
Abstract
A novel method based on ultrasonic-assisted aqueous two-phase extraction (UAATPE) coupled with solidifying organic drop-dispersible liquid–liquid microextraction (SOD-DLLME) was developed for simultaneous determination of nine mycotoxins (aflatoxins of B1, B2, G1, G2, and M1, ochratoxin A, zearalenone, deoxynivalenol and patulin) in medicinal and edible foods by high-performance liquid chromatography (HPLC) with diode array detector (DAD) and fluorescence detector (FLD) in series. Using an aqueous two-phase system (ATPS) of acetonitrile and (NH4)2SO4 as the extractant, the effects of the ATPS composition, extraction temperature and time were investigated respectively to extract the mycotoxins from the samples. Also, SOD-DLLME conditions including type and volume of extractant and dispersant were optimized by the single factor experiments. The optimum conditions were as follows: the ATPS composition of 34.0% acetonitrile concentration (w/w) and 22.0% (NH4)2SO4 concentration (w/w), pH 6.0, extraction temperature 40 °C, ultrasonic time 10 min for UAATPE; 1-dodecanol 600 μL as extractant, acetonitrile 1.0 mL as dispersant, and vortex-assisted time 1.0 min for SOD-DLLME. By means of HPLC–DAD-FLD detection, nine mycotoxins had good linearity in the range of 0.5 − 200.0 ng/mL (R2 ≥ 0.9991). LODs and LOQs were in the range of 0.01563 − 0.5161 ng/mL and 0.05210 − 1.720 ng/mL, respectively. The average recoveries and intra-day and inter-day precisions were 82.77 − 103.2%, 1.1 − 3.4% and 1.5 − 4.3%, respectively. The proposed method was successfully applied to simultaneous determination of multiple mycotoxins in black bean, black sesame, lotus seed, apricot kernel and litchi, demonstrating the presence of five different mycotoxins in these samples.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Medicinal and edible foods such as fruits, seeds, pulp and/or peel/pericarp from dietary plants and traditional medicinal herbs play an important role in human nutrition and health because of their nutritional properties and bioactive principles (Huang et al. 2010; Shi and Jiang 2020; Que et al. 2017a. 2017b). Black beans, black sesame, lotus seeds and apricot kernel as the archetypal medicinal and edible plants are widely applied in China (Chinese Pharmacopoeia Commission 2020a). Litchi produced in South China possesses a sweet odor of rose, delicious taste, and good nutritional value, its dried fruit exhibited multiple bioactivities such as hypoglycemic, anticancer, antibacterial, anti-hyperlipidemic, anti-platelet, and antiviral (Ibrahim and Mohamed 2015). Taking into account the important medicinal and edible values, these foods are favored by consumers in south China, and prepare delicious foods with various local flavors and health functions. However, owing to unsanitary conditions during planting, harvesting, processing, storage and transportation, they will be polluted to different degrees. Especially seeds and fruits are susceptible to mycotoxins contamination caused by various fungi with moldy growth and spoilage in a tropical climate (Paterson and Lima 2010; Yang et al. 2020). After ingestion, mycotoxins will cause several diseases in humans and animals (Tripathy et al. 2014; Thanushree et al. 2019). Therefore, mycotoxins contamination is very harmful to human and animal, and persistent efforts have been devoted to controlling and monitoring mycotoxins. So far, more than 400 mycotoxins have been identified from a variety of foods and agricultural commodities worldwide (Yang et al. 2020). Among them, aflatoxins (AFs), ochratoxin A (OTA), deoxynivalenol (DON), patulin (PAT), zearalenone (ZEN) are quite common contaminants occurring jointly in a wide range of foods (the chemical structures are presented in Fig. S1) (Yang et al. 2020). Due to high toxicity of these compounds, many countries and organizations have formulated the maximum limits (MLs) in foodstuffs, feeds, and other matrices intended for human or animal consumption (European Commission 2006; National Standard of the People’s Republic of China National 2017; Chinese Pharmacopoeia Commission 2020b). In recent years, monitoring the mycotoxins in medicinal herbs has been paying more and more attention (Wang et al. 2020; Liu et al. 2016, 2019, 2018; Sun et al. 2018; Cho et al. 2019). Especially in South China, the foregoing medicinal and edible foods as major ingredients are very prone to occurrence of mycotoxins contamination in the moist climate. Moreover, multiple mycotoxins may be found in the same product because a single species of fungi can produce several toxic metabolites, even several species of fungi can be simultaneously present and produce different toxins (Tripathy et al. 2014; Thanushree et al. 2019; Yang et al. 2020). Up to now, there have been a few related reports about monitoring the mycotoxins in these medicinal and edible foods.
In order to control the mycotoxins in foodstuffs, a reliable analytical method is essential to obtain accurate data for establishment of the regulatory limits and evaluation of the infection risk. In the last few years, strenuous efforts have been devoted to develop analytical methodologies for the effective determination of mycotoxins, particularly multi-mycotoxins methods (Yang et al. 2020; Cho et al. 2019; Liu et al., 2019, 2018; Rahmani et al. 2009). Among them, high-performance liquid chromatography-tandem mass spectrometry (HPLC–MS/MS) and HPLC with FLD or DAD are preferred methods of mycotoxins analysis (Yang et al. 2020; Chinese Pharmacopoeia Commission 2020b; Rahmani et al. 2009; General Administration of Quality Supervision 2016). However, sample pretreatment is fundamental and indispensable steps for accurate and sensitive determination of trace mycotoxins in complex sample matrices (Yang et al. 2020; Chinese Pharmacopoeia Commission 2020b; Sun et al. 2018; Cho et al. 2019; Liu et al. 2019; Liu et al. 2018; Rahmani et al. 2009; General Administration of Quality Supervision 2016). Usually, extraction of mycotoxins from solid samples is the first step in sample preparation, followed by clean-up procedures to improve the sensitivity and specificity of detection methods (Chinese Pharmacopoeia Commission 2020b; Liu et al. 2019, 2018; Rahmani et al. 2009; General Administration of Quality Supervision 2016). Most of protocols use acetonitrile/methanol or acetonitrile–water/methanol–water mixture as a solvent to extract mycotoxins from samples. In addition to filtration and centrifugation, further clean-up steps are needed to improve sensitivity and selectivity (Yang et al. 2020; Chinese Pharmacopoeia Commission 2020b; Liu et al. 2019; Liu et al. 2018; Rahmani et al. 2009; General Administration of Quality Supervision 2016). The different sample pretreatment techniques have been developed to match a solvent extraction step for purification and enrichment of trace mycotoxins in various sample matrices such as solid-phase extraction (SPE) (Dong et al. 2019), dispersive solid-phase extraction (DSPE) (Wang et al. 2020), liquid–liquid extraction (LLE) (Liu et al. 2016; Romera et al. 2018), solid-phase microextraction (SPME) (Andrade and Lancas 2017), QuEChERS (quick, easy, cheap, effective, rugged and safe) (Liu et al. 2018; Colli et al. 2020), immunoaffinity column (IAC) (Liu et al. 2019, 2018; Rahmani et al. 2009), magnetic solid phase extraction (MSPE) (Zhao et al. 2020), and dispersive liquid–liquid micro-extraction (DLLME) (Yang et al. 2020), have been developed to match a solvent extraction step for purification and enrichment of trace mycotoxins in diverse sample matrices. With the function of biphasic extraction, aqueous two-phase extraction (ATPE) can achieve separation of the different components in two phases based on the difference of surface properties, charge action and various forces such as van der Waals force, hydrophobic bond, hydrogen bond and ionic bond (Assis et al. 2020; Iqbal et al. 2016). Compared to single solvent extraction, an ATPS can selectively extract target analytes into the top or bottom phase, reducing the interference of sample matrix. An ATPS composed of two phase-components (e.g. polymer–polymer, polymer-salt, and alcohol-salt, etc.) has an option to adjust the extraction performance by controlling the composition of the phase-forming components, expanding a wide range of extractants (Mai et al. 2020; Zhou et al. 2018). Moreover, water-rich phases would boost the penetration of the extractants in the food matrix. An acidic solution can break the strong bonds between the analytes and other food components such as protein and sugars, thus enhancing the extraction efficiency (Yang et al. 2020; Rahmani et al. 2009). For example, the green ATPSs formed by polyethylene glycol and salt as alternatives to organic solvents and/or toxic reagents have exhibited high extraction capacity to mycotoxins, keeping a good connection with the subsequent clean-up procedures (Soares et al. 2014, 2017). DLLME is superior to others in respect of simple configuration, rapid operation, lower costs and high enrichment efficiency. It is frequently combined with clean-up techniques such as microwave-assisted extraction (MAE), QuEChERS and DSPE, etc. (Sajid and Alhooshani 2018; Wang et al. 2018; Trevisan et al. 2017; Farajzadeh et al. 2019). For mycotoxins or other analytes in solid samples, most cases are usually subjected to single-phase solvent extraction with acetonitrile or acetonitrile–water mixture prior to DLLME procedures, inevitably resulting in more complex process (Rausch et al. 2020; Yazdanfaret al. 2019). Therefore, novel or modified methods are needed to simplify extra steps or reduce unnecessary errors. ATPE combining with DLLME is a resolution to avoid this situation because of integrating extraction with purification and enrichment in a one-step procedure.
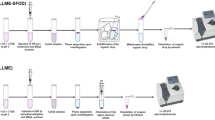
Although high-sensitive HPLC–MS/MS has been used for determination of multiple mycotoxins in different species of functional and medicinal herbs, measurement recoveries and uncertainty had a wide fluctuation due to matrix effect (Sun et al. 2018; Cho et al. 2019; Liu et al. 2019, 2018; Rahmani et al. 2009). Compared with HPLC–MS/MS, which is not easily accessible for most laboratories, HPLC is simple with low cost, and easy to operate. In this work, a method of combining APTE with DLLME was developed for simultaneous extraction and enrichment of the mycotoxins in black beans, black sesame, lotus seeds, apricot kernel and litchi by HPLC coupled to series connection of DAD and FLD, so that the mycotoxins in a wide range of the sample matrices can be quantitatively determined to evaluate contamination levels. To this end, an ATPS of acetonitrile/(NH4)2SO4 was used for UAATPE, and followed by screening of a modified DLLME system to connect with UAATPE procedure for simultaneous extraction and enrichment of the mycotoxins in the samples. Accordingly, UAATPE conditions including the composition of the ATPS, pH, extraction temperature and time were investigated to enhance extraction of the mycotoxins. In the DLLME process, key factors such as extractant and extractant volume, dispersant and dispersant volume, vortex-assisted time were investigated to maximize extraction recoveries of the mycotoxins. Based on the UV and fluorescence characteristics of the mycotoxins before and after derivatization, HPLC conditions were optimized through in-series DAD and FLD detection. Finally, UAAPTE-DLLME coupled with HPLC–DAD-FLD would be applied to simultaneous determination of the mycotoxins in medicinal and edible foods (See Fig. 1).

The process of UAATPE-DLLME coupled to HPLC–DAD-FLD for analysis of the mycotoxins in medicinal and edible foods
Materials and Methods
Chemicals and Materials
Methanol, acetonitrile of HPLC grade were obtained from Merck Ltd. (Germany). Ethanol, n-hexane, ammonium sulfate and n-hexane were purchased from Zhiyuan Chemical Reagent Co., Ltd. (Tianjin, China). Chloroform, carbon tetrachloride, dimethyl carbonate, dimethyl carbonate, methyl salicylate, ethyl salicylate, 1-dodecanol and concentrated hydrochloric acid were obtained from Guangzhou Chemical Reagent Factory (Guangzhou, China). Trifluoroacetic acid (TFA) was purchased from Maclean Biochemical Technology Co., Ltd. (Shanghai, China). The standards of aflatoxins (AFs including AFG1, AFG2, AFB1, AFB2, AFM1), ochratoxin A (OTA), zearalenone (ZEN), deoxynivalenol (DON) and patulin (PAT) were obtained from Sigma-Aldrich (St. Louis, MO, USA).
At least 500 g of dried samples of black beans, black sesame seeds, lotus seeds and apricot kernel were respectively purchased from Qingping Market in Guangzhou, between March 2017 and April 2018. A year-old dried litchi was collected from farmers in Dongguan, and the pulp was used as the sample to be tested after the pericarp and the core were removed. All samples were pulverized, sealed in plastic packages, and then stored in desiccators at room temperature. Analysis of samples was completed in 30 days.
Instrumentations
HPLC analysis of the mycotoxins was performed on a Shimadzu LC-20AD chromatograph equipped with RF-20A FLD detector and SPD-20A DAD detector (Shimazu Co., Ltd., Japan); LabSolution Version 5.92 software was used for calculation and quantification (Shimazu Co., Ltd., Japan). UAATPE was carried out on a KQ-2200B ultrasonic device with frequency and temperature controller (Kunshan Ultrosonic Instrument Co., Ltd., China); DLLME extraction was carried out on a HD-2500 multi-tube vortex mixer (Hangzhou Youning Instrument Co., Ltd., China); After the vortex, centrifugation of the resulting mixture was performed in a TD6-Benchtop centrifuge (Hunan Pingfan Science and Technology Co., Ltd., China), respectively; The mycotoxins were derived in the DK-98-II thermostatic water bath (Tianjin Taisite Instrument Co., Ltd., China); The top phase collected from UAATPE was blown to dryness under a gentle stream of nitrogen gas by ZGDCY-12 Termovap sample concentrator (Shanghai Zigui Instrument Co., Ltd., China).
Preparation of Standard Solutions
0.1 mg/mL of the stock standard solutions of AFs (AFG1, AFG2, AFB1, AFB2, AFM1), OTA, ZEN, DON and PAT was prepared in acetonitrile. The stock solutions were stored at 4 °C before use. According to the requirements of the experiment, 0.1 − 10 µg/mL of working standard solutions were freshly prepared by diluting and mixing each stock solution with acetonitrile step by step.
UAATPE and SOD-DLLME procedure
1.0 g of sample powder or 0.1 mL of mixed standard solution of mycotoxins was taken into a glass tube, then 1.256 g (NH4)2SO4 and 2.51 mL water was added, respectively. After dissolving, 2.49 mL acetonitrile was added to form an ATPS, and the mixture was placed in the ultrasonic bath for extraction of mycotoxins for 10 min at 40 °C and 40 kHz with the output power of 200 W. Then, allowed it to stand until two phases be separated. 1.0 mL of the top phase, 600 µL of 1-dodecanol and 4 mL of water were added into a centrifuge tube in turn. The mixture was vigorously vortexed for 1 min to allow the complete extraction. After being centrifuged at 2000 rpm for 5 min, the mixture was placed in a refrigerator for solidification of the top phase at 4 °C for about 10 min. Afterward, the bottom phase was removed by a syringe with needle, the solidified top phase was kept at room temperature until thawed for the following derivatization and HPLC analysis.
Derivatization of Mycotoxins
100 µL of TFA was added to the above extract liquefied. The mixture was subjected to derivatization of mycotoxins in a water bath at 40 °C for 20 min, and then allowed to separate into layers. The top layer was collected and passed through 0.22 μm membrane for HPLC analysis.
HPLC Analysis with in-series DAD and FLD
The quantification of the mycotoxins in the sample solution was conducted by HPLC coupled to in-series FLD and DAD. Using the mixture of 0.5% acetic acid–water (v/v, A) and 50% methanol–acetonitrile (v/v, B) as the mobile phase, the mycotoxins in the above sample solution was chromatographically separated on a Phenomenex Gemini C18 column (250 mm × 4.6 mm, 5 µm) according to the following gradient elution: 0–7 min, 25% B; 7–15 min, 25–70% B; 15–25 min, 70% B; 25–35 min, 70–100% B. Injection volume, flow rate and column temperature were 10 µL, 1.0 mL/min, and 40 °C, respectively. Detection was performed with DAD and FLD detectors, connected in series, and the FLD detection procedure was as followed: 0–20 min, excitation wavelength of 360 nm and emission wavelength of 430 nm; 20–25 min, excitation wavelength of 275 nm and emission wavelength of 440 nm; 25–35 min, excitation wavelength of 333 nm and emission wavelength of 460 nm.
Results and discussion
Optimization of UAATPE conditions
Screening of an ATPS
The ATPS made by a short-chain solvent and a salt, offers the advantages of low viscosity, easy demixing, convenient to subsequent processing and solvent recycling, so ethanol with nontoxicity and acetonitrile with chromatographic use were used as phase-forming components to construct an ATPS with (NH4)2SO4. According to the phase formation diagrams (Zhang et al. 2015), the ATPSs of ethanol/(NH4)2SO4 and acetonitrile/(NH4)2SO4 were prepared, respectively, for screening of an appropriate biphasic extraction system. Thus, the effects of two ATPSs on extraction of the mycotoxins analyzed were investigated under the same conditions of the ATPS composition and the phase ratio.
Both two ATPSs were spontaneously formed while mass composition at 17.0% (w/w) of (NH4)2SO4 and 24.0% (w/w) of acetonitrile or ethanol, but nine mycotoxins exhibited diverse extraction performances in two ATPSs. The results demonstrated that nine mycotoxins preferred to be extracted into the rich-acetonitrile/ethanol top phase, whereas some matrix components with great polarity were left in the bottom phase. As a result, the interferences from the sample matrix could be eliminated or partially removed. From Fig. 2a, acetonitrile/(NH4)2SO4 system obviously had higher recoveries than that of the ATPS made by ethanol and (NH4)2SO4. As shown in Fig. 2b, the recoveries of nine mycotoxins were in the range of 71.59−87.61% with acetonitrile/(NH4)2SO4 system as the extractant while at 1:1 of the phase ratio (e.g. the volume ratio of the top phase to bottom phase). However, the recoveries of all mycotoxins for ethanol/(NH4)2SO4 system were lower than 55.16%. Therefore, acetonitrile/(NH4)2SO4 system as biphasic extractant was suitable for extraction of nine mycotoxins.

The effects of two ATPSs at the same composition [17.0% (NH4)2SO4 concentration (w/w) and 24.0% acetonitrile/ethanol concentration (w/w)] (a) and the phase ratio at 1:1 [20.0% (NH4)2SO4 concentration (w/w) and 37.0% acetonitrile, and 18.0% (NH4)2SO4 concentration (w/w), 25.0% ethanol concentration] (b) on the recoveries of the mycotoxins
Effect of composition of the ATPS
The biphasic extraction property of an ATPS to the mycotoxins depends on the ATPS composition of phase-forming components, which mean to dictate the polarity and the volume of the top or bottom phase (Zhang et al. 2015). Accordingly, the effect of the composition of the ATPS on extraction of the mycotoxins was investigated by controlling the amounts of acetonitrile and (NH4)2SO4. According to the phase diagram, there is a wider range of formation of an ATPS while at 15.0% of (NH4)2SO4 concentration (w/w). Thus, the influence of acetonitrile concentration on extraction of the mycotoxins was firstly investigated, followed by investigation of (NH4)2SO4 concentration.
As shown in Fig. 3a, the recoveries of the mycotoxins increased with the increase of acetonitrile concentration, but decreased at higher than 34.0% of acetonitrile concentration (w/w). Subsequently, (NH4)2SO4 concentration was further investigated when kept at 34.0% of acetonitrile concentration (w/w). From Fig. 3b, the recoveries of the mycotoxins increased with the increase of (NH4)2SO4 concentration, and almost reached maximum values at 22.0% (NH4)2SO4 concentration (w/w). The above results demonstrated that the ATPS composition should be controlled at 34.0% of acetonitrile concentration (w/w) and 22.0% of (NH4)2SO4 concentration (w/w) to transfer the mycotoxins to the top phase rather than the bottom phase.

The effects of acetonitrile concentration (a), (NH4)2SO4 concentration (b), pH (c), extraction temperature (d), extraction time (e) and different concentration (f) on the recoveries of the mycotoxins
Effect of pH
Since the mycotoxins are susceptible to acidity, different pH will affect their species or forms, resulting in change of partition coefficient in an UAATPE process even decomposition. The effect of pH on extraction of mycotoxins was investigated in the range of pH 3.0 − 7.0. From Fig. 3c, the recoveries of the mycotoxins increased gradually with the increase of pH, and reached the highest values at pH 6.0 except for OTA and PAT. Under the condition of strong acid, some mycotoxins were slightly decomposed, the other’s mycotoxins may lead to the protonation. As a result, the partition coefficient of the mycotoxins became smaller in the top phase with relatively small polarity, and their extraction recoveries decreased. Comprehensively, all mycotoxins could achieve higher recoveries, thus pH 6.0 was selected for the following experiments.
Effect of temperature and time
Extraction temperature and time are key factors that influence extraction of target analytes. Appropriate temperature and time can provide enough energy to improve extraction recoveries of the mycotoxins. Also, the effects of extraction temperature and time on the recoveries of the mycotoxins were investigated in the range of 20 − 60 °C and 5 − 25 min, respectively. As shown in Fig. 3d, the recoveries of the mycotoxins increased with the increase of extraction temperature, and then decreased at more than 40 °C. From Fig. 3e, the impaction of extraction time on the recoveries was relatively smooth and steady, taking 10 min of extraction time for the mycotoxins was enough to reach higher recoveries. Therefore, extraction temperature and time were set at 40 °C and 10 min as the best option for extraction of the mycotoxins.
Effect of mycotoxins concentration
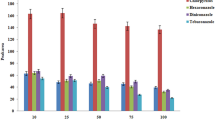
In addition to the extraction performance of the ATPS, the limited ATPS volume influences the extraction capacity of the mycotoxins. In order to link to the subsequent DLLME process, the effects of different concentrations of the mycotoxins on extraction of the mycotoxins were investigated at three levels according to the above conditions optimized. The three levels of the mixed standard solutions consisted of low, medium and high concentrations of the mycotoxins from 25.0 to 500 ng/mL according to detection sensitivity of each mycotoxin. From Fig. 3f, the recoveries of the mycotoxins at three levels ranged between 73.71 and 97.54%. In the results as a whole, the low concentration achieved slightly higher recoveries than that of medium and high concentration, but there was no significant difference while using 5 mL of the ATPS. The results indicated that the extraction capacity of the ATPS could recover the mycotoxins in the above concentration range.
Finally, the optimized extraction conditions for the mycotoxins were summarized as follow: the ATPS composition of 34.0% acetonitrile concentration (w/w) and 22.0% of (NH4)2SO4 concentration (w/w), pH 6.0, 40 °C of extraction temperature, 10 min of ultrasonic time. With 5 mL of the ATPS as the extractant, nine mycotoxins ranged in the concentration from 25.0 ng/mL to 500 ng/mL could be extracted to the top phase with higher recoveries.
Optimization of DLLME conditions
Effect of extractants
In the DLLME process, selection of an appropriate extractant is key to preconcentrate the target analytes. Based on the physiochemical properties of the mycotoxins, chloroform, carbon tetrachloride, dimethyl carbonate, methyl salicylate, ethyl salicylate and 1-dodecanol were used as the extractant to extract these mycotoxins (Sajid and Alhooshani 2018). The two formers are often used as DLLME extractant, the latter four solvents with nontoxicity are attempted to serve the mycotoxins. As shown in Fig. 4a, chloroform, methyl salicylate, ethyl salicylate, and 1-dodecanol exhibited the higher recoveries to the mycotoxins, and illustrated different phase-separation behaviors (See Fig. S2). Relatively, methyl salicylate, ethyl salicylate, and 1-dodecanol were less toxic than chloroform. However, the viscosity of the salicylates located to the bottom phase of the ATPS was too high for separation and operation, and the chromatographic peaks would make serious interference to detection of DON. Considering the lower density and higher melting point, 1-dodecanol located at the top phase could be solidified at low temperature or in ice water, which was also more to be conveniently collected by solidification of floating organic drop and two-phase separation before and after derivatization. Thus, 1-dodecanol could be selected as the solidifying extractant for modification of the common DLLME process [named solidifying organic drop solidifying organic drop-dispersible liquid–liquid microextraction (SOD-DLLME)] in next experiments.

The effects of extractant (a), extractant volume (b), dispersant (c) and dispersant volume (d) on the recoveries of the mycotoxins
Effect of extractant volume
In order to ensure full enrichment of target analytes, the effect of 1-dodecanol volume was further investigated in range of 400 − 900 μL. From the results in Fig. 4b, the recoveries of the mycotoxins were improved with increase of 1-dodecanol, and then reached relatively stable values. The results illustrated that 600 μL of 1-dodecanol could be sufficient to extract nine mycotoxins. Thus, 600 μL of 1-dodecanol was chosen for the following experiments.
Effect of dispersants
In the SOD-DLLME process, an appropriate dispersant can homogenize two immiscible solutions for increase of the contact area between the extractant and the aqueous sample, resulting in target analytes that are extracted from the sample to finely homogeneous droplets. To disperse the extractant to the aqueous sample, methanol, ethanol, acetonitrile, and acetone were screened for dispersion of 1-dodecanol. The results in Fig. 4c demonstrated that acetonitrile could have achieved higher recoveries for the mycotoxins. Moreover, acetonitrile as both the suitable dispersant and the phase-forming component could directly connect to the above UAATPE process without being blown to dryness (Chen et al. 2020). Therefore, acetonitrile was selected as the dispersant in the following experiment.
Effect of dispersant volume
To effectively disperse the extractant to the aqueous sample, the moderate volume of the dispersant must be adopted for complete dispersion so as to ensure high extraction capacity, because the dispersant volume is too small or large to attain higher recoveries for target analytes due to incomplete dispersion or dissolution loss of the extractant. Due to the rich-acetonitrile, the top phase from UAATPE as the dispersant can be directly linked to SOD-DLLME procedure to avoid extra sample preparation steps such as evaporation or concentration and re-dissolution, etc. Accordingly, the effect of the top phase volume on the recoveries of the mycotoxins was investigated while 1-dodecanol was kept at 600 μL. As shown in Fig. 4d, the recoveries of the mycotoxins increased with increase of the dispersant volume, and illustrated the maximum value at 1.0 mL of the rich-acetonitrile top phase. Consequently, 1.0 mL of the top phase solution was selected as the dispersant for SOD-DLLME enrichment of the mycotoxins.
In addition, the effects of vortex-assisted time and NaCl added on the recoveries of the mycotoxins were also investigated. The results showed that the recoveries of the mycotoxins had no significant change between 1 and 5 min without addition of NaCl. This might be because the top phase had been saturated by (NH4)2SO4. In summary, SOD-DLLME optimum conditions were 600 μL of 1-dodecanol as the extractant, 1.0 mL of the rich-acetonitrile top phase as the dispersant solvents, 1 min of vortex-assisted time, respectively.
Method performance
HPLC separation and DAD-FLD detection
The mixed standard solution and the above extract were subjected to TFA derivatization, and the top layer was used for HPLC analysis with DAD and FLD in series. As shown in Fig. 4, nine common mycotoxins were well separated within 35 min with the mixture of 50% methanol–acetonitrile (v/v, A) and 0.5% acetic acid–water (v/v, B) as mobile phase in gradient elution, the resolution (Rs) between any two peaks was more than 1.5, and the theoretical plates were more than 3000. Based on UV absorption spectra, PAT, AFM1, DON, ZEN, and OTA could be detected by DAD at 277, 360, 218, 276, and 334 nm, respectively [See Fig. 5a]. Under the excitation of the optimized wavelengths, fluorescence intensity of AFM1, ZEN, OTA, AFG1, AFB1, AFG2, and AFB2 could be detected by FLD through TFA derivatization [See Fig. 5b], and AFM1, ZEN and OTA exhibited higher sensitivity than DAD detection. Therefore, using HPLC coupled to in-series connection of DAD and FLD improved high-throughput detection of the mycotoxins, and could meet the requirements of simultaneous determination of nine mycotoxins in real samples.

HPLC chromatograms of mixed standard solution of the mycotoxins by DAD detection (a) and FLD detection (b): 50 ng/mLof AFB1, AFB2, AFG1, AFG2 and 125 ng/mL of PAT, AFM1, DON, ZEN, OTA
Linearity, LOD and LOQ
To evaluate quantification performance of the proposed method, a series of different concentrations of the mixed standard solutions were prepared by diluting the stock standard solutions, respectively, and treated by the UAATPE-DLLME procedures described above. After being subjected to FTA derivatization, the mycotoxins were determined by HPLC coupled with DAD and FLD in series. Data obtained were fitted by linear regression according to the correlation between the peak area (A) and the concentration (C, ng/mL), and the results are shown in Table 1. Meanwhile, the limit of detection (LOD) and the limit of quantification (LOQ) were calculated according to signal-to-noise ratio of 3:1 (S/N = 3) and 10:1 (S/N = 10), respectively.
From Table 1, the results demonstrated that there was good linearity in the ranges of 0.5 − 100.0 ng/mL with correlation coefficients (R2) higher than 0.9991. LODs and LOQs were in the range of 0.01563 − 0.5161 ng/mL and 0.05210 − 1.720 ng/mL, respectively. DLLME enrichment factors (EFs) were ranged from 12.2 to 21.1, exhibiting stronger enrichment ability to nine mycotoxins. The EF value of each mycotoxin was calculated by the following equation: EF = Cb/C0 (C0 and Cb were its initial concentrations and preconcentration concentration, respectively). By means of FLD detection, five aflatoxins, ZEN, and OTA could reach higher sensitivity instead of DAD.
Accuracy, precision, and stability
UAATPE-DLLME as sample pretreatment was coupled to HPLC–DAD-FLD for investigation of accuracy, precision, and stability. Accordingly, the recovery experiments were accomplished by spiking samples at 20.0 ng/g of nine mycotoxins standards to assess the accuracy of the proposed method. According to experimental results of six repetitions, precision and stability were evaluated by the intra-day and inter-day precisions with relative standard deviations (RSD). As shown in Table 2, the recoveries of nine mycotoxins ranged from 82.77 to 103.2%, and the RSDs of intra-day and inter-day precisions for these mycotoxins were 1.1 to 5.6% and 1.5 − 4.9%, respectively. The results revealed that the method achieved high accuracy, credible repeatability, and satisfactory stability, and suggested that the method could withstand the influence of the sample matrices.
Real sample analysis
According to the process in Fig. 1, the developed method was applied to the analysis of black beans, black sesame, lotus seed, apricot kernel, and litchi pulp to check the presence of the mycotoxins in medicinal and edible foods. Before HPLC analysis, the samples crushed were subjected to UAAPTE/SOD-DLLME procedures and FTA derivatization successively (See Fig. S3). Furthermore, the recovery experiments of five spiked samples were accomplished so as to further validate the accuracy. The results obtained from the analysis of five samples were summarized in Table 3.
As shown in Table 3 five different mycotoxins in the range of 2.364 − 5.714 μg/kg were observed from the above samples. The recoveries and RSDs of nine mycotoxins were respectively 82.31 − 104.3% and 1.8% − 4.2%, suggesting that the method was more accurate and reliable. Among them, AFB1 was detected respectively from lotus seed and apricot kernel, AFB1 and DON were found in black bean, and litchi pulp was contaminated by AFB1 and AFB2. Even more, AFG1, AFB2, DON, and ZEN were simultaneously observed in black sesame. The above results also demonstrated that these samples were easily exposed to the contamination of the mycotoxins due to the relative higher temperature and humidity environment in South China. However, monitoring of the mycotoxins is not regulated equally all over the world. Thus, it is imperative to strengthen control of mycotoxin contamination in order to ensure safety and quality of these foods.
Conclusions
In this work, ultrasonic-assisted UAATPE combining with SOD-DLLME was developed for extraction, purification, and enrichment of nine mycotoxins in medicinal and edible foods, followed by simultaneous determination using HPLC coupled to DAD and FLD in series. Compared to LLE method with mono-phase solvent, UAATPE/SOD-DLLME can not only extract target analytes from the samples but also remove impurities in the extract through twice successive biphasic extractions. Acetonitrile as the ATPS phase-forming component and the dispersive solvent could directly connect UAATPE with SOD-DLLME, and 1-dodecanol as extractant was easily solidified at 4 °C or in ice water. As a result, the sample preparation and preconcentration procedures were greatly simplified, the solidified extract was conveniently collected before and after FTA derivatization, and the sensitivity and selectivity of the mycotoxins were significantly improved. Using HPLC coupled to in-series connection of DAD and FLD improved high-throughput detection of the mycotoxins (Chinese Pharmacopoeia Commission 2020b; General Administration of Quality Supervision 2016; Shi et al. 2018), achieving simultaneous determination of nine mycotoxins in black bean, black sesame, lotus seed and apricot kernel and litchi. The proposed method was proved to be simple, feasible and reliable. AFB1, AFB2, AFG1, DON, and ZEN were respectively detected from the above samples, revealing that they were easily exposed to the contamination of the mycotoxins in South China. The results provided the evidence for legislation of monitoring the mycotoxins in medicinal and edible foods.
References
Andrade MA, Lancas FM (2017) Determination of Ochratoxin A in wine by packed in-tube solid phase microextraction followed by high performance liquid chromatography coupled to tandem mass spectrometry. J Chromatogr A 1493:41–48. https://doi.org/10.1016/j.chroma.2017.02.053
Assis RC, Mageste AB, Lemos LRD, Orlando RM, Rodrigues GD (2020) Application of aqueous two-phase system for selective extraction and clean-up of emerging contaminants from aqueous matrices. Talanta 223:121697. https://doi.org/10.1016/j.talanta.2020.121697
Chen Z, Li Q, Yang T, Zhang Y, He M, Zeng H, Mai X, Liu Y, Fan H (2020) Sequential extraction and enrichment of pesticide residues in Longan fruit by ultrasonic-assisted aqueous two-phase extraction linked to vortex-assisted dispersive liquid-liquid microextraction prior to high performance liquid chromatography analysis. J Chromatogr A 1619:460929. https://doi.org/10.1016/j.chroma.2020.460929
Chinese Pharmacopoeia Commission (2020a) Pharmacopoeia of the People’s Republic of China (Vol. 1), China Medicine Science and Technology Press, Beijing, pp. 210, 255, 285, 359, 360.
Chinese Pharmacopoeia Commission, Pharmacopoeia of the People's Republic of China (Vol. 4): 2351 determination of mycotoxins. China Medical Science and Technology Press, Beijing, 2020b, pp. 280–285.
Cho H, Suh J, Feng S, Eom T, Kim J, Hyun S, Kim J, Wang Y, Han S (2019) Comprehensive analysis of multi-class mycotoxins in twenty different species of functional and medicinal herbs using liquid chromatography-tandem mass spectrometry. Food Control 96:517–526. https://doi.org/10.1016/j.foodcont.2018.10.007
Colli LD, Elliott C, Finnan J, Grant J, Arendt EK, McCormick SP, Danaher M (2020) Determination of 42 mycotoxins in oats using a mechanically assisted QuEChERS sample preparation and UHPLC-MS/MS detection. J Chromatogr B 1150:122187. https://doi.org/10.1016/j.jchromb.2020.122187
Dong H, Xian Y, Xiao K, Wu Y, Zhu L, He J (2019) Development and comparison of single-step solid phase extraction and QuEChERS clean-up for the analysis of 7 mycotoxins in fruits and vegetables during storage by UHPLC-MS/MS. Food Chem 274:18471–18479. https://doi.org/10.1016/j.foodchem.2018.09.035
European Commission (2006) Commission Regulation (EC) No 401/2006 of 23 February 2006 laying down the methods of sampling and analysis for the official control of the levels of mycotoxins in foodstuffs. https://eur-lex.europa.eu/legal-content/EN/TXT/?uri=CELEX%3A02006R0401-20140701&qid=1626492676557. Accessed 17 July 2021.
Farajzadeh MA, Sohrabi H, Mohebbi A (2019) Combination of modified Quechers extraction method and dispersive liquid-liquid microextraction as an efficient sample preparation approach for extraction and preconcentration of pesticides from fruit and vegetable samples. Food Anal Method 12(2):4–543. https://doi.org/10.1007/s12161-018-1384-x
General Administration of Quality Supervision (2016) Inspection and Quarantine of the People's Republic of China, Determination of mycotoxins in Chinese herbal medicine for import and export (SN/T 4604-2016). Standards Press of China, Beijing, pp 1-13
Huang W, Cai Y, Corke H, Sun M (2010) Survey of antioxidant capacity and nutritional quality of selected edible and medicinal fruit plants in Hong Kong. J Food Compos Anal 23:510–517. https://doi.org/10.1016/j.jfca.2009.12.006
Ibrahim SRM, Mohamed GA (2015) Litchi chinensis: medicinal uses, phytochemistry, and pharmacology. J Ethnopharmacol 174:492–513. https://doi.org/10.1016/j.jep.2015.08.054
Iqbal M, Tao Y, Xie S, Zhu Y, Chen DM, Wang X, Huang L, Peng D, Sattar A, Shabbir MAB, Hussain HI, Ahme S, Yuan Z (2016) Aqueous two-phase system (ATPS): an overview and advances in its applications. Biol Proced Online 18:18. https://doi.org/10.1186/s12575-016-0048-8
Liu C, Qin J, Dou X, Yang M, Sun X (2018) Extrinsic harmful residues in Chinese herbal medicines: types, detection, and safety evaluation. Chin Herb Med 10:117–136. https://doi.org/10.1016/j.chmed.2018.02.002
Liu H, Luo J, Kong W, Liu Q, Hu Y, Yang M (2016) UFLC-ESI-MS/MS analysis of multiple mycotoxins in medicinal and edible Areca Catechu. Chemosphere 150:176–183. https://doi.org/10.1016/j.chemosphere.2016.02.032
Liu Q, Xiao C, Liu H, Hu Y, Guo W, Kong W (2019) Sensitive assessment of multi-class mycotoxins residue in Atractylodis Rhizome. Ind Crop Prod 127:1–10. https://doi.org/10.1016/j.indcrop.2018.10.026
Mai X, Liu Y, Tang X, Wang L, Lin Y, Zeng H, Luo L, Fan H, Li P (2020) Sequential extraction and enrichment of flavonoids from Euonymus alatus by ultrasonic-assisted polyethylene glycol-based extraction coupled to temperature induced cloud point extraction. Ultrason Sonochem 66:105073. https://doi.org/10.1016/j.ultsonch.2020.105073
National Standard of the People's Republic of China National (2017) Food Safety Standard-Maximum Levels of Mycotoxins in Food (GB2761–2017). National Health Commission of the People's Republic of China & National Medical Products Administration, Beijing, pp1–8.
Paterson RRM, Lima N (2010) How will climate change affect mycotoxins in food? Food Res Int 43:1902–1914. https://doi.org/10.1016/j.foodres.2009.07.010
Que L, Yang G, Li Y, Shan F, Chen M, Huang L (2017) Overview of revision of the catalogue of the substances traditionally considered as both food and Chinese medicine. Chin Pharm J 52(7):521–524 (In Chinese). https://doi.org/10.11669/cpj.2017.07.001
Rahmani A, Jinap S, Soleimany F (2009) Qualitative and quantitative analysis of mycotoxins. Compre Rev Food Sci Food Saf 8:202–251. https://doi.org/10.1111/j.1541-4337.2009.00079.x
Rausch AK, Brockmeyer R, Schwerdtle T (2020) Development and validation of a QuEChERS-based liquid chromatography tandem mass spectrometry multi-method for the determination of 38 native and modified mycotoxins in cereals. J Agric Food Chem 68(16):4657–4669. https://doi.org/10.1021/acs.jafc.9b07491
Romera D, Mateo EM, Mateo-Castro R, Gómez JV, Gimeno-Adelantado JV, Jiménez M (2018) Determination of multiple mycotoxins in feedstuffs by combined use of UPLC-MS/MS and UPLC-QTOF-MS. Food Chem 267:140–148. https://doi.org/10.1016/j.foodchem.2017.11.040
Sajid M, Alhooshani K (2018) Dispersive liquid-liquid microextraction based binary extraction techniques prior to chromatographic analysis: a review. TrAc Trends Anal Chem 108:167–182. https://doi.org/10.1016/j.trac.2018.08.016
Shi H, Li S, Bai Y, Prates LL, Lei Y, Peiqiang Yu (2018) Mycotoxin contamination of food and feed in China: occurrence, detection techniques, toxicological effects and advances in mitigation technologies. Food Control 91:202–215. https://doi.org/10.1016/j.foodcont.2018.03.036
Shi Z, Jiang D (2020) Study advances in safety of medicine and food homology of traditional Chinese medicine. J Hunan Uni Chin Med 40(6):772–777 (In Chinese).https://doi.org/10.3969/j.issn.1674-070X.2020.06.026
Soares RRG, Azevedo AM, Fernandes P, Chu V, Conde JP, Aires-Barros MR (2017) A simple method for point-of-need extraction, concentration and rapid multi-mycotoxin immunodetection in feeds using aqueous two-phase systems. J Chromatogr A 1511:15–24. https://doi.org/10.1016/j.chroma.2017.07.004
Soares RRG, Novo P, Azevedo AM, Fernandes P, Chu V, Conde JP, Aires-Barros MR (2014) Aqueous two-phase systems for enhancing immunoassay sensitivity: simultaneous concentration of mycotoxins and neutralization of matrix interference. J Chromatogr A 1361:67–76. https://doi.org/10.1016/j.chroma.2014.08.007
Sun S, Yao K, Zhao S, Zheng Y, Liang D, Ke Y, Jiang H (2018) Determination of aflatoxin and zearalenone analogs in edible and medicinal herbs using a group-specific immunoaffinity column coupled to ultra-high-performance liquid chromatography with tandem mass spectrometry. J Chromatogr B 1092:228–236. https://doi.org/10.1016/j.jchromb.2018.06.012
Thanushree MP, Sailendri D, Yoha KS, Moses JA, Anandharamakrishnan C (2019) Mycotoxin contamination in food: an exposition on spices. Trends Food Sci Tech 93:69–80. https://doi.org/10.1016/j.tifs.2019.08.010
Trevisan MTS, Owen RW, Calatayud-Vernich P, Breuer A, Picó Y (2017) Pesticide analysis in coffee leaves using a quick, easy, cheap, effective, rugged and safe approach and liquid chromatography tandem mass spectrometry: optimization of the clean-up step. J Chromatogr A 1512:98–106. https://doi.org/10.1016/j.chroma.2017.07.033
Tripathy V, Basak BB, Varghese TS, Saha A (2014) Residues and contaminants in medicinal herbs—a review. Phytochem Lett 14:67–78. https://doi.org/10.1016/j.phytol.2015.09.003
Wang K, Xie X, Zhang Y, Huang Y, Zhou S, Zhang W, Lin Y, Fan H (2018) Combination of microwave-assisted extraction and ultrasonic-assisted dispersive liquid-liquid microextraction for separation and enrichment of pyrethroids residues in Litchi fruit prior to HPLC determination. Food Chem 240:1233–1242. https://doi.org/10.1016/j.foodchem.2017.08.061
Wang N, Duan C, Li S, Geng X, Ding K, Guan Y (2020) Aqueous extraction followed by dispersive solid phase extraction with in situ derivatization for the determination of aflatoxins in traditional Chinese medicine. J Chromatogr A 1618:460894. https://doi.org/10.1016/j.chroma.2020.460894
Yang Y, Li G, Wu D, Liu J, Li X, Luo P, Hu N, Wang H, Wu Y (2020) Recent advances on toxicity and determination methods of mycotoxins in foodstuffs. Trends Food Sci Tech 96:233–252. https://doi.org/10.1016/j.tifs.2019.12.021
Yazdanfar N, Shamsipur M, Ghambarian M (2019) Simultaneous extraction of 32 polychlorinated biphenyls by using magnetic carbon nanocomposite based dispersive microextraction, subsequent dispersive liquid-liquid microextraction with two miscible stripping solvents, and quantitation by GC-μECD. Microchim Acta 186:178. https://doi.org/10.1007/s00604-019-3235-x
Zhang W, Zhu D, Fan H, Liu X, Wan Q, Wu X, Liu P, Tang J (2015) Simultaneous extraction and purification of alkaloids from Sophora Flavescens Ait. by microwave-assisted aqueous two-phase extraction with ethanol/ammonia sulfate system. Sep Purif Technol 141:113–123. https://doi.org/10.1016/j.seppur.2014.11.014
Zhao Y, Yuan Y, Bai X, Liu Y, Wu G, Yang F, Liao X (2020) Multi-mycotoxins analysis in liquid milk by UHPLC-Q-Exactive HRMS after magnetic solid-phase extraction based on PEGylated multi-walled carbon nanotubes. Food Chem 305:125429. https://doi.org/10.1016/j.foodchem.2019.125429
Zhou S, Wu X, Huang Y, Xie X, Lin Y, Fan H, Luo ZW, Tang TZ (2018) Microwave-assisted aqueous two-phase extraction of alkaloids from Radix Sophorae Tonkinensis with an ethanol/Na2HPO4 system: Process optimization, composition identification and quantifification analysis. Ind Crop Prod 122:316–328. https://doi.org/10.1016/j.indcrop.2018.06.004
Funding
This work was supported by the Science and Technology Planning Project of Guangdong Province (No. 2016A040403119) and Guangdong Academy of Sciences Special Funding Projects for Construction of Domestic First-Class Research Institutions (2020GDASYL-20200102007), China.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
This article does not contain any studies with human participants or animals performed by any of the authors.
Consent for publication
Not applicable.
Conflict of interest
All authors declare that they have no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Pi, J., Jin, P., Zhou, S. et al. Combination of Ultrasonic-assisted Aqueous Two-phase Extraction with Solidifying Organic Drop-dispersive Liquid–liquid Microextraction for Simultaneous Determination of Nine Mycotoxins in Medicinal and Edible Foods by HPLC with In-series DAD and FLD. Food Anal. Methods 15, 428–439 (2022). https://doi.org/10.1007/s12161-021-02134-w
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12161-021-02134-w