
Abstract
Ion mobility spectrometry is an analytical method for identification and quantification of gas-phase analytes in the ppbv-pptv range. Traditional ionisation methods suffer from low sensitivity (UV light), lack of long-term stability (partial discharge), or legal restrictions when radioactive sources are used. A miniaturised helium plasma was applied as ionisation source in an ion mobility spectrometer (IMS). Experiments were carried out to compare plasma IMS with β-radiation IMS. It could be demonstrated that the plasma IMS is characterised by higher sensitivity and selectivity than β-radiation ionisation. Plasma IMS is approximately 100 times more sensitive than the β-radiation IMS. Furthermore, variable sensitivity can be achieved by variation of the helium flow and the electric field of the plasma, and variable selectivity can be achieved by changing the electric field of the IMS. The experimental arrangement, optimisation of relevant conditions, and a typical application are presented in detail.

Micro-plasma used in ion mobility spectrometry
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Ion mobility spectrometry is an analytical method for detection of gas-phase analytes at ambient pressure and temperature. Broad application of the method took place in the nineteen-sixties for military use first (detection of chemical warfare agents) and for security purpose (security at airports, detection of explosives and drugs) later on [1, 2]. In recent years ion mobility spectrometry is increasingly in demand for civilian applications such as process control [3–5], food quality and safety [6–8], biological applications [9, 10] and medical diagnosis [11–13].
The principle of the method is detection of the ion mobility of gas-phase analytes in a particular gas [1, 2]. Analyte molecules are ionised directly by use of UV light leading to positive ions only. When using ionisation sources like β-radiation or partial discharge, the analyte molecules are ionised via so-called reactant ions. Those sources ionise the available gas (most common are nitrogen or synthetic air) leading to positive protonated water clusters and, when synthetic air is available, negative oxygen ions also. When an analyte is also introduced, the analyte molecules are ionised by charge transfer from the reactant ions. Furthermore, the reactant ion peak (RIP)—which results from those well known ions—can be used as an internal standard with regard to ion mobility and to sensitivity.
After ionisation, the resulting ions are accelerated in a weak electric field in the direction of a detector. During their drift they collide with the available so-called drift gas and are therefore decelerated, to an extent depending on the frequency of collisions. Therefore, in the ideal case, different ions reach the detector totally separated. The drift time over a known drift distance in a particular electric field is a measure of their mobility in the available gas. This is characteristic of the ion and depends on its mass, shape, and charge. The mobility is normalised for pressure and temperature resulting in the so-called reduced ion mobility K 0. For analysis of rather complex and, especially, humid samples, pre-separation techniques, for example gas-chromatographic or multi-capillary columns (MCC), are applied in addition [14] to avoid clustering of the ions with water molecules and to obtain the retention time as additional information for the identification of analytes.
Obviously, the ionisation source used in an ion mobility spectrometer is an essential aspect of the sensitivity (ionisation yield) of the method and the experimental arrangement required to operate the source influences the mobility and stability of the equipment itself (e.g. additional power supply). Traditional ionisation sources [1] and their characteristics are summarised in Table 1. It is obvious that β-radiation sources are optimally capable as they need no additional power supply while guaranteeing high long-term stability, high sensitivity, and high ionisation efficiency. The only disadvantage is the restriction of transport and location due to legal regulations.
In order to provide an ionisation source with sensitivity and stability comparable with those of β-radiation sources and simultaneously avoid the use of radioactive material, a miniaturised helium plasma [15] was applied as ionisation source to an ion mobility spectrometer (IMS). Experiments were carried out to compare the sensitivity, selectivity, and stability of the plasma source with those of traditional radioactive sources. The investigations were focussed on plasma IMS compared with β-radiation IMS which presently is by far the most common ionisation method.
Experimental
The ion mobility spectrometers used for the presented investigations were developed and built at ISAS (Institute for Analytical Sciences, Dortmund, Germany). The IMS were operated with an electric field strength of 330 V cm−1. The drift distance was 12 cm and the shutter grid opening time was 300 μs. The drift gas flow was 220 mL min−1 and the sample gas flow was 110 mL min−1 nitrogen. For the breath analyses synthetic air was used as drift and sample gas with the same flow rates. The signal of the Faraday plate detector was amplified by a factor of 11 nA V−1. The drift time spectra were detected with a length of 100 ms and a resolution of 50 kHz by usbADC3, a data-acquisition board developed at ISAS. The IMS were equipped with a 63Ni β-radiation source (550 MBq) and another one with a UV lamp (10.6 eV). For the breath analyses, where rapid pre-separation was required, a multi-capillary column (MCC OV-5, Sibertech, Novosibirsk, Russia) was also used. These columns consist of ∼1000 glass capillaries and enable high sample flow (up to 150 mL min−1) and rapid separation (∼500 s for 20 cm column length operated at 30°C constant temperature). Samples were introduced into the MCC via a 10-mL sample loop.
The plasma used for ionisation in the IMS–a dielectric barrier discharge with a plasma cone outside the electrode region–consists of a capillary with an inner diameter of 500 μm and an outer diameter of 1.2 mm. Silver cables of 500 μm diameter are wrapped around the capillary forming electrodes with a separation distance of 12 mm. The helium applied to the plasma at atmospheric pressure had a purity of 99.999% and an impurity of 3 ppmv nitrogen. The plasma and its spectroscopic characterisation are described in detail elsewhere [15]. The ions from the helium plasma initiate an ion-reaction chain leading to the same reactant ions as available from traditional β-radiation ionisation (protonated water clusters and O2 − ions). When applied to the IMS, the plasma was operated with a He flow of 330 mL min−1, and high-voltage pulses of 4 kV and 35 kHz were applied to the electrodes.
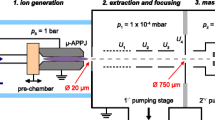
For comparison of a plasma IMS with traditional instruments, the plasma was enclosed in a Teflon tube and implemented in the IMS radial to the gas flow, as shown in Fig. 1. This arrangement resulted in a higher yield of ions than implementation axial to the gas flow for the same instrumentation. Furthermore, in the axial arrangement ions could enter the drift region even when the shutter grid was closed, due to the high flow velocity of the helium in the capillary. All experiments were carried out with the same amplifier and data acquisition board, to avoid corruption of the results by the characteristics of electronic components, and they were repeated several times to guarantee reproducible results.

Schematic diagram of an ion mobility spectrometer with the micro plasma implemented radial to the drift gas flow
The chemicals (puriss. p.a., Sigma-Aldrich) used for the typical application on the detection of analytes and for the exponential dilution experiments carried out for determination of detection limits were:
-
2-ethyl-1-hexanol: alcohol, C8H18O, molecular weight: 130.23, CAS registry number: 104-76-7, (diluent, solvent)
-
2-nonanone: ketone, C9H18O, molecular weight: 142.24, CAS registry number: 821-55-6, (formed by oxidation of fatty acids, e.g. during frying)
-
methyl laurate: ester, C13H26O2, molecular weight: 214.34, CAS registry number: 111-82-0, (e.g. in adhesives)
The analytes were chosen, on the basis of their known characteristics, to cover a broad range of ion mobilities (Table 2).
A mass spectrometer (Thermo Finnigan LCQ Classic) was used to characterise the ions provided by the IMS ionisation chamber. The chamber was coupled to the MS inlet using a sample gas flow of 100 mL min−1 nitrogen and a make up gas of 500 mL min−1.
Results and discussion
To obtain information about the ion chemistry taking place in the plasma IMS compared with traditional ionisation methods, the plasma source, a UV-lamp (10.6 eV) and a 63Ni β-radiation source were coupled to a quadrupole mass spectrometer. Although the experimental setup needs further optimisation, the preliminary findings were promising. The expected reactant ions, protonated water clusters with 2–4 water molecules could be observed for both plasma and radioactive source. As expected, the ions produced by the plasma seem to initiate a reaction chain leading to the same reactant ions as in β-radiation ionisation [1, 15]. When analytes were introduced, protonated monomer and dimer ions could be observed for acetone (Fig. 2), limonene, and nonanone. As no fragment ions were observed, it can be expected that ionisation is mainly a consequence of proton transfer from the reactant ions to the analyte molecules and not of direct ionisation by the plasma source. Only at extremely high concentrations could different ions and fractions be observed, unlike for the traditional ionisation sources, but in the mass-spectrometry only. However, those results need further validation and will be published separately.

Mass spectra of acetone ions provided by the plasma-IMS ionisation chamber. Protonated monomer (MH+) and dimer (2MH+) ions could be identified
As the reactant ions from both plasma and β-radiation ionisation are expected to be the same, the drift-time spectra were normalised to the ion mobility of the reactant ions (K 0,RIP = 2.06 cm2 V−1 s−1 in synthetic air, K 0,RIP = 2.02 cm2 V−1 s−1 in nitrogen). The spectra are presented with 1/K 0 axes which are proportional to the drift time but normalised to the IMS characteristics and to temperature and pressure.
The reactant ion peaks (RIP) for both plasma and β-radiation IMS (Fig. 3. have the same shape with the small water-cluster peak left from the RIP. The faster decrease of the slope of the plasma RIP and the tailing of the β-radiation RIP can be explained by different characteristics of the shutter grid of both spectrometers (same design, but possibly small deviation e.g. of the grid distance). The same effect can be observed from the analyte signals (see below). The RIP of the plasma IMS is more than three times higher, representing the higher ionisation yield of the plasma source. The minor signals at 0.52 and 0.54 V s cm−2 in the spectrum are expected to derive from contamination of the plasma IMS, most probably from the materials used to fix the plasma capillary in the Teflon tube.

Reactant ion peaks (RIP) of plasma and β-radiation IMS
Detection of analytes (Fig. 4) was carried out using selected substances from which already in medium concentrations (lower ppbv) a monomer and a dimer ion could be observed. Furthermore they cover a broad range of ion mobilities (Table 2; from 0.91 to 1.47 cm2 V−1 s−1). For 2-ethyl-1-hexanol a monomer and a dimer ion are observed with the β-radiation IMS. Due to the higher ionisation yield of the plasma IMS and the associated higher ion concentration in the IMS even a trimer ion can be observed with this instrument. Similar observations were made for other analytes such as methyl laurate and 2-nonanone. For all three analytes the same ion mobilities were observed for the monomer and dimer ion peaks detected with β-radiation and with plasma IMS. The ion mobilities of the detected ions are summarised in Table 2.

Ion mobility spectra of 2-ethyl-1-hexanol detected with plasma and β-radiation IMS. Attention is directed to the normalised signal height—the real RIP is 3.3 times higher for the plasma IMS than for the β-radiation IMS
It has to be kept in mind that the spectrum of the β-radiation IMS in Fig. 3 is normalised to the height of the RIP of the plasma IMS. The small ammonia signal left from the RIP is due to impurities in the drift gas and generally rises with increasing humidity. The real β-radiation RIP maximum is 3.3 times smaller. As already observed from the RIP only, the higher ionisation yield of the plasma source is the reason for the higher plasma IMS signal. As a consequence, the sensitivity of the plasma IMS is expected to be significantly higher. Detection limits of both plasma IMS and β-radiation IMS were determined by exponential dilution of a initial concentration (lower ppm range) from the same permeation source. The experiment was repeated three times with good reproducibility. The standard deviation of the ion mobility was less than 0.5% and that of signal height was less than 5%.
For 2-nonanone the detection limits were 1000 pptv using the β-radiation ionisation source and 10 pptv with the plasma IMS. As the signal height of the RIP is only 3.3 times higher using the plasma source, the enormously higher sensitivity of the plasma-IMS can be explained by the influence of the positive high-voltage pulses of the plasma power supply which could cause higher acceleration of the ions towards the shutter grid, thus leading to a greater amount of ions entering the drift region.
As the number of available reactant ions is the only measure of the sensitivity of an IMS, those results can be translated to any other analyte detectable by IMS, independent on the particular sensitivity for this analyte. Similar to β-radiation, the linear range covers two orders of magnitude, starting with a monomer ion peak for low concentration and an additional dimer ion peak for increasing concentration.
The high sensitivity of the plasma IMS compared to the β-radiation IMS can be expressed by the signal-to-noise-ratio (Fig. 5a). The high signal enables a decrease of the electric field in the drift region starting from the commonly used 330 V cm−1. As an increasing drift time is the result, the losses of ions e.g. to the walls of the drift tube also increases, and the signal decreases. For the β-radiation IMS the RIP disappears in the signal noise at 275 V cm−1, the signal of the plasma IMS can be observed down to 205 V cm−1.

Signal-to-noise ratio (a) and resolution (b) of the plasma IMS compared with the β-radiation IMS
To find out if the longer drift time could be used to improve the selectivity of the IMS, the resolution (drift time/width at half maximum) was calculated for both IMS and for decreasing electric fields (Fig. 5b). It was found, that the resolution is constant for the β-radiation IMS until the RIP disappears in the signal noise at 275 V cm−1—the decrease of the peak height is balanced by the increase of the drift time. In contrast, the resolution shows a significant increase with decreasing electric field for the plasma IMS. This effect could be used to balance sensitivity and selectivity according to particular applications (lower sensitivity with higher selectivity and vice versa). The reason for the different behaviour of β-radiation and plasma IMS resolution is most probably the influence of the high-voltage supply of the plasma, which results in a slightly higher electric field in the ionisation region. Therefore, the acceleration of the ions towards the shutter grid is higher than in the β-radiation chamber and more ions can enter the drift region, especially for low electric fields of the IMS, because the difference between drift field and plasma voltage is higher.
To demonstrate the suitability of the plasma IMS even for challenging applications, the instrument was used for breath analysis after consumption of a particular candy. The sample (10 mL via a sample loop) of exhaled breath was introduced into a multi-capillary column which was directly connected to the ionisation region of the plasma IMS. The resulting topographic plot or ion mobility chromatogram (signal height vs. ion mobility and retention time) of the positive ions, as obtained from data evaluation software developed at ISAS, is presented in Fig. 6.

Ion mobility chromatogram of a 10-mL sample of exhaled breath after consumption of candy. The peaks of limonene and dl-menthol, as identified from a calibration carried out earlier, are indicated. The colour indicates the peak height (yellow, high; blue, medium; white, zero). For the position of both reticles, the spectra at the chosen retention time (bottom) and the chromatogram at the chosen 1/K 0 (right) are displayed
By a calibration carried out earlier, limonene monomer and dimer and four dl-menthol peaks could be identified. Both analytes were also found in the headspace of the candy. Many other peaks can be observed in the diagram but they are not identified yet. However, they are not present in breath without the previous consumption of the candy and therefore can be considered as a direct effect of that consumption.
In the negative ion mode, reactant ions could be observed as well as analyte ions, similar to β-radiation ionisation. However, the positive pulsed high voltage of the plasma disturbs the negative drift field in the ionisation region and therefore the sensitivity was poor. This will be solved with a different experimental setup and will be subject of future work.
The plasma used for the presented experiments was used over a period of almost one year and for many experiments it was operated continuously for more than 10 h with no negative effects on the ionisation yield. Furthermore, the plasma source was stable over the investigation period of one year.
Conclusions and outlook
The miniaturised plasma was found to be a stable and efficient ionisation source and was, for this reason, ideal for ion mobility spectrometry. The sensitivity (about 100 times greater, as found from the detection limits for 2-nonanone), selectivity, and resolution of the plasma IMS were significantly higher than those of a comparable β-radiation IMS. Furthermore the high ionisation yield of the plasma IMS enables variation of the electric field in the drift region and use of shorter opening time of the shutter grid; both approaches result in an increase of selectivity. A further advantage of the plasma IMS is the variable sensitivity achieved by variation of the He flow and of the high voltage of the plasma. The disadvantage of the additional power supply for the plasma is minimised as such equipment is already available in a low-cost miniaturised version.
Overall, a miniaturised plasma as ionisation source for ion mobility spectrometry is a more than adequate alternative to radioactive and other ionisation sources (e.g. photoionisation). In fact, due to the high and variable sensitivity and selectivity it has greater potential for many applications.
However, some work still has to be done to provide similar sensitivity and selectivity in the negative-ion mode by help of a modified setup of drift field, plasma voltage, and detector. Use of He only as plasma gas will make it difficult to use a plasma IMS as a mobile instrument, but work on the development of a plasma operated with a different gas is already in progress—to enable mobility by avoiding the need for a helium supply on the one hand and to reduce the operating costs on the other.
The focus of work in the near future will be on detailed mass spectroscopic investigations of the ion chemistry in the ionisation region of the plasma IMS. Furthermore the experimental setup of the high voltage supply has to be optimised to avoid disturbances of the drift field, especially in the negative-ion mode.
References
Eiceman GA, Karpas Z (2005) Ion mobility spectrometry, 2nd edn. CRC Press, London, UK
Baumbach JI, Eiceman GA (1999) Appl Spectrosc 53:338A
Baumbach JI (2006) Anal Bioanal Chem 384:1059
Matsaev VT, Gumerov MF, Chilipenko LL, Kozlov NN (2002) Int J Ion Mobility Spectrom 5(3):115–118
Roehl JE (1991) Appl Spectrosc Rev 26(1–2):1
Vautz W, Zimmermann D, Hartmann M, Baumbach JI, Nolte J, Jung J (2006) Food Addit Contam 23:1064
Vautz W, Baumbach JI, Jung J (2006) J Inst Brewing 112:157
Raatikainen O, Reinikainen V, Minkkinen P, Ritvanen T, Muje P, Pursiainen J, Hiltunen T, Hyvoenen P, von Wright A, Reinikainen S-P (2005) Anal Chim Acta 544(1–2):128
Kaddis CS, Lomeli SH, Yin S, Berhane B, Apostol MI, Kickhoefer VA, Rome LH, Loo JA (2007) J Am Soc Mass Spectrom 18(7):1206–1216
Kolehmainen M, Ronkko P, Raatikainen O (2003) Anal Chim Acta 484(1):93
Baumbach JI, Westhoff M (2006) Spectrosc Eur 18:22
Ruzsanyi V, Baumbach JI, Sielemann S, Litterst P, Westhoff M, Freitag L (2005) J Chromatogr A 1084:2145
Karpas Z, Chaim W, Gdalevsky R, Tilman B, Lorber A (2002) Anal Chim Acta 474(1–2):115
Vautz W, Sielemann S, Baumbach JI (2004) Anal Chim Acta 513:393
Michels A, Tombrink S, Vautz W, Miclea M, Franzke J (2007) Spectrochim Acta B 62B(11):1208
Acknowledgements
Financial support by the Bundesministerium für Bildung und Forschung and the Ministerium für Wissenschaft und Forschung des Landes Nordrhein-Westfalen is gratefully acknowledged. The dedicated work of Luzia Seifert, Susanne Krois, and Antje Michels was indispensable for the success of the investigations, as also was the support of Michael Schilling for the coupling of IMS and MS.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Vautz, W., Michels, A. & Franzke, J. Micro-plasma: a novel ionisation source for ion mobility spectrometry. Anal Bioanal Chem 391, 2609–2615 (2008). https://doi.org/10.1007/s00216-008-2181-y
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00216-008-2181-y