
Abstract
For the first time, an electromembrane extraction combined with a HPLC procedure using diode array and fluorescence detection has been developed for the determination of seven widely used fluoroquinolones (FQs): marbofloxacin, norfloxacin, ciprofloxacin, danofloxacin, enrofloxacin, gatifloxacin and grepafloxacin. The drugs were extracted from acid aqueous sample solutions (pH 5), through a supported liquid membrane consisting of 1-octanol impregnated in the walls of a S6/2 Accurel® polypropylene hollow fiber, to an acid (pH 2) aqueous acceptor solution inside the lumen of the hollow fiber. The main operational parameters were optimized, and extractions were carried out in 15 min using a potential of 50 V. Enrichment factors of 40–85 have been obtained using only 15 min of extraction time versus 330 min used in a previously proposed hollow-fiber liquid-phase microextraction procedure. The procedure allows low detection and quantitation limits of 0.005–0.07 and 0.007–0.15 μg L−1, respectively. The proposed method was successfully applied to the FQs analysis in urban wastewaters.

EME for HPLC determination of fluoroquinolones in wastewater
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
The analysis of complex samples and analyte detection or quantitation at very low levels are nowadays two of the main analytical problems. The analytical complexity increases in those cases where both problems are present; important developments were carried out in clean-up procedures in order to resolve those analytical limitations. Traditional liquid–liquid extraction (LLE) procedures have evolved towards liquid-phase microextraction (LPME) ones [1–3] which offer efficient clean-ups, and allow, in general, high preconcentrations with low solvent consumptions, according to the trends to a “Green Analytical Chemistry”.
LPME has been accomplished either by extraction into small water immiscible drops of organic solvent, hanging (single-drop microextraction) [4, 5] or dispersed (dispersive liquid–liquid microextraction) [6, 7], or into small volumes of acceptor solution inside the lumen of porous hollow fibers (hollow fiber liquid-phase microextraction, HF-LPME) [8]. To date, many articles have been published using HF-LPME configurations for the determination of many compounds belonging to different families, and they have been reviewed recently [8]. However, in some cases, the best results were obtained using long extraction times, even up to 6 h [9, 10].
The ionic nature of several of the interesting analytes in liquid extraction procedures led some authors to propose the use of electrical fields to enhance and to manipulate LLE procedures. Early attempts of analytical LLE driven by external electrical fields (1-15 kV d.c.) were reported by Van der Vlis et al. between 1994 and 1996 [11–13]. In 2005, Arrigan et al. proposed another approach to LLE driven by electrical potential with the development of “electrochemically modulated LLE of ions” [14–16] where the analytes were extracted from a flowing aqueous phase of a flow injection system through a stationary organogel phase (pseudo-liquid) using electrical potentials in the −1 to +1 V range. The electromembrane extraction (EME) concept was introduced for the first time in 2006 by Pedersen-Bjergaard and Rasmusen [17, 18]. In this concept, the mass transfer across the organic solvent immobilized within the pores of a porous hollow fiber (supported liquid membrane, SLM) was accomplished by application of an electrical potential difference as the driving force from two electrodes placed outside the fiber and inside the lumen. This recent sample preparation principle was shown to be a fast and very selective method that provides excellent sample clean-up. One year later, the same authors published an article demonstrating that the flux of analytes across the membrane can be described using a mathematical model based on the Nernst–Planck equation [19]. Up to date, a reduced number of articles related to the use of electromembrane extraction for analytical purposes have been published. They describe the extraction and analysis of acid [20, 21] and, mainly, basic drugs [22–26], even both kinds of drugs simultaneously [27, 28]. Two reviews covering this item, among others HF-LPME procedures, have been also published [8, 29].
Fluoroquinolones (FQs) constitute a large and growing family of antibacterial agents. The precursor of this family, nalidixic acid, was synthesized and commercialized in the 1960s. From this date, and by the modification of this basic structure, successive generations of FQs have been developed, seeking a wider spectrum activity and higher bioavailability. This way, most recent FQs (“fourth generation”), as gatifloxacin, moxifloxacin or trovafloxacin (among others), show a broad activity against both Gram-positive and Gram-negative bacteria through inhibition of their DNA gyrase and topoisomerase IV [30].
In general, FQs are administered to humans or animals by oral ingestion or parental injection with urinary, respiratory or cutaneous infections. FQs can appear as a pollutant by its excretion by treated patients (mainly through urine in their unmodified form) or as a residue in animal tissues (or other products as milk, eggs, etc.) for human consumption. This could lead to the apparition of bacterial strains with resistance to these antibiotics, hindering future treatments [31]. This makes the development of sensitive methods for the determination of FQs at the low levels found in environmental or foodstuff samples a major issue.
There is a wide literature concerning the determination of FQs, as can be seen in the number of reviews published under this topic. From the point of view of the analytical technique applied, recent reviews survey the application of liquid chromatography [32, 33], spectrophotometry [34], luminescence (as spectrofluorimetry and chemiluminescence) [35] and electrochemical methods [36]. With respect to the nature of the studied samples, recent reviews discuss the determination of FQs in environmental waters [37–39] and biological samples and pharmaceuticals [40–42].
The methods of analysis are generally coupled with diverse clean-up procedures like off-line solid-phase extraction (SPE) [43–49], on-line SPE [50, 51], pressurized liquid extraction [52–54] or molecularly imprinted polymer extraction [55, 56], single-drop liquid–liquid microextraction [42], ultrasound-assisted ionic liquid dispersion liquid–liquid microextraction [38, 39]. All these extraction approaches are, generally, time-consuming, require tedious handling and someone needs the consumption of appreciable quantities of organic solvents. The analytical techniques used for the later FQ determination are HPLC and CE using diverse detection systems (DAD, FLD, MS). With respect to the use of hollow fiber in LPME procedures, there is only one article that describes the use of hollow-fiber liquid-phase microextraction (HF-LPME) for the FQs extraction and the later HPLC-DAD/FLD determination [10] using extraction times of 330 min.
In the present work, we study an EME system utilizing electrical potential as driving force (EME) for the extraction of some FQs. This study aims to achieve enrichment factors as high as those that we have previously obtained using HF-LPME but with the purpose to get the best results employing relatively short extraction times and smaller volumes of the acceptor solution. The simplicity of the extraction process and analysis procedure, including the use of aqueous solutions only, makes the developed method as an attractive alternative to other methods, and the low organic solvent consumption to several microlitres versus other clean-up/preconcentration alternatives is also an interesting aspect to consider.
Materials and methods
Chemicals and reagents
All chemicals were of analytical-reagent grade or better. All solutions and dilutions were prepared with ultrapure water from a Milli-Q Plus water purification system (Millipore, Billerica, MA, USA). MRB, NRF, CPR, DNF, ENR, GTF, GRP, dihexyl ether and 1-octanol were purchased from Fluka-Sigma-Aldrich (Madrid, Spain), and the rest of the products were obtained from Merck (Darmstadt, Germany). Table 1 shows the chemical structures and pKa values for the studied fluoroquinolones.
Methanolic working solutions of MRB, NRF, DNF, ENR, GTF and GRP were daily prepared by adequate dilutions from methanolic 100 μg mL−1 stock solutions, and CPR were prepared from methanolic/water 1:1 100 μg mL−1 stock solutions. S6/2 Accurel® polypropylene hollow fiber (1,800 μm i.d., 450 μm wall thickness and 0.2 μm pore size) was purchased from Membrana (Wuppertal, Germany).
Chromatographic conditions
The chromatographic separation was performed at 10 º C using a LaChrom® VWR-Hitachi (Barcelona, Spain) with a quaternary L-2130 pump. For detection, HPLC was equipped with a L-7455 diode array detector (DAD) and a L-7485 fluorescence detector (FLD). For injection, a LaChrom Elite® L-2200 Autosampler was used. Separations were carried out using an LichroCART® 75–4 Purosphere® STAR RP-18 3 μm (75 × 4.0 mm i.d.; VWR, Darmstadt, Germany) preceded by a guard column Kromasil® 100 Å, C18, 5 μm, (15 × 4.6 mm i.d.; Scharlab S.L., Barcelona, Spain). Separations were carried out at 10 °C using a homemade thermostated column holder (VWR Refrigerated Circ Model 1160S, West Chester, PA, USA).
The mobile phase consisted of 0.1 % formic acid (pH 2.6; component A) and acetonitrile (component B) at a flow rate of 0.8 mL min−1. An initial composition 86–14 % (A–B) was used in isocratic mode for 8 min, and then a linear elution gradient was programmed from 86 to 20 % A for another 12 min. Three minutes were waited between injections which allowed re-equilibration of the column to the initial conditions.
Supported liquid membrane preparation and extraction procedure
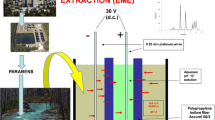
The electrical equipment consisted in a d.c. power supply model Power Source 300 V (VWR International, West Chester, PA, USA) with programmable voltage in the range 2–300 V, providing currents in the range 4–500 mA. Simple platinum wires with a diameter of 0.25 mm were used as electrodes in the sample and acceptor solutions with an average inter-electrode distance of 2 mm which resulted in an electrical field of 50 V cm−1 (for a typical 10 V d.c.). As sample compartment, 10-mL glass vials were used with a height of 48 mm and with an internal diameter of 20 mm (VWR International, West Chester, PA, USA). The sample solution was stirred with a FB15107 Magnetic Stirrer (Fisher Scientific, Pittsburgh, PA, USA).
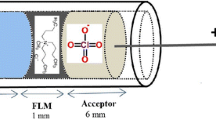
S6/2 hollow fibers were cut into 17-mm pieces, washed with acetone in an ultrasonic bath and dried; the fiber was closed in the lower end by thermal and mechanical pressure, whereas the upper end was connected to a piece of 8-mm length from a pipette tip of polypropylene (Rodelab, Seville, Spain) as a guiding tube. The fiber was soaked with 1-octanol for 5 s to impregnate the pores, and the excess of organic solvent was removed with a medical wipe. The lumen of the prepared fiber piece was filled with 30 μL of acceptor phase (pH 2 aqueous solution) using a HPLC syringe, and the positive electrode was placed in the acceptor solution. Hollow fiber with acceptor solution was placed into the 10-mL sample solution (pH 5), and voltage (50 V) was applied for 15 min during the sample stirring at 750 rpm.
After extraction, the fiber was taken out, and the acceptor phase was extracted using a HPLC syringe and placed into a HPLC microvial to be injected (20 μL) into the HPLC system through the autosampler unit.
Preparation of wastewater samples
Wastewater samples were obtained from “EDAR Punta Umbría-AQUALIA” Wastewater Treatment Plant (WWTP) which is located in Punta Umbría, Huelva, Spain. The WWTP essentially receives urban wastewaters. The capacity of this WWTP is 140,000 inhabitants, and the discharged flow is 21,250 m3/day. Single samples from the influent (raw water, WWR) after the primary sedimentation tank (WW1) and the effluent (treated water after anaerobic digestion, WWT) were analysed.
All samples were filtered through a GDU1 glass fibre filter bed (10–1 μm; Whatman, Mainstone, UK) and through Pall Nylaflo™ nylon membrane filter 0.45 μm ( Pall Corporation, Ann Arbor, MI, USA) and adjusted to pH 4 with HCl. Filtered samples were stored in the dark at 4 °C prior to electromigration procedure. Prior to the EME extraction procedure, nitrogen was bubbled for 10 min into the water samples at pH 2 to eliminate dissolved carbonate, and then, the pH was fixed at 5.
Results and discussion
Chromatographic conditions
Chromatographic separation and detection were optimised in a previous paper [10]. The mobile phase consisted of 0.1 % formic acid and acetonitrile. Different gradient elution conditions were tested searching for the shortest time of analysis without sacrificing peak shape. The gradient elution program described in the previous section was the best option in terms of time of analysis, shape of the peaks and reproducibility. Table 2 shows the monitoring wavelengths for DAD and FLD detection, the retention times and the corresponding standard deviations for the analyzed compounds. To confirm the identification of the substances, the UV and the fluorescence spectra of the chromatographic peaks of samples and the standard solution were compared.
Optimization of experimental conditions for electromembrane extraction
The optimization of the experimental extraction parameters was carried out step by step. In a first experiment, the nature of the SLM was investigated. Based on the pKa values for the analytes, the donor solution (containing all compounds in concentration 5 μg mL−1 each) was fixed at pH 4 to ensure all FQs were protonated, the acceptor phase was fixed at pH 2, the voltage was fixed at 40 V and the extraction was stirring at 600 rpm for 15 min. Extractions were accomplished with dihexyl ether, 2-nitrophenyl octyl ether (NPOE), 1-heptanol, benzyl alcohol, cyclohexanol, pippermint oil, 1-ethyl-2-nitrobenzene, 1-octanol and 1:1, 1:2, 1:3 and 1:5 dihexyl ether : 1-octanol mixtures as the SLM. These solvents were selected as test solvents based on the described experiences in the bibliography and on our earlier experience from HF-LPME and EME procedures, and represented different types of solvent properties [8]. Dihexyl ether, NPOE, peppermint oil and 1-ethyl-2-nitrobenzene could not be used as supported liquid membrane because they produce an interruption in the electrical conduction. Enrichment factors (E f) were calculated as the ratio between the final concentration in the acceptor phase and its initial value in the donor phase. Table 3 shows the E f obtained for these preliminary tests and, as can be seen, 1-octanol resulted in the highest enrichment factors for the extractions and was consequently used throughout the rest of the study. The rest of the solvents and mixtures only lead to recoveries of 3−12 % with respect to the 1-octanol ones. Additionally, an impregnation time of 5 s was established as optimum.
The inter-electrode distance was also checked between 1 and 5 mm, and an optimum distance of 2 mm was obtained (Table 3). Lower and higher inter-electrode distances lead to a general decrease in the analytes’ extraction.
Once organic solvent was optimized, the donor and acceptor solution were tested. Substances to be extracted must be in positive form in the donor phase to cross the organic liquid membrane. Due to the analysed FQs pKa values, donor aqueous solutions within 3–8 pH range were tested using 10-mL aqueous solutions containing 1 μg mL−1 MRB, NRF, CPR and GTF, and 0.5 μg mL−1 DNF, ENR and GRP, that were extracted at 40 V and a speed stirring of 750 rpm during 15 min. As can be seen in Fig. 1a, enrichment factor decreases for pH values lower than 5 and upper than 6 for all analytes, so pH 5 aqueous solutions were selected as donor phase. The effect of the acceptor phase pH was checked using pH 5 aqueous samples containing all analytes studied, and as acceptor phase, aqueous HCl solutions with pH values in the 1–5 range were tested (Fig 1b). The enrichment factors decrease significantly at pH values above 6 and remain practically constant for pH values between 1 and 2, so a pH value about 2 is the best option in order to obtain the best sensitivity for all the analysed compounds.

Optimization of experimental electromembrane extraction conditions. a Donor phase pH, b acceptor phase pH, c voltage, d time and e stirring speed (mean values, n = 3; 1 μg mL−1 MRB, NRF, CPR and GTF; 0.5 μg mL−1 DNF, ENR and GRP; for experimental details, see text)
These data are according to pKa values of fluoroquinolones, so at pH values below pKa2, FQs are as positive, amphoteric or neutral species, and at very low pH values, they are mainly in their positive forms [57, 58]. These facts are according to their electromigration through the SLM from the pH 5 donor phase to the pH 2 acceptor phase where the negative electrode was placed.
Using the selected donor and acceptor phases, the influence of the voltage was checked between 20 and 200 V (Fig 1c) obtaining the best results at 50 V. Finally, the time and stirring speed were optimized keeping the pH of the donor and acceptor solutions and voltage value at the optimum values. The time stirring was tested between 5 and 30 min (Fig 1d) obtaining the best enrichment factor after 15 min extraction for all compounds; after this time, the enrichments decrease for all the compounds. The stirring speed was checked in the range 300–900 rpm (Fig 1e) obtaining the best results at 750 rpm. So, a stirring time of 15 min at 750 rpm was selected as optimum value for the extractions.
Additionally, some tests concerning the effect of the ionic strength of the donor phase in the electromigration process were carried out. Sodium sulphate and sodium chloride concentrations between 0.1 and 1.5 M were checked. No significant effects were observed with this experimental parameter so salting out was not considered for further experiences. It is remarkable that sodium sulphate concentrations over 1 M produce the burning of the fiber. On the other hand, we have checked that, in the experimental conditions, if no electrical power is applied, the extraction of the analytes is null, which shows that there is no liquid-phase microextraction (LPME) contribution to the proposed electromembrane extraction procedure.
Linearity, sensitivity, precision and robustness for the proposed procedure
Linearity of the response function was studied from external calibration. A ten-point (in triplicate) calibration curve was constructed using a least-square linear regression analysis of standard mixtures of the analytes at different concentrations. Using the selected EME conditions, aqueous (Milli-Q water) pH 5 solutions with different analyte concentrations were submitted to the EME procedure and analysed according to the described HPLC procedure. Peak areas are proportional to concentrations in the donor phase. A linear relationship was obtained with correlation coefficients r ≥ 0.999, and the calibration curves obtained showed no changes over the course of 1 month.
Detection and quantitation limits were calculated as the minimum concentration of an analyte giving peaks whose signal-to-noise ratio is 3 and 10, respectively. Table 4 shows the enrichment factors and the detection (LOD) and quantitation limits obtained, using diode array and fluorescence detectors for all analysed compounds. As can be seen, the high enrichment values obtained for all the analytes allow their determination at low concentration levels, and the detection limits were in the 5–70 ng L−1 range.
To evaluate the repeatability and the intermediate precision, aqueous samples at three concentrations levels, 102, 103 and 105 ng L−1, of each compound (in triplicate) were subjected to the entire analytical procedure and measured in one single day and 1 day per week during 2 months, respectively. Intermediate precision was performed using the prediction of actual concentrations from the validation standards selected for the analytical assay in the m × p × n design (m = analytical levels, p = days and n = replications). From the corresponding ANOVA, the intermediate precision was computed [59]. The repeatability, expressed as relative standard deviation, was in the range 1.5−4.4 %. Intermediate precision, also expressed as relative standard deviation, was in the range 1.8–5.1 %.
Wastewater samples’ analysis
To check the suitability of the proposed procedure, the described wastewater samples were spiked with all the FQs at three concentration levels: 150, 500, and 5,000 ng L−1, and submitted to the proposed procedure. Recoveries in the 51−85 % range were obtained. In general, the better results were obtained for the cleaner wastewater samples. Simultaneously, a spiked tap water sample was also extracted and analysed showing recoveries around 85 % for all the FQs, so we thought that it was possible that CO3 2− provokes interference similar to those observed in a previous HF-LPME work developed in our group [60].
For the elimination of the CO3 2−, nitrogen was bubbling 10 min into the sample at pH 2 prior to EME, and this pretreatment procedure was applied to the same spiked water samples above described. The results obtained are depicted in Table 5, showing that, after their CO3 2− elimination, extraction recoveries increase above 80 % in most of the analysed samples, showing only values around 70 % for MRB and NRF in the more complex raw wastewater sample; in this conditions, tap water samples show recoveries around 100 % for all the analysed FQs.
Wastewater samples described in the “Materials and methods” section were directly submitted, after carbonate elimination, to the EME proposed procedure and analysed. Some of the fluoroquinolones were detected in the samples analysed (CPR and DNF in WW1 and ENR in WWR), and CPR and DNF were measured in the sample WWR (22 and 29 ng L−1, respectively).
Figure 2 shows representative DAD and FLD chromatograms obtained from blank, spiked and original (only FLD) raw wastewater (WWR) samples that were selected as the more complex wastewater. Spiked chromatograms show well-defined peaks corresponding only to the spiked substances.

Diode array (DAD) and fluorescence (FLD) chromatograms from blank (a, b), spiked (500 ng L−1) (c, d) and original (e) raw urban wastewater samples using the proposed EME procedure
The excellent recoveries, preconcentration and clean-up obtained imply a great advantage over other sample treatment procedures, like traditional SPE ones, which could justify the alternative use of the proposed EME procedure. Additionally, the proposed EME-HPLC-DAD/FLD procedure competes favourably with more expensive HPLC-MS procedures.
Conclusions
The present work shows the suitability of the proposed electromembrane extraction procedure for the analysis of fluoroquinolones in complex matrices as urban wastewaters. The fluoroquinolones were efficiently extracted across the supported liquid membrane in the acceptor phase, under an electrical field as driving force. The proposed EME procedure allows a highly sensitive determination of seven widely used fluoroquinolones.
The obtained LODs are of the same order of magnitude of the traditional SPE-LC-MS approach, which is very interesting once DAD and FLD detectors are used. EME extraction time, handling and cost, including solvent consumptions, are also interesting compared with more usual SPE procedures.
The extracts obtained from urban wastewaters are very clean, and they can be directly injected into the chromatographic system allowing excellent baselines. Similar or better results were obtained compared to other extraction procedures, and the extraction time decreases from 5.5 h to 15 min with similar and very low detection limits versus an earlier HF-LPME procedure described [10].
References
Auduson G (1986) Aqueous/aqueous extraction by means of a liquid membrane for sample cleanup and preconcentration of amines in a flow system. Anal Chem 58:2714–2723
Thordarson E, Pálmarsdóttir S, Mathiasson L, Jönsson JÅ (1996) Sample preparation using a miniaturized supported liquid membrane device connected online to packed capillary liquid chromatography. Anal Chem 68:2559–2563
Pedersen-Bjergaard S, Rasmussen KE (1999) Liquid–liquid–liquid microextraction for sample preparation of biological fluids prior to capillary electrophoresis. Anal Chem 71:2650–2656
Jeannot MA, Cantwell F (1996) Solvent microextraction into a single drop. Anal Chem 68:2236–2240
Liu HG, Dasgupta PK (1996) Analytical chemistry in a drop. Solvent extraction in a microdrop. Anal Chem 68:1817–1821
Afzali D, Ghanbarian M, Mostafavi A, Shamspur T, Ghaseminezhad S (2012) A novel method for high preconcentration of ultra trace amounts of B1, B2, G1 and G2 aflatoxins in edible oils by dispersive liquid–liquid microextraction after immunoaffinity column clean-up. J Chromatogr A 1247:35–41
Najafi NM, Tavakoli H, Abdollahzadeh Y, Alizadeh R (2012) Comparison of ultrasound-assisted emulsification and dispersive liquid–liquid microextraction methods for the speciation of inorganic selenium in environmental water samples using low density extraction solvents. Anal Chim Acta 714:82–88
Bello-López MA, Ramos-Payán M, Ocaña JA, Fernández-Torres R, Callejón M (2012) Analytical applications of hollow fiber liquid phase microextraction (HF-LPME): a review. Anal Lett 45:1–27
Ramos-Payán M, Bello-López MA, Fernandez-Torres R, Villar-Navarro M, Callejón-Mochón M (2011) Hollow fiber-based liquid phase microextraction (HF-LPME) for a highly sensitive HPLC determination of sulfonamides and their main metabolites. J Chromatogr B 879:197–204
Ramos-Payán M, Bello-López MA, Fernandez-Torres R, Ocaña-Gonzalez JA, Callejón-Mochón M (2011) Hollow fiber-based liquid phase microextraction (HF-LPME) as a new approach for the HPLC determination of fluoroquinolones in biological and environmental matrices. J Pharm Biomed Anal 55:332–341
van der Vlis E, Mazereeuw M, Tjaden UR, Irth H, van der Greef J (1994) Combined liquid–liquid electroextraction and isotachophoresis as a fast online focusing step in capillary electrophoresis. J Chromatogr A 687:333–341
van der Vlis E, Mazereeuw M, Tjaden UR, Irth H, van der Greef J (1995) Combined liquid–liquid electroextraction-isotachophoresis for loadability enhancement in capillary zone electrophoresis-mass spectrometry. J Chromatogr A 712:227–234
van der Vlis E, Mazereeuw M, Tjaden UR, Irth H, van der Greef J (1996) Development of a needle device for online electroextraction–liquid chromatography. J Chromatogr A 741:13–21
Berduque A, Sherburn A, Ghita M, Dryfe RAW, Arrigan DWM (2005) Electrochemically modulated liquid–liquid extraction of ions. Anal Chem 77:7310–7318
Berduque A, Arrigan DWM (2006) Selectivity in the coextraction of cation and anion by electrochemically modulated liquid–liquid extraction. Anal Chem 78:2717–2725
Berduque A, Brien JO, Alderman J, Arrigan DWM (2008) Microfluidic chip for electrochemically-modulated liquid|liquid extraction of ions. Electrochem Commun 10:20–24
Pedersen-Bjergaard S, Rasmussen KE (2006) Electrokinetic migration across artificial liquid membranes. J Chromatogr A 1109:183–190
Gjerstad A, Rasmussen KE, Pedersen-Bjergaard S (2006) Electrokinetic migration across artificial liquid membranes: tuning the membrane chemistry to different types of drug substances. J Chromatogr A 1124:29–34
Gjerstad A, Rasmussen KE, Pedersen-Bjergaard S (2007) Simulation of flux during electro-membrane extraction based on the Nernst–Planck equation. J Chromatogr A 1174:104–111
Ramos-Payán M, Bello-López MA, Fernandez-Torres R, Villar-Navarro M, Callejón-Mochón M (2011) Electromembrane extraction (EME) and HPLC determination of non-steroidal anti-inflammatory drugs (NSAIDs) in wastewater samples. Talanta 85:394–399
Balchen M, Gjerstad A, Rasmussen KE, Pedersen-Bjergaard S (2007) Electrokinetic migration of acidic drugs across a supported liquid membrane. J Chromatogr A 1152:220–225
Dominguez NC, Gjelstad A, Molina-Nadal A, Jensen H, Petersen NJ, Hansen SH, Rasmussen KE, Pedersen-Bjergaard S (2012) Selective electromembrane extraction at low voltages based on analyte polarity and charge. J Chromatogr A 1248:48–54
Davarani SSH, Najarian AM, Nojavan S, Tabatabaei M (2012) Electromembrane extraction combined with gas chromatography for quantification of tricyclic antidepressants in human body fluids. Anal Chim Acta 725:51–56
Jamt RE, Gjelstad A, Eibak LE, Oiestad EL, Christophersen AS, Rasmussen KE, Pedersen-Bjergaard S (2012) Electromembrane extraction of stimulating drugs from undiluted whole blood. J Chromatogr A 1232:27–36
Fotouhi L, Yamini Y, Molaei S, Seidi S (2011) Comparison of conventional hollow fiber based liquid phase microextraction and electromembrane extraction efficiencies for the extraction of ephedrine from biological fluids. J Chromatogr A 1218:8581–8586
Eibak LEE, Gjerstad A, Rasmussen KE, Pedersen-Bjergaard S (2011) Exhaustive electromembrane extraction of some basic drugs from human plasma followed by liquid chromatography–mass spectrometry. J Pharm Biomed Anal 57:33–38
Basheer C, Lee J, Pedersen-Bjergaard S, Rasmussen KE, Lee HK (2010) Simultaneous extraction of acidic and basic drugs at neutral sample pH: a novel electro-mediated microextraction approach. J Chromatogr A 1217:6661–6667
Seidi S, Yamini Y, Rezazadeh M, Esrafili A (2012) Low-voltage electrically-enhanced microextraction as a novel technique for simultaneous extraction of acidic and basic drugs from biological fluids. J Chromatogr A 1243:6–13
Gjelstad A, Taherkhani H, Rasmussen KE, Pederson-Bjergaard S, Majors R (2011) Hollow-fiber liquid-phase microextraction in the three-phase mode—practical consideration. LC-GC Europe 24(12):633634
Blondeau MJ (2004) Fluoroquinolones: mechanism of action, classification, and development of resistance. Surv Ophthalmol 49:S73–S78
Pedersen K, Wedderkopp A (2003) Resistance to quinolones in Campylobacter jejuni and Campylobacter coli from Danish broilers at farm level. J Appl Microbiol 94:111–119
Sousa J, Alves G, Abrantes J, Fortuna A, Falcao A (2012) Analytical methods for determination of new fluoroquinolones in biological matrices and pharmaceutical formulations by liquid chromatography: a review. Anal Bioanal Chem 403:93–129
Samanidou V, Christodoulou E, Papadoyannis I (2005) Advances in chromatographic analyses of fluoroquinolones in pharmaceuticals and biological samples—a review article. Current Pharm Anal 1:283–308
Kaur K, Kumar A, Malik AK, Singh B, Rao ALJ (2008) Spectrophotometric methods for the determination of fluoroquinolones: a review. Crit Rev Anal Chem 38:2–18
Kuldeep K, Baldev S, Ashok Kumar M (2011) Chemiluminescence and spectrofluorimetric methods for determination of fluoroquinolones: a review. Anal Lett 44:1602–1639
Radi AE (2010) Application of electrochemical methods for analysis of fluoroquinolones antibacterial agents and fluoroquinolones–DNA interactions. Open Chem and Biomed Meth Journal 3:27–36
Speltini A, Sturini M, Maraschi F, Profumo A (2010) Fluoroquinolone antibiotics in environmental waters: sample preparation and determination. J Sep Sci 33:1115–1131
Parrilla MM, Parrilla P, Martínez M, Gil MD (2012) Determination of eight fluoroquinolones in groundwater samples with ultrasound-assisted ionic liquid dispersive liquid–liquid microextraction prior to high-performance liquid chromatography and fluorescence detection. Anal Chim Acta 748:20–27
Yan H, Wang H, Qin X, Liu B, Du J (2011) Ultrasound-assisted dispersive liquid–liquid microextraction for determination of fluoroquinolones in pharmaceutical wastewater. J Pharm Biomed Anal 54:53–57
Kulapina EG, Barinova OV, Kulapina OI, Utc IA, Snesarev SV (2009) Current methods of antibiotic determination in biological samples and drugs (review). Antibiotiki i Khimioterapiya 54:53–60
Schneider M, Marilyn J (2009) Methods for the analysis of fluoroquinolones in biological fluids. Bioanalysis 1:415–435
Gao W, Chen G, Chen Y, Zhang X, Yin Y, Hu Z (2011) Application of single drop liquid–liquid–liquid microextraction for the determination of fluoroquinolones in human urine by capillary electrophoresis. J Chromatogr B 879:291–295
Herrera-Herrera AV, Hernandez-Borges J, Rodriguez-Delgado MA (2009) Fluoroquinolone antibiotic determination in bovine, ovine and caprine milk using solid-phase extraction and high-performance liquid chromatography-fluorescence detection with ionic liquids as mobile phase additives. J Chromatogr A 1216:7281–7287
Li-Li M, Chang-ShenG WH, Jian S, Xing-Wang Z, Yue-Fei R, Yu-Qiu W (2010) Simultaneous extraction and determination of eighteen fluoroquinolone, tetracycline and sulfonamide antibiotics from soils using solid-phase extraction and liquid chromatography-tandem mass spectrometry. Fenxi Huaxue 38:21–26
Jianzhong S, Liming G, Fei X, Qinxiong R, Xi X, Xiaowei L, Shuangyang D (2010) Simultaneous determination of fluoroquinolones, tetracyclines and sulfonamides in chicken muscle by UPLC-MS-MS. Chromatographia 71:383–388
Tang Q, Yang T, Tan X, Luo J (2009) Simultaneous determination of fluoroquinolone antibiotic residues in milk sample by solid-phase extraction-liquid chromatography-tandem mass spectrometry. J Agric Food Chem 57:4535–4539
Lombardo-Aguei M, Gamiz-Gracia L, Garcia-Campana AM, Cruces-Blanco C (2010) Sensitive determination of fluoroquinolone residues in waters by capillary electrophoresis with laser-induced fluorescence detection. Anal Bioanal Chem 396:1551–1557
Wang S, Mu H, Bai Y, Zhang Y, Liu H (2009) Multiresidue determination of fluoroquinolones, organophosphorus and N-methyl carbamates simultaneously in porcine tissue using MSPD and HPLC-DAD. J Chromatogr B 877:2961–2966
Sturini M, Speltini A, Pretali L, Fasani E, Profumo A (2009) Solid-phase extraction and HPLC determination of fluoroquinolones in surface waters. J Sep Sci 32:3020–3028
Jing-Fang H, Bo L, Qiong-Wei Y, Yu-Qi F (2006) Determination of fluoroquinolones in eggs using in-tube solid-phase microextraction coupled to high-performance liquid chromatography. Anal Bioanal Chem 384:1228–1235
Mitami K, Kataoka H (2006) Determination of fluoroquinolones in environmental waters by in-tube solid-phase microextraction coupled with liquid chromatography–tandem mass spectrometry. Anal Chim Acta 562:16–22
Rodriguez E, Villoslada F, Moreno-Bondi MC, Marazuela MD (2010) Optimization of a pressurized liquid extraction method by experimental design methodologies for the determination of fluoroquinolone residues in infant foods by liquid chromatography. J Chromatogr A 1217:605–613
Lillenberg M, Yurchenko S, Kipper K, Herodes K, Pihl V, Sepp K, Lohmus R, Nei L (2009) Simultaneous determination of fluoroquinolones, sulfonamides and tetracyclines in sewage sludge by pressurized liquid extraction and liquid chromatography electrospray ionization-mass spectrometry. J Chromatogr A 1216:5949–5954
Carretero V, Blasco C, Pico Y (2008) Multi-class determination of antimicrobials in meat by pressurized liquid extraction and liquid chromatography–tandem mass spectrometry. J Chromatogr A 1209:162–173
Ligang C, Xiaopan Z, Yang X, Xiaobo D, Xin S, Lei S, Hui W, Qi Z, Aimin Y, Hanqi Z (2010) Determination of fluoroquinolone antibiotics in environmental water samples based on magnetic molecularly imprinted polymer extraction followed by liquid chromatography–tandem mass spectrometry. Anal Chim Acta 662:31–38
Hanwen S, Guangyu L, Fengxia Q (2009) Water-compatible molecularly imprinted solid phase extraction-HPLC for determination of fluoroquinolones residues in chicken. Shipin Kexue (Beijing, China) 30:151–154
Babic S, Horvat AJM, Mutavdzic D, Kastelan-Macan M (2007) Determination of pKa values of active pharmaceutical ingredients. Trends in Anal Chem 26(11):1043–1061
Sun J, Sakai S, Tauchi Y, Deguchi Y, Chen J, Zhang R, Morimoto K (2007) Determination of lipophilicity of two quinolone antibacterials, ciprofloxacin and grepafloxacin, in the protonation equilibrium. European J of Pharm and Biopharm 54(1):51–58
Gonzalez AG, Herrador MA (2007) A practical guide to analytical method validation including measurement uncertainty and accuracy profiles. Trends Anal Chem 26:227–238
Villar-Navarro M, Ramos-Payán M, Pérez-Bernal JL, Fernández-Torres R, Callejón-Mochón M, Bello-López MA (2012) Application of three phase hollow fiber based liquid phase microextraction (HF-LPME) for the simultaneous HPLC determination of phenol substituting compounds (alkyl-, chloro- and nitrophenols). Talanta 99:55–61
Acknowledgments
This work was supported by the Project CTM2009-12858-C02-01 from the “Dirección General de Investigación y Gestión del Plan Nacional de I + D + i (Ministerio de Educación y Ciencia) Spain. María Ramos-Payán is grateful to the University of Seville for personal funding through the “Fundación Cámara” program. We are grateful to Enrique Castro Pérez from MP for kindly supplying the analysed wastewater samples from “EDAR Punta Umbría-AQUALIA” Wastewater Treatment Plant located in Punta Umbría, Huelva, Spain.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Ramos-Payán, M., Villar-Navarro, M., Fernández-Torres, R. et al. Electromembrane extraction (EME)—an easy, novel and rapid extraction procedure for the HPLC determination of fluoroquinolones in wastewater samples. Anal Bioanal Chem 405, 2575–2584 (2013). https://doi.org/10.1007/s00216-012-6664-5
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00216-012-6664-5