
Abstract
A miniaturized dispersive liquid–liquid microextraction (DLLME) procedure coupled to liquid chromatography (LC) with fluorimetric detection was evaluated for the preconcentration and determination of thiamine (vitamin B1). Derivatization was carried out by chemical oxidation of thiamine with 5 × 10−5 M ferricyanide at pH 13 to form fluorescent thiochrome. For DLLME, 0.5 mL of acetonitrile (dispersing solvent) containing 90 μL of tetrachloroethane (extraction solvent) was rapidly injected into 10 mL of sample solution containing the derivatized thiochrome and 24% (w/v) sodium chloride, thereby forming a cloudy solution. Phase separation was carried out by centrifugation, and a volume of 20 μL of the sedimented phase was submitted to LC. The mobile phase was a mixture of a 90% (v/v) 10 mM KH2PO4 (pH 7) solution and 10% (v/v) acetonitrile at 1 mL min−1. An amide-based stationary phase involving a ligand with amide groups and the endcapping of trimethylsilyl was used. Specificity, linearity, precision, recovery, and sensitivity were satisfactory. Calibration graph was carried out by the standard additions method and was linear between 1 and 10 ng mL−1. The detection limit was 0.09 ng mL−1. The selectivity of the method was judged from the absence of interfering peaks at the thiamine elution time for blank chromatograms of unspiked samples. A relative standard deviation of 3.2% was obtained for a standard solution containing thiamine at 5 ng mL−1. The esters thiamine monophosphate and thiamine pyrophosphate can also be determined by submitting the sample to successive acid and enzymatic treatments. The method was applied to the determination of thiamine in different foods such as beer, brewer’s yeast, honey, and baby foods including infant formulas, fermented milk, cereals, and purees. For the analysis of solid samples, a previous extraction step was applied based on an acid hydrolysis with trichloroacetic acid. The reliability of the procedure was checked by analyzing a certified reference material, pig’s liver (CRM 487). The value obtained was 8.76 ± 0.2 μg g−1 thiamine, which is in excellent agreement with the certified value, 8.6 ± 1.1 μg g−1.
Similar content being viewed by others
Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
The water-soluble B group vitamins include many compounds of different chemical structure and biological roles, which are essential for the health of adults and, especially, children. In foodstuffs, thiamine (vitamin B1) occurs in its free form or as the mono- and pyrophosphate esters bound to proteins. Thiamine pyrophosphate, the physiologically active form of thiamine, functions as a coenzyme in the carbohydrate metabolism. Although it is extremely widespread in small amounts, only a few foodstuffs, generally those that are rich in carbohydrate, can be regarded as good sources. Examples are legume seeds and the germ of cereal grains, cereal products, vegetables, meat, and milk products [1]. There is renewed interest in vitamin measurement due to the popularity of fortified foods or dietary supplements, in which vitamins are usually added in a single chemical form.
Thiamine cannot be differentiated from its esters by the usual analytical techniques such as the fluorimetric method proposed by the AOAC [2]. It is important to know the speciation of thiamine because of the different activities and stabilities of the thiamine esters, a task usually accomplished by liquid chromatography [3]. Procedures for the determination of thiamine in foods using reversed-phase or ion pair chromatography [4–43] have been proposed, and several reviews have also been published [44–47]. Detection has been carried out by UV spectrophotometry for samples that contain sufficient amounts of thiamine, while a fluorescence derivatization reaction based on the oxidation of thiamine to thiochrome or mass spectrometry has been used to determine small concentrations.
Emerging methods for food matrices tend towards efficient and miniaturized techniques that share the priorities of green chemistry with respect to the environment through the use of chemical processes that do not produce residues and which use low amounts of safe solvents for dissolving or extracting analytes [48].
Microextraction techniques [49, 50] represent a relevant way for the miniaturization of the analytical laboratory [51], the selective extraction of compounds being based on differences in their physical–chemical characteristics (molecular weight, charge, solubility, polarity, and volatility). Liquid-phase microextraction (LPME) includes several miniaturized techniques based on the extraction of analytes in a liquid phase using very low amounts of organic solvents [52]. Dispersive liquid–liquid microextraction (DLLME) is a very simple and rapid extraction method, based on the use of a ternary component solvent system, which has been applied to the extraction and preconcentration of both organic and inorganic compounds from aqueous samples [52–54]. The low consumption of time and organic solvents are two of the main advantages of this technique, which can be included in the group of clean chemistry procedures. A procedure has been proposed for the determination of thiamine using DLLME without chromatographic separation [55].
In the present study, a miniaturized sample treatment procedure based on DLLME coupled to a reversed-phase LC technique using an amide-based stationary phase is proposed for determining thiamine (T), thiamine monophosphate (TMP), and thiamine pyrophosphate (TPP). Detection was performed by precolumn fluorescence derivatization using a system involving the oxidation to the corresponding highly fluorescent thiochromes. The procedure was applied to the determination of thiamine and its esters in different type of foods: beer, brewer’s yeast, honey, and baby foods including infant formulas, fermented milk, cereals, and purees. The main significance of this work is that, at the best of our knowledge, this is the first time that these vitamins have been determined using green chemistry principles avoiding the use of high amounts of solvents and the generation of residues coupled to liquid chromatography.
Materials and methods
Chemicals
T, TMP, and TPP were obtained from Sigma-Aldrich (St. Louis, MO, USA). Individual stock solutions of the compounds (1,000 μg mL−1) were prepared in pure water and stored in darkness at −10 °C. Working standard solutions were freshly prepared in pure water and stored at 4 °C. Alkaline phosphatase (ALKP) from Bovine intestinal mucosa (Sigma-Aldrich) was dissolved in 10 mM Tris buffer (pH 7.4), and takadiastase from Aspergillus oryzae (Fluka) was used solid. Potassium ferricyanide, sodium hydroxide, potassium dihydrogen phosphate, and phosphoric acid (85%) were purchased from Fluka (Buchs, Switzerland) and Merck (Darmstadt, Germany). Chromatographic quality carbon tetrachloride, tetrachloroethane dichloromethane, chloroform, undecanone, undecanol, decanol, acetone, acetonitrile, and methanol were obtained from Sigma. Water used was previously purified in a Milli-Q system (Millipore, Bedford, MA, USA).
Instrumentation
The LC system consisted of an Agilent 1100 (Agilent, Waldbronn, Germany) quaternary pump (G1311A) operating at room temperature with a flow rate of 1 mL min −1. The solvents were degassed using an online membrane system (Agilent 1100, G1379A). The fluorescence detector was an Agilent FLD (Agilent 1100, G1321A) operating at an excitation wavelength of 375 nm and an emission wavelength of 438 nm. The analytical column used for the reversed-phase technique was a Discovery RP-AmideC16 (15 cm × 0.46 cm × 5 μm) (Supelco, Bellefonte, PA, USA). Aliquots of 20 μL were injected manually using a Model 7125-075 rheodyne injection valve (Rheodyne, Berkeley, CA, USA).
To filter the samples, Econofilter 25 nylon filters (0.45 μm) (Agilent) were used. An EBA 20 (Hettich, Tuttlingen, Germany) centrifuge was used at the maximum speed supported by the conical glass tubes, 4,000 rpm. The ultrasonic processor UP 200 H (Hielscher Ultrasonics GmbH, Germany) was used for the hydrolysis step. A laboratory-made system built in the Central Laboratory Service of the University of Murcia, consisting of a drilled block equipped with an electronic temperature control system, was used to heat the tubes for hydrolysis.
Samples
The samples were different type of foods rich in thiamine (beer, brewer’s yeast, honey, and baby foods including infant formulas, fermented milk, cereals, and purees). The method was validated using a reference material, pig’s liver CRM 487 supplied by the Community Bureau of Reference, BCR (Belgium).
All operations were performed in subdued light. The beer samples were diluted with water in a 1:1 proportion (5 mL beer and 5 mL water) and directly submitted to extraction. The honey samples were prepared by weighing 1 g and diluting up to 10 mL with water. For the analysis of solid samples, a previous extraction step was applied based on an acid hydrolysis by weighing different amounts of sample (0.1 g of brewer’s yeast, 2 g of fermented milk, 0.2 g of infant formula or cereals) and adding 4 mL water and 2 mL of 3% m/v trichloroacetic acid. After mixing for 15 min, the mixture was centrifuged for 10 min at 6,000 rpm, and the supernatant was diluted up to 10 mL with water and filtered.
Enzymatic hydrolysis
Samples were submitted to successive acid and enzymatic hydrolysis steps, as described by the analytical methods committee [44]. Amounts of 0.1–2 g were weighed into an amber 15-mL screw cap glass tube with conical bottom, and 6 mL of 0.1 M hydrochloric acid was added. The suspension was homogenized by using an ultrasonic processor for 30 s (40% amplitude, 0.5 cycles) and then heated at 90 °C for 30 min. When the suspension was cold, the pH was adjusted to 7 using 1 M sodium hydroxide, and 25 units of ALKP were added. The sample was incubated in a block with magnetic stirring at 55 °C for 2 h in the absence of light. Then, 1 mL of 50% (w/v) trichloroacetic acid was added, and the mixture was again heated at 90 °C for 10 min. The sample was cooled and centrifuged at 4,000 rpm for 10 min. For carrying out the derivatization reaction, the supernatant was made up to 10 mL with water, and 2 mL of 2 M sodium hydroxide solution (pH 13) and 50 μL of 0.01 M potassium ferricyanide were added for derivatization and subsequently DLLME-LC. The certified reference sample was analyzed in the same way but using the enzyme takadiastase (0.02 g at pH 4) following the procedure proposed by the BCR.
DLLME procedure
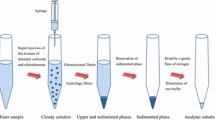
For DLLME, a 10-mL aliquot of the standard or the sample was placed in a 15-mL screw cap glass tube with conical bottom. For carrying out the derivatization reaction, volumes of 2 mL of a 2 M sodium hydroxide solution (pH 13) and 50 μL of 0.01 M potassium ferricyanide were added, and the mixture was vortex-shaken. Sodium chloride was added to reach a 24% m/v concentration. Then, 0.5 mL of acetonitrile (dispersing solvent) containing 90 μL of tetrachloroethane (extraction solvent) was rapidly injected into the sample solution using a micropipette, and the mixture was again gently shaken manually for several seconds. A cloudy solution consisting of very fine droplets of tetrachloroethane dispersed into the sample solution was formed, and the derivatized thiochromes were extracted into the fine droplets. After centrifugation for 1 min at 4,000 rpm, the extraction solvent was sedimented at the bottom of the conical tube (volume about 40 μL). Twenty microliters of the sedimented phase was removed with a microsyringe and injected into the LC. The calibration curve was obtained by least-squares linear regression analysis of the peak area versus thiamine concentration in nanograms per milliliter using six levels in duplicate experiments.
Results and discussion
Derivatization reaction
The oxidation of thiamine to thiochrome is a very efficient, simple, and fast derivatization reaction, giving a highly fluorescent derivative. An alkaline medium is needed for the reaction, and the pH effect was studied in the 6–13 range using a sodium hydroxide solution. The sensitivity continuously increased up to pH 13 (obtained by adding 2 mL of 2 M NaOH solution), which was selected. When the influence of the oxidant concentration was studied, the fluorescence increased to reach a maximum up to 2 × 10−4–7 × 10−4 M and decreased for higher concentrations; a 5 × 10−4 M concentration was selected.
Chromatographic separation
Thiamine and its esters have an ionic character and can interact with the silanol groups of the conventional ODS columns. Separation using the amide-based stationary phase with the endcapping of trimethylsilyl is advantageous with respect to other reversed-phases because the peaks are much narrower and column life is longer due to the simplicity of the mobile phase, avoiding the need for ion-pairing reagents. The optimal composition of the mobile phase was studied using several phosphate buffers at pH values ranging between 4 and 8, and phosphate concentrations in the 10–50 mM range. The addition of acetonitrile was assayed to decrease vitamin retention. The best separation was achieved using a mixture of 90% v/v 10 mM KH2PO4 buffer (pH 7) and 10% v/v acetonitrile. The flow rate was 1 mL min−1.
DLLME parameters
The parameters affecting the DLLME procedure, such as the type and volume of both the extraction and disperser solvents, salt addition, and centrifugation time, were optimized. For this purpose, 10 mL of an aqueous solution or a sample containing a thiamine concentration of about 100 ng mL−1 and the derivatized reagents were submitted to DLLME, and a 20-μL aliquot of the settled phase was injected into the LC.
The extraction solvent must have high extraction capability, a higher density than water, low solubility in water, and good chromatographic behavior. Thus, carbon tetrachloride (CCl4), chloroform (CHCl3), dichloromethane (CH2Cl2), and 1,1,2,2-tetrachloroethane (C2H2Cl4) were assayed using 100 μL of the extraction solvent and 0.5 mL acetone as the disperser solvent. The sedimented phase was not discernible when using dichloromethane due to its high solubility in water. Figure 1a shows the results obtained. The best extraction efficiency was obtained using tetrachloroethane as extraction solvent.

Influence of different extraction solvents (a) and disperser solvents (b) on the extraction of thiochrome by DLLME. Concentration of thiamine, 100 ng mL−1
The influence of the C2H2Cl4 volume was studied in the 40 to 120 μL range. Peak areas increased with increasing extraction solvent volumes in the range 40–90 μL. On further increasing the volume of the extraction solvent, the peak areas decreased as a consequence of dilution and so 90 μL was selected. The volume of the sedimented phase was 40 ± 10 μL after extraction and centrifugation, which means a preconcentration factor close to 250.
The disperser solvent must be miscible in the extraction solvent and the aqueous phase. Acetone, methanol, and acetonitrile were assayed by rapidly injecting 0.5 mL of each disperser containing 90 μL of C2H2Cl4 into the aqueous solution. The extraction efficiency was highest when using acetonitrile (Fig. 1b), and this was therefore selected.
The volumes assayed for the disperser solvent were 0.25–2 mL, containing, in all cases, the extraction solvent volume at the previously optimized value. The extraction efficiency increased up to 0.5 mL and then decreased with higher volumes, as the solubility of thiochrome probably increases in water, and so the extraction efficiency decreases. Highest sensitivity was attained with 0.5 mL of acetonitrile.
Sodium chloride was added to the aqueous phase to increase its ionic strength, thus reducing the solubility of thiochrome and increasing the solubility in the organic phase. The effect of the amount of sodium chloride on the extraction efficiency was studied between 0% and 24% m/v. The peak area increased with increasing salt concentration in all the ranges studied, and a 24% m/v concentration was selected.
Extraction time in DLLME is defined as the time between injecting the mixture of disperser and extraction solvents and before starting the centrifugation step. As expected, no differences in sensitivity were attained in the interval 30 s to 5 min, demonstrating that DLLME is practically time-independent, one of its most important advantages. Therefore, the mixture was shaken for a few seconds and then centrifuged, the centrifugation step being the most time-consuming. Nevertheless, the centrifugation time and speed necessary to disrupt the cloudy solution and collect the sedimented phase were evaluated. The centrifugation time was varied between 1 and 4 min, and extraction efficiency continuously decreased with longer times, and a time of 1 min is recommended. The centrifugation speed was modified in the 1,000–6,000 rpm range, and sensitivity increased up to 4,000 rpm, decreasing for higher values. Thus, the maximum speed recommended for the glass conic tubes used, 4,000 rpm, was selected.
Comparison with other LPME procedures
The preconcentration procedure using DLLME was compared with other LPME methods. Thus, the use of directly suspended drop microextraction (DSDME), in which a symmetrical rotated flow field is created by a stir bar placed on the bottom of a cylindrical vessel containing the sample solution was assayed. For this, a 10-mL sample solution containing the derivatized thiochrome was stirred until a vortex was obtained. Then, 100 μL of an extraction solvent (undecanone, 1-undecanol, or decanol) were incorporated, and the mixture stirred for 30 min. The organic phase was removed with a microsyringe, and an aliquot of 20 μL was injected into the LC. Best results were obtained using 1-undecanol, but the extraction efficiency was lower and the extraction time longer than when using DLLME. The ultrasound-assisted emulsification microextraction (USAEME) technique was also tried using a 10-mL aliquot containing the derivatized thiochrome to which 100 μL of the extraction solvent (1-undecanol or tetrachloroethane) were added and the mixture submitted to an ultrasonic probe for 2 min. A solution consisting of very fine droplets of tetrachloroethane dispersed into the sample solution was formed. After centrifugation for 1 min at 4,000 rpm, the extraction solvent was sedimented at the bottom of the conical tube and an aliquot was injected into the LC. However, the volume of organic solvent recovered in these conditions was too low, and the USAEME technique was also discarded. DLLME was confirmed to be the most suitable LPME technique for the purpose here reported. Sample throughput was about 4 samples h−1 and was very favorable compared with the other LPME systems.
Optimization of enzymatic hydrolysis
For the determination of free thiamine in food samples, it must be released from the proteic bindings. For this, a step of hot acid digestion using trichloroacetic acid was chosen because this does not destroy the phosphate esters. However, for the extraction of total thiamine, acid digestion was followed by enzymatic hydrolysis to achieve dephosphorylation of the phosphate esters using ALKP, which is a hydrolase enzyme responsible for removing phosphate groups from many types of molecules. Finally, trichloroacetic acid was added to precipitate the proteins. The parameters influencing the hydrolysis, such as pH, concentration of enzyme and substrate, and both temperature and time of the incubation step, were optimized.
As can be seen in Fig. 2a, the hydrolysis was more effective at pH 7 for both substrates, TMP and TPP. The optimal enzyme concentration was studied between 5 and 30 units, and the sensitivity continuously increased throughout the range studied for TMP, while a maximum value was obtained for TPP using 25 units (Fig. 2b). When the effect of substrate concentration was analyzed (Fig. 2c), the signal increased for higher concentrations of both TMP and TPP; however, linearity was also achieved for concentrations lower than 1 μg mL−1. The incubation temperature was varied in the 30–70 °C range and, as shown in Fig. 2d, the signals were higher for both esters up to 55 °C after which they rapidly decreased, and so this value was chosen. Finally, the incubation time was also seen to be an important factor affecting enzymatic hydrolysis. The effect of this variable was studied between 1 and 8 h (Fig. 2e), a time of 2 h being selected as a compromise between the maximum signal and the total analysis time.

Influence of the experimental variables affecting the extraction efficiency of thiochrome by DLLME. Concentration of thiamine, 100 ng mL−1
Analytical characteristics of the method
The method was validated for linearity, detection and quantification limits, selectivity, accuracy, and precision. The calibration curve using DLLME was obtained by least-squares linear regression analysis of the peak area versus thiamine concentration using six levels in duplicate experiments. Table 1 shows the results obtained. The limit of detection (LOD, calculated as three times the standard error of the estimate) was 0.09 ng mL−1. The selectivity of the method was judged from the absence of interfering peaks at the thiamine elution time for blank chromatograms of different unspiked samples. No matrix compounds existed that might give a false-positive signal in the blank samples. The repeatability was calculated by using the relative standard deviation from a series of ten consecutive DLLME-LC analyses of one aqueous standard solution containing thiamine at 5 ng mL−1 and a beer sample containing 520 ng g−1. Relative standard deviations (RSD) of 3.2% and 5.6%, respectively, were obtained.
The matrix effect was studied by comparing the slopes of aqueous standards and standard additions calibration graphs for the different samples. Values were 0.257 for cereals, 0.456 for beer, 0.033 for honey, 0.136 for puree, 0.082 for infant formula, and 0.205 mL ng−1 for fermented milk, obtained by plotting concentration (at six different levels) against peak area and following linear regression analysis. The presence of a matrix effect was confirmed because “p” values obtained from application of a paired t test were lower than 0.05. Consequently, calibration and analysis of all the samples must be performed by the standard additions method.
The validation parameters obtained for the thiamine esters using the enzymatic hydrolysis and DLLME procedure are also shown in Table 1.
Analysis of samples and validation of the method
Thiamine is very labile in neutral to basic solutions but not in acidic solutions. Loss of thiamine may occur for mildly acidic to neutral or basic foods upon heat treatment, but more acidic foods would not suffer this loss. The greatest losses during domestic cooking as well as in commercial food processing occur when the vitamin is leached into the cooking water. The vitamin is necessary to break up carbohydrates, and its main sources are cereals and wholemeal grain. Beer is a natural beverage manufactured from malted barley, water, yeast, and hops, and contains all of the important vitamins of the B group that come from malt, increase during barley germination, and remain during toasting. Consequently, samples of different type of foods rich in thiamine as beer, brewer’s yeast, honey, and baby foods including infant formulas, fermented milk, cereals, and purees were analyzed.
For the solid samples, an acid hydrolysis using trichloroacetic acid was used because, in this way the phosphate esters are not affected. The results showed that the commercial brewer’s yeast analyzed contained the higher levels of thiamine. Cereals and infant formula also contained a high amount of the vitamin, since the products are commercialized in the dry form. The samples of fermented milk contained lower levels and the lowest concentrations were found in the samples of beer, honey, and infant puree (Table 2). To test the accuracy of the proposed method, five different foods were fortified and analyzed by the optimized method, taking into account the known analyte contents for these samples. The results for thiamine are presented in Table 3. Figure 3 shows typical chromatographic profiles obtained using DLLME-LC for non-spiked samples of brewer’s yeast and two baby foods, namely cereals mixture and fermented milk in the selected conditions. Similar chromatograms were obtained for the other samples.

Elution profiles obtained for different food samples by DLLME-LC–fluorescence
The food samples were then submitted to both acid and enzymatic hydrolysis to examine the presence of thiamine esters. A comparison was made on the efficiency of the enzymes ALKP and takadiastase for dephosphorylation of the esters in the cereals mixture sample, and the results obtained (micrograms per gram thiamine) were 4.15 without enzyme, 4.05 with takadiastase, and 4.35 with ALKP, showing that all the thiamine is present in the free form. The values obtained were also in accordance with the contents reported by the manufacturer, which were 4.0 μg g−1 thiamine for both cereals mixture and cereals without gluten samples. For the infant formula, values of 4.97 without enzyme, 5.03 with takadiastase, and 5.12 with ALKP were obtained, which is again in accordance with the 4.7 μg g−1 thiamine content reported by the manufacturer. Similar results were obtained for the rest of the samples, confirming the presence of the thiamine in the free form and the non-existence of esters.
The reliability of the method was further checked by using a certified reference material, pig’s liver (CRM 487). The value obtained was 8.76 ± 0.2 μg g−1 thiamine, which is in excellent agreement with the certified value, 8.6 ± 1.1 μg g−1. The statistical study using the paired t test showed that there was no significant difference (95% confidence interval) between the result obtained and the certified value. Such data also confirm the efficacy of the extraction procedure for recovering both free supplemented and endogenous thiamine in foods.
Conclusion
The miniaturized preconcentration procedure based on DLLME was seen to be an excellent alternative for the analysis of thiamine in different foods at low concentrations using LC-fluorescence. In addition of achieving low detection limits, very low quantities of solvent were used so that the procedure can be described as environmentally friendly. The possibility of determining thiamine esters after a suitable enzymatic treatment is also noteworthy.
References
Coultate TP (1989) Food. The chemistry of its components, 2nd edn. Royal Society of Chemistry, London
Association of Official Analytical Chemists (1984) In: Williams S (ed) Official methods of analysis. Association of Official Analytical Chemists, Arlington, VA, p 836
Van Niekerk PJ (1988) Determination of vitamins. In: Macrae R (ed) HPLC in food analysis. Academic, London, p 156
Goldschmidt RJ, Wolf WR (2010) Anal Bioanal Chem 397:471–481
Lebiedzińska A, Marszall ML, Kuta J, Szefer P (2007) J Chromatogr A 1173:71–80
Tang XY, Cronin DA, Brunton NP (2006) J Food Compos Anal 19:831–837
Marszall ML, Lebiedzińska A, Czarnowski W, Szefer P (2005) J Chromatogr A 1094:91–98
Chienthavorn O, Smith RM, Saha S, Wilson ID, Wright B, Taylor SD, Lenz EM (2004) J Pharm Biomed Anal 36:477–482
Sánchez-Machado DI, López-Cervantes J, López-Hernández J, Paseiro-Losada P (2004) J Chromatogr Sci 42:117–120
Viñas P, López-Erroz C, Balsalobre N, Hernández-Córdoba M (2003) J Agric Food Chem 51:3222–3227
Viñas P, López-Erroz C, Balsalobre N, Hernández-Córdoba M (2003) J Chromatogr A 1007:77–84
Li K (2002) Biomed Chromatogr 16:504–507
Woollard DC, Indyk HE (2002) J Assoc Off Anal Chem Int 85:945–951
Esteve MJ, Farre R, Frigola A, García-Cantabella JM (2001) J Agric Food Chem 49:1450–1454
Viñas P, López-Erroz C, Balsalobre N, Hernández-Córdoba M (2001) J Chromatogr B 757:301–308
Yamanaka K, Matsuoka M, Banno K (1996) J Chromatogr A 726:237–240
Blanco D, Llaneza MB, Gutierrez MDA (1996) J Liq Chromatogr Relat Technol 19:2155–2164
Gehring TA, Cooper WM, Holder CL, Thompson HC Jr (1995) J Assoc Off Anal Chem Int 78:307–309
Barna E, Dworschak E (1994) J Chromatogr A 668:359–363
Yamanaka K, Horimoto S, Matsuoka M, Banno K (1994) Chromatographia 39:91–96
Ollilainen V, Vahteristo L, Uusi-Rauva A, Varo P, Koivistoinen P, Huttunen J (1993) J Food Compos Anal 6:152–165
Alyabis AM, Simpson KL (1993) J Food Compos Anal 6:166–171
Ohta H, Maeda M, Nogata Y, Yoza KI, Takeda Y, Osajima Y (1993) J Liq Chromatogr 16:2617–2629
Chase GW Jr, Landen WO Jr, Soliman AGM, Eitenmiller RR (1993) J Assoc Off Anal Chem Int 76:1276–1280
Bognar A (1992) Fresenius J Anal Chem 343:155–156
Chase GW Jr, Landen WO Jr, Eitenmiller RR, Soliman AGM (1992) J Assoc Off Anal Chem Int 75:561–565
Otles S (1991) Z Lebensm Unters Forsch 193:347–350
Nicolas EC, Pfender K (1990) J Assoc Off Anal Chem 73:792–798
Vidal-Valverde C, Reche A (1990) Z Lebensm Unters Forsch 19:313–318
Dawson KR, Unklesbay NF, Hedrick HB (1988) J Agric Food Chem 36:1176–1179
Lavigne C, Zee JA, Simard RE, Gosselin C (1987) J Chromatogr A 410:201–205
Vanderslice JT, Huang MHA (1986) J Micronutr Anal 2:189–199
Ayi BK, Yuhas DA, Moffett KS, Joyce DM, DeAngelis NJ (1985) J Assoc Off Anal Chem 68:1087–1092
Wills RBH, Wimalasiri P, Greenfield H (1985) J Micronutr Anal 1:23–29
Wimalasiri P, Wills RBH (1985) J Chromatogr A 318:412–416
Wehling RL, Wetzel DL (1984) J Agric Food Chem 32:1326–1331
Mauro DJ, Wetzel DL (1984) J Chromatogr A 299:281–287
Augustin J (1984) J Assoc Off Anal Chem 67:1012–1015
Fellman JK, Artz WE, Tassinari PD, Cole CL, Augustin J (1982) J Food Sci 47:2048–2050
Kamman JF, Labuza TP, Warthesen JJ (1980) J Food Sci 45:1497–1499
Kimura M, Fujita T, Nishida S, Itokawa Y (1980) J Chromatogr A 188:417–419
Ang CYW, Moseley FA (1980) J Agric Food Chem 28:483–486
Hemming BC, Gubler CJ (1980) J Liq Chromatogr Relat Technol 3:1697–1712
Analytical Methods Committee (2000) Analyst 125:353–360
Lynch PLM, Young IS (2000) J Chromatogr A 881:267–284
Hollman PCH, Slangen JH, Wagstaffe PJ, Faure U, Southgate DAT, Finglas PM (1993) Analyst 118:481–488
Abdel-Kader ZM (1992) Food Chem 43:393–397
Anastas PT, Warner JC (2000) Theory and practice. Oxford University Press, Green Chemistry
Ramos L, Ramos JJ, Brinkman UATh (2005) Anal Bioanal Chem 381:119–140
Cuadros-Rodriguez L, Almansa-Lopez EM, Garcia-Campana AM, Gonzalez-Casado A, Egea-González FJ, Garrido-Frenich A, Martinez-Vidal JL (2005) Talanta 66:1063–1072
Mitra S (ed) (2003) Sample preparation techniques in analytical chemistry. Wiley-Interscience, New Jersey
Nerín C, Salafranca J, Aznar M, Batlle R (2009) Anal Bioanal Chem 393:809–833
Rezaee M, Assadi Y, Hosseini MRM, Aghaee E, Ahmadi F, Berijani S (2006) J Chromatogr A 1116:1–9
Bosch Ojeda C, Sánchez Rojas F (2009) Chromatographia 69:1149–1159
Zeeb M, Ganjali MR, Norouzi P (2010) Microchim Acta 168:317–324
Acknowledgments
The authors acknowledge Comunidad Autónoma de la Región de Murcia (CARM, Fundación Séneca, Project 15217/PI/10) and the Spanish MICINN (Project CTQ2009-08267/BQU) for financial support. M. Bravo-Bravo acknowledges a fellowship from Fundación Séneca, CARM. Samples of baby foods were kindly supplied by Hero España S.A.
Author information
Authors and Affiliations
Corresponding author
Additional information
Published in the special issue Euroanalysis XVI (The European Conference on Analytical Chemistry) with guest editor Slavica Ražić.
Rights and permissions
About this article
Cite this article
Viñas, P., López-García, I., Bravo-Bravo, M. et al. Dispersive liquid–liquid microextraction coupled to liquid chromatography for thiamine determination in foods. Anal Bioanal Chem 403, 1059–1066 (2012). https://doi.org/10.1007/s00216-012-5804-2
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00216-012-5804-2