−1 in the MIP. The weak combination bands from 1635 to 1259 cm −1 and the sharp band at 1159 cm−1, specifically on the MIP* spectra, indicated the presence of an aromatic ring of the template, which was removed successfully by washing MIP* with methanol–acetic acid (9:1, v/v).
The results of the cross-selectivity assessment of the MIP are shown in Table 2. The following observations indicate the successful imprinting effect on the polymer and the selectivity of the MIP: (1) the higher values of K d for MA in MIP than in NIP (distribution coefficients of 198 vs. 59 and 189 vs. 48 for the binary mixtures of MA/tt-MA and MA/HA, respectively); (2) the higher K d values for MA in MIP compared to those of the interferents (distribution coefficients of 198 vs. 28.5 for tt-MA and 189 vs. 99 for HA); (3) the higher values of k for MA in MIP than in NIP, which indicated the good ability of MIP to discriminate between MA and the interferents (selectivity coefficient of 6.95 vs. 0.63 for tt-MA and 1.91 vs. 0.55 for HA); and (4) the calculated kʹ, which evaluates the imprinting effectiveness of MIP. The values of kʹ for both binary mixtures (11.03 and 3.47) were greater than 1, confirming the successful imprinting effect of the MIP.
The evaluation of the recovery of MA in the presence of interferents (tt-MA and HA) using MIMEPS showed that the recoveries of MA, tt-MA, and HA were 89.6, 18, and 29%, respectively. The higher recovery of MA can be attributed to the presence of selective sites in the MIP structure.
Optimization of MEPS procedure
For the MEPS procedure, approximately 4 mg of the MIP was packed manually between two polyethylene frits inside a 250-µL syringe. The various parameters that affect the extraction efficiency of MEPS were optimized, including the number of extraction cycles, sample volume, type and volume of the washing solution, type and volume of the elution solution, and the pH of the sample. All procedures were performed in triplicate.
Extraction cycles and sample volume
Figure 2a shows the effects of the number of extraction cycles and the sample loading volumes on the recovery of MA. Five extraction cycles (2, 4, 6, 8, and 10 cycles) and three sample volumes (25, 50, and 100 µL) were investigated. We used sample loadings of 10 µL/s measured by a chronometer to ensure better interactions between the analyte and MIP (Abdel-Rehim 2011). The recovery increased from 22.4% (2 × 25 µL sample loading) to 91.8% (8 × 100 µL sample loading). Increasing the number of sample loadings from 8 × 100 µL to 10 × 100 µL did not result in any additional increase in the extraction recovery. Thus, the sample loading of 8 × 100 µL was selected for the rest of the optimization process.

Effects of a the number of extraction cycles and sample volume; b type and volume of the washing solution; c type and volume of the elution solution; and d pH on MIMEPS performance
Washing solutions
An appropriate washing solution must be chosen to remove the unwanted components from the sorbent, minimize the loss of the analyte, and achieve a clean extract. In this study, 1 × 100 µL of water and different aqueous solutions of methanol (water–methanol 9:1, 8:2, and 7:3, v/v) were investigated to remove the interferences and to assess their effects on the recovery of the analyte (Fig. 2b). When water–methanol mixtures (8:2 and 7:3, v/v) were used, some losses of the analyte were noted. However, when water–methanol (9:1, v/v) or water was used, no losses of the analyte were observed. Therefore, an aliquot of 1 × 100 µL of water was selected as the washing solution for further experiments.
Elution solutions
The elution step is critically important for displacing the target analyte from the solid sorbent at the lowest possible volume. In this study, we investigated the efficacy of several elution solutions for the recovery of MA, i.e., ethanol, ethanol–acetic acid (1:1, v/v), methanol, methanol–acetic acid (1:1, v/v), and methanol–HCl (1:1, v/v) (Fig. 2c). Among these solutions, methanol–acetic acid (1:1, v/v) provided the highest extraction recoveries. Further optimization indicated that, among the different ratios of methanol–acetic acid (i.e., 9:1, 8:2, and 7:3, v/v), the ratio of 8:2 (v/v) was the most effective in eluting the analyte (Fig. 2c). The recovery of MA increased from 39.8 to 91.6% as the volume of the elution solution increased from 1 × 50 µL to 2 × 100 µL. However, recoveries remained constant when the volume of the elution solution increased from 2 × 100 µL to 4 × 100 µL. Therefore, an aliquot of 2 × 100 µL of methanol–acetic acid (8:2, v/v) was chosen as the eluent.
pH of the sample
The pH is a critical parameter to achieve reproducible data. Therefore, the effect of pH on the recovery of MA was investigated in the pH range of 2–10 (Fig. 2d). Hydrochloric acid or sodium hydroxide solution was used to adjust the pH to the desired values. Figure 2d shows that good extraction recoveries occurred from pH values ranging from 2 to 5 with the highest recovery at pH 2.
Carryover
Carryover is a well-known problem in bioanalysis. In MEPS, since a small amount of a sorbent is used, it can be easily and effectively cleaned between injections to avoid carryover (Abdel-Rehim 2011; Moein et al. 2015a; Soleimani et al. 2017). Therefore, MEPS is not disposable as compared to single-use SPE columns. Therefore, it is critical to choose a suitable cleanup solvent between injections to reduce or eliminate the possibility of carryover. In this study, carryover was investigated by analyzing a blank urine sample after injection of the highest standard (20 µg/mL). We investigated several wash–discard cycles with the elution solution, i.e., 1 × 150, 2 × 150, 4 × 150, and 6 × 150 µL, followed by the washing solution. The 4 × 150 µL wash–discard cycles essentially eliminated carryover by reducing the level to only 0.3%, and the 6 × 150 µL wash–discard cycles gave approximately the same result. Thus, the use of 4 × 150 µL wash–discard cycles with the elution solution, followed by 4 × 150 µL of the washing solution, was considered as a suitable procedure to avoid carryover.
Method validation
Figure 3 shows the representative chromatograms of a blank urine sample, a spiked urine sample with MA at LOQ, a urine sample from a worker who had been exposed to EB, and the chromatogram of a blank sample spiked with MA, HA, and tt-MA at 2 µg/mL concentrations. No interfering peaks were observed at the retention time of MA when the standards, the QC samples, and the samples of EB-exposed workers were analyzed (Figs. 3a–c). In addition, in the presence of the interferents (HA and tt-MA), as described in the section of Study of the imprinting effect and selectivity of the MIP, no overlap was observed in the chromatogram (Fig. 3d), indicating good selectivity and the ability of the MIMEPS to remove interfering compounds and to enrich the MA to sufficient purity. A calibration curve was constructed for six concentrations ranging from 0.2 to 20 µg/mL. Under the optimized MIMEPS–HPLC–UV conditions, a close relationship between concentration and peak area was observed in the range we studied (R 2 = 0.9994) (Table 3). The accuracy and precision were determined at three QC samples (0.2, 1, and 10 µg/mL). The accuracy of the method was within the range of −8.4 to −11.1%. Intra- and inter-day precisions (RSD%) varied from 3.6 to 4.7% and 3.8 to 5.4%, respectively, which indicated that the MIMEPS–HPLC–UV method had acceptable precision and accuracy (Table 3).

HPLC chromatograms of a blank urine sample of a non-smoker who did not drink alcoholic beverages, b the same blank sample spiked at LOQ, c urine sample of an EB-exposed worker, and d a blank urine sample spiked with MA, HA, and tt-MA (2 µg/mL each) after the MIMEPS procedure
LOD and LOQ were estimated to be 0.06 and 0.2 µg/mL, respectively. The precision and accuracy of the LOQ concentration were 5.2 and −11.1%, respectively. Extraction recovery was determined by five replicates of the QC samples. The recovery values ranged from 88.9 to 91.6% (Table 3).
The method application for urine samples
The optimized MIMEPS–HPLC–UV procedure was applied for the determination of MA in urine samples from eight male workers who had been exposed to EB in a petrochemical plant. The urine samples were analyzed in triplicate. The results of the analyses are shown in Online Resource 3. The concentrations of MA measured in the workers’ urine samples were in the range of 1.0–14.7 µg/mL with %RSD <5.2.
Comparison of methods
Online Resource 4 provides a comparison of the results acquired using our technique with the results of other previously published methods for analyzing MA in urine samples. Sample preparation is still a bottleneck in the analytical techniques that use the SPE or LLE methods for the determination of MA in urine samples (Inoue 1995; Mao et al. 1996; Ohashi et al. 2006; Wang et al. 2006; Lee et al. 2010) as well as in the techniques that involve the derivatization step before GC and/or LC analysis (Ohashi et al. 2006; Wang et al. 2006; Lee et al. 2010). The MIMEPS–HPLC–UV method provided good recovery of MA, and the LOD and LOQ were similar or better than those reported in other published studies. In addition to having similar sensitivity to other methods, our procedure offers several advantages, including the lowest consumption of the solvent, the lowest amount of sorbent material, the shortest sample preparation time (about 5 min), high selectivity, and reuse of the sorbent for as many as 70 extractions. Therefore, the MIMEPS–HPLC–UV method can be a promising alternative for the determination of tt-MA in urine samples.
Conclusions
In our study, for the first time, a new sample preparation technique using the combination of MIP and MEPS was introduced for the determination of MA in urine samples. The MIMEPS–HPLC–UV method we used is selective, sensitive, fast, inexpensive, and both user friendly and environmentally friendly. Since MIMEPS is faster, simpler, and uses much smaller volumes of organic solvents than LLE and conventional SPE columns, it is suggested as an alternative to these methods for the biomonitoring of workers who are exposed to EB and/or styrene.
Change history
08 January 2018
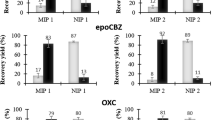
In the original publication of the article, there is an error in Fig. 2c. The authors would like to correct this error and the corrected version of Fig. 2c is as follow.
References
Abdel-Rehim M (2011) Microextraction by packed sorbent (MEPS): a tutorial. Anal Chim Acta 701:119–128. doi:10.1016/j.aca.2011.05.037
Agency for Toxic Substances and Disease Registry (2010a) Toxicological profile for styrene, US Department of Health and Human Services, Atlanta, GA. https://www.atsdr.cdc.gov/toxprofiles/TP.asp?id=421&tid=74. Accessed 10 July 2017
Agency for Toxic Substances and Disease Registry (2010b) Toxicological profile for ethylbenzene, US Department of Health and Human Services, Atlanta, GA. https://www.atsdr.cdc.gov/toxprofiles/tp.asp?id=383&tid=66. Accessed 10 July 2017
American Conference of Governmental Industrial Hygienists (2016) Threshold Limit Values and Biological Exposure Indices. Cincinnati (OH)
Andrade FN, Santos-Neto AJ, Lancas FM (2014) Microextraction by packed sorbent liquid chromatography with time-of-flight mass spectrometry of triazines employing a molecularly imprinted polymer. J Sep Sci 37:3150–3156. doi:10.1002/jssc.201400616
Asgari A, Bagheri H, Es-haghi A, AminiTabrizi R (2017) An imprinted interpenetrating polymer network for microextraction in packed syringe of carbamazepine. J Chromatogr A 1491:1–8. doi:10.1016/j.chroma.2017.02.033
Bahrami A (2001) Distribution of volatile organic compounds in ambient air of Tehran. Arch Environ Health 56:380–383. doi:10.1080/00039890109604472
Bahrami A, Ghamari F, Yamini Y, Ghorbani Shahna F, Moghimbeigi A (2017) Hollow fiber supported liquid membrane extraction combined with HPLC-UV for simultaneous preconcentration and determination of urinary hippuric acid and mandelic acid. Membranes 7:1–13. doi:10.3390/membranes7010008
Chen L, Wang X, Lu W, Wu X, Li J (2016) Molecular imprinting: perspectives and applications. Chem Soc Rev 45:2137–2211. doi:10.1039/C6CS00061D
Collins DE, Richey FA Jr. (1992) Synthetic organic chemicals. Riegel’s handbook of industrial chemistry, 9th edn. Springer, New York, pp 800–862
Daryanavard SM, Jeppsson-Dadoun A, Andersson LI, Hashemi M, Colmsjö A, Abdel-Rehim M (2013) Molecularly imprinted polymer in microextraction by packed sorbent for the simultaneous determination of local anesthetics: lidocaine, ropivacaine, mepivacaine and bupivacaine in plasma and urine samples. Biomed Chromatogr 27:1481–1488. doi:10.1002/bmc.2946
Dopico-Garcio MS, Cela-Perez C, Lopez-Vilarino JM, Gonzalez-Rodriguez MV, Barral-Losada LF (2011) An approach to imprint irganox 1076: potential application to the specific migration test in olive oil. J Appl Polym Sci 119:2866–2874. doi:10.1002/app.32964
Du W, Lei C, Zhang S, Bai G, Zhou H, Sun M et al (2014) Determination of clenbuterol from pork samples using surface molecularly imprinted polymers as the selective sorbents for microextraction in packed syringe. J Pharm Biomed Anal 91:160–168. doi:10.1016/j.jpba.2013.12.022
Food and Drug Administration. U.S. Department of Health and Human Services (2001) Guidance for industry: bioanalytical method validation. https://www.fda.gov/downloads/Drugs/Guidance/ucm070107.pdf Accessed 10 July 2017
Fumes BH, Andrade FN, dos Santos Neto AJ, Lancas FM (2016) Determination of pesticides in sugarcane juice employing microextraction by packed sorbent followed by gas chromatography and mass spectrometry. J Sep Sci 39:2823–2830. doi:10.1002/jssc.201600077
Hung CY, Huang HH, Hwang CC (2005) Chiral separations of mandelic acid by HPLC using molecularly imprinted polymers. Eclética Química 30:67–73. http://www.scielo.br/scielo.php?script=sci_arttext&pid=S010046702005000400009&lng=en&tlng=en. Accessed 2 sept 2017
IARC (2002) Agents reviewed by the IARC Monographs. Volumes 1–118. Lyon, France: International Agency for Research on Cancer. http://monographs.iarc.fr/ENG/Classification/latest_classif.php Accessed 10 July 2017
IARC (2006) Agents reviewed by the IARC Monographs. Volumes 1–118. Lyon, France: International Agency for Research on Cancer. http://monographs.iarc.fr/ENG/Classification/latest_classif.php. Accessed 10 July 2017
Inoue O, Seiji K, Kudo S, Jin C, Cai SX, Liu SJ et al (1995) A sensitive HPLC method for determination of mandelic acid in urine, and its application to biological monitoring of ethylbenzene-exposed Chinese workers. Int J Occup Environ Health 1:245–251. doi:10.1179/oeh.1995.1.3.245
Lee J, Kim M, Ha M, Chung BC (2010) Urinary metabolic profiling of volatile organic compounds in acute exposed volunteers after an oil spill in Republic of Korea. Biomed Chromatogr 24:562–568. doi:10.1002/bmc.1328
Manini P, Andreoli R, Poli D, De Palma G, Mutti A, Niessen WMA (2002) Liquid chromatography/electrospray tandem mass spectrometry characterization of styrene metabolism in man and rat. Rapid Commun Mass Spectrom 16:2239–2248. doi:10.1002/RCM.848
Mao IF, Chen ML, Lo EW (1996) Simultaneous determination of urinary metabolites of toluene, xylene, styrene and ethyl benzene by solid-phase extraction technique and high-performance liquid chromatographic/photo diode array detection. Int J Environ Anal Chem 94:1–9. doi:10.1080/03067319608028330
Moein MM, El-Beqqali A, Javanbakht M, Karimi M, Akbari-adergani B, Abdel-Rehim M (2014) On-line detection of hippuric acid by microextraction with a molecularly-imprinted polysulfone membrane sorbent and liquid chromatography–tandem mass spectrometry. J Chromatogr A 1372:55–62. doi:10.1016/j.chroma.2014.10.061
Moein MM, Abdel-Rehim A, Abdel-Rehim M (2015a) Microextraction by packed sorbent (MEPS). Trends Anal Chem 67:34–44. doi:10.1016/j.trac.2014.12.003
Moein MM, Abdel-Rehim A, Abdel-Rehim M (2015b) On-line determination of sarcosine in biological fluids utilizing dummy molecularly imprinted polymers in microextraction by packed sorbent. J Sep Sci 38:788–795. doi:10.1002/jssc.201401116
Ohashi Y, Mamiya T, Mitani K, Wang B, Takigawa T, Kira S, Kataoka H (2006) Simultaneous determination of urinary hippuric acid, o-, m- and p-methylhippuric acids, mandelic acid and phenylglyoxylic acid for biomonitoring of volatile organic compounds by gas chromatography–mass spectrometry. Anal Chim Acta 566:167–171. doi:10.1016/j.aca.2006.03.018
Paci E, Pigini D, Caporossi L, Rosa M, Santoro A, Sisto R, Papaleo B, Tranfo G (2013) Matrix effect in the quantitative determination of mandelic and phenylglyoxylic acid in urine samples by HPLC–MS/MS with isotopic dilution. Curr Anal Chem 9:439–446(8) http://www.ingentaconnect.com/content/ben/cac/2013/00000009/00000003/art00013. Accessed 2 sept 2017
Pereira J, Câmara JS, Colmsjö A, Abdel-Rehim M (2014) Microextraction by packed sorbent: an emerging, selective and high-throughput extraction technique in bioanalysis. Biomed Chromatogr 28:839–847. doi:10.1002/bmc.3156
Prieto A, Schrader S, Bauer C, Möder M (2011a) Synthesis of a molecularly imprinted polymer and its application for microextraction by packed sorbent for the determination of fluoroquinolone related compounds in water. Anal Chim Acta 685:146–152. doi:10.1016/j.aca.2010.11.038
Prieto A, Vallejo A, Zuloaga O, Paschke A, Sellergen B, Schillinger E et al (2011b) Selective determination of estrogenic compounds in water by microextraction by packed sorbents and a molecularly imprinted polymer coupled with large volume injection-in-port-derivatization gas chromatography–mass spectrometry. Anal Chim Acta 703:41–51. doi:10.1016/j.aca.2011.07.007
Qiao F, Sun H, Yan H, Row KH (2006) Molecularly imprinted polymers for solid phase extraction. Chromatographia 64:625–634. doi:10.1365/s10337-006-0097-2
Smith RM (2003) Before the injection—modern methods of sample preparation for separation techniques. J Chromatogr A 1000:3–27. doi:10.1016/S0021-9673(03)00511-9
Soleimani E, Bahrami A, Afkhami A, Ghorbani-shana F (2017) Determination of urinary trans, trans-muconic acid using molecularly imprinted polymer in microextraction by packed sorbent followed by liquid chromatography with ultraviolet detection. J Chromatogr B 1061–1062:65–71. doi:10.1016/j.jchromb.2017.07.008
Šperlingová I, Dabrowská L, Stránský V, Tichý M (2004) A rapid HPLC method for the determination of carboxylic acids in human urine using a monolithic column. Anal Bioanal Chem 378:536–543. doi:10.1007/s00216-003-2289-z
Tarley CRT, Andrade FN, Oliveira FM, Corazza MZ, Azevedo LFM, Segatelli MG (2011) Synthesis and application of imprinted polyvinylimidazole-silica hybrid copolymer for Pb2+ determination by flow-injection thermospray flame furnace atomic absorption spectrometry. Anal Chim Acta 703:145–151. doi:10.1016/j.aca.2011.07.029
Wang JZ, Wang XJ, Tang YH, Shen SJ, Jin YX, Zeng S (2006) Simultaneous determination of mandelic acid enantiomers and phenylglyoxylic acid in urine by high-performance liquid chromatography with precolumn derivatization. J Chromatogr B 840:50–55. doi:10.1016/j.jchromb.2006.04.037
Acknowledgements
This work was supported by the Hamadan University of Medical Sciences, Vice Chancellor for Research Affairs (Grant Number 9410015467).
Author information
Authors and Affiliations
Corresponding author
Additional information
A correction to this article is available online at https://doi.org/10.1007/s00204-018-2153-8.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Soleimani, E., Bahrami, A., Afkhami, A. et al. Selective determination of mandelic acid in urine using molecularly imprinted polymer in microextraction by packed sorbent. Arch Toxicol 92, 213–222 (2018). https://doi.org/10.1007/s00204-017-2057-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00204-017-2057-z