
Abstract
Ion mobility spectrometry, originally used to detect chemical warfare agents, explosives and illegal drugs, is now frequently applied in the field of process analytics. The method combines both high sensitivity (detection limits down to the ng to pg per liter and ppbv/pptv ranges) and relatively low technical expenditure with a high-speed data acquisition. In this paper, the working principles of IMS are summarized with respect to the advantages and disadvantages of the technique. Different ionization techniques, sample introduction methods and preseparation methods are considered. Proven applications of different types of ion mobility spectrometer (IMS) used at ISAS will be discussed in detail: monitoring of gas insulated substations, contamination in water, odoration of natural gas, human breath composition and metabolites of bacteria. The example applications discussed relate to purity (gas insulated substations), ecology (contamination of water resources), plants and person safety (odoration of natural gas), food quality control (molds and bacteria) and human health (breath analysis).
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Ion mobility spectrometry was a technique originally applied for the detection of trace compounds within a gas, for example gaseous pollutants in air. More than 70,000 units are in service worldwide, and most of these are used to detect chemical warfare agents, explosives or illegal drugs [1–4]. Ion mobility spectrometry combines both high sensitivity (detection limits down to the ng/L-range to pg/L-range, ppbv-range and pptv-range), and relatively low technical expenditure with high-speed data acquisition. The time required to acquire a single spectrum is in the range of 20–50 ms. Using gas chromatographic preseparation, total analysis times of a few minutes are achievable.
The working principle is based on the drift of ions at ambient pressure under the influence of an external electric field (for more details see the following section). The mean free path of the ions is smaller than in mass spectrometry, as are the dimensions of the instrument. Therefore, an ion swarm drifting under such conditions experiences a separation process that is based on ions with different masses or structures having different drift velocities. Collecting these ions on a Faraday plate delivers a time-dependent signal corresponding to the mobility of the arriving ions. This ion mobility spectrum contains information on the nature of the different trace compounds present in the sample gas.
Ion mobility spectrometry (IMS) is suitable for process control, but due to the occurrence of ion-molecule reactions and relatively poor resolution of the species formed, it is generally not used to identify unknown compounds, but it is being increasingly applied to cases where the processes involved are known. The fact that ambient air can be used as the carrier gas—in contrast to the pure nitrogen or helium used in gas chromatographic detectors normally—and that a vacuum is not necessary has helped IMS to gain increasing popularity in the field of process analysis [5–13]. Here, the term “ambient” indicates that cleaned surrounding air, not just bottled synthetic air, can be used, especially for field and process applications.
In the present paper, proven applications of different kinds of IMS will be discussed in detail: monitoring of gas insulated substations, contamination in water, odoration of natural gas, human breath composition and metabolites of bacteria.
IMS is used to monitor the quality of the SF6 gas used in gas insulated substations, in order to safeguard the energy distribution process [14–16]. Rapid on-site monitoring of ground and surface water contamination is also considered here [17]. Inspecting the odoration of natural gas, especially for sulfur-free odorants, plays an important role in the process of converting sulfur-containing odorants to those that do not contribute to global atmospheric warming.
The combination of headspace solid phase microextraction (HS–SPME) with fast chromatographic preseparation by means of multicapillary columns (MCC) coupled to an IMS equipped with a 10.6 eV photoionisation source was applied to the rapid on-site monitoring of ground and surface water contamination. Under field conditions, water contaminants were directly detectable down to the upper μg/L range.
The next two examples deal with metabolomics, which is related to the analysis of low molecular compounds involved in cell physiology and their influence on cell functions. Metabolites of bacteria and molds are detectable using IMS, and this can be applied to analyze biotechnological processes as well exhaled air [12]. By analyzing exhaled breath, early diagnosis and therapeutical control can be attained in cases of lung cancer, inflammations and illnesses.
In addition, volatile organic compounds related to different bacteria can be directly investigated by IMS. So-called IMS chromatograms (peak height diagrams) give different signatures for different kinds of bacteria, which allows rapid recognition of the bacteria.
These examples cover both technical processes (gas insulated substations and odoration of natural gas) and life processes (bacteria, molds and humans).
The subject of process analysis is discussed, starting with purely industrial production and then moving on to address living matter. Applications of IMS to analyze aggregation states, damaged surfaces (gas insulated switch gears), water supervision and gas analytics are all described. Applications of IMS therefore relate to gas or water purity (gas insulated substations, water contamination), ecology (contamination of water resources, sulfur-free odorants and unwanted release of SF6 into the surrounding atmosphere), plants and person safety (gas insulated substations and odoration of natural gas), food quality control (molds and bacteria) and human health (breath analysis).
Working principles of ion mobility spectrometers
The term ion mobility spectrometry refers to a method that characterizes chemical substances via their gas phase ion mobility [1, 2, 18–21]. Normally, the drift time of ion swarms formed using suitable ionization sources and electrical shutters are measured and the drift length and the electric field strength are held fixed (see Fig. 1). As well as showing the working principles of IMS, the photograph in Fig. 1 shows a miniaturized IMS, which allows to get a feel for the outer dimensions of the ionization and drift region of an IMS.

Working principle of the ion mobility spectrometer; from [1]
Different methods are used to ionize the gas in IMS. β-radiation sources like 63Ni [22–28] and 3H [29, 30] are usually used, but 241Am sources are also used to ionize gaseous analytes. Sometimes UV light [31–37] or electrical discharges [38–47] are applied.
Generally, ion mobilities are characteristic properties of analytes. Thus, databases of ion mobility values allow the identification of gaseous analytes. The drift velocity of the ion is related to the electric field strength (the driving force) and the ion mobility constant. The mobility is proportional to the inverse of the drift time, which is measured at fixed drift length. Theoretical considerations show that the mobility is related to the collision rate of the ion with gas molecules (reduced mass), the temperature, the dimensions of the ion (structural dependencies), and the collision integral, which is influenced by the size of the ion or molecule, its structure and polarizability. Because of the structural dependencies, it is possible to separate isomers due to their different structures, even though they have the same masses [2].
To perform IMS, the high voltage supply needs to deliver voltages of 100–10,000 V to establish a strong electric field in the drift tube, and to keep this field as constant as possible. The ions can drift appreciably under the influence of a static electric field of this size away from the direction of gas flow. The shutter used (usually a Bradbury–Nielsen shutter) has an opening time that is normally fixed between ~10 μs and ~1 ms. Ideally the final spectrum consists of clearly separated peaks. The nA or pA current measured is converted into a signal voltage which is then converted by commercial A-D cards into digital signals for further analysis. Because of the very small size of the signal (the current), it is necessary to shield the IMS to protect it against external electromagnetic disturbances. Other factors that influence the signal, like the gas flow rate (normally in the mL/min range) and the temperature in the ionization and drift region, must be controlled [1].
The carrier gas transports sample molecules into the ionisation region. Direct ionisation may occur in this region (when using UV lamps for instance, which are available in emission energies of between 8.5 and 11.7 eV, with 10.6 eV being the most common); chemical ionization may be induced by collisions of the analyte with ionized carrier gas molecules produced by applying radioactive ionization sources; or fragmentation may occur due to partial or corona discharges.
Gas flows from the Faraday plate towards the ionization region (this is called drift gas, see Fig. 1) to make sure that only ions (no uncharged particles) enter the drift region of the IMS. Thus, charge transfer reactions that result in the ionization of different analytes only occur in the ionization region, never in the drift region. If the shutter is held closed, all analyte molecules—neutrals and ions—will be passed into the gas outlet. When the shutter is opened, a swarm of ions enters the drift region. After several collisions with the surrounding gas molecules they rapidly reach a steady drift velocity [1, 2].
The type of ions produced will differ depending on the ionization technique. If nitrogen or air is used as carrier gas, the carrier gas molecules are directly ionized by the β-particles. Positive carrier gas ions and free electrons become available. These primary positive ions (called reaction ions) undergo different chemical reactions with the analyte molecules to form “product”: proton transfer, nucleophilic attachment, hydride abstraction and other processes. The electrons become attached to the sample molecules, forming negative ions (via electrophilic attachment, resonant attachment and dissociative attachment). Charge transfer and proton abstraction can also occur. Positive and negative ions are often formed simultaneously. The charge transfer process is limitated by the total number of ions available, which is reflected in the reaction ion peak. Due to the fact that increasing numbers of dimer ions are formed as the analyte concentration increases [2], the spectra obtained are sometimes rather complex and difficult to interpret.
In the case of UV ionization, all analytes with ionization energies lower or equal to the energy of the UV light from the lamp can be ionized. Only positive ions are formed directly from UV ionization . However, if photolamps with different radiation energies are used, a broad spectrum of organic compounds can be ionized. If mixtures of substances are investigated, charge transfer reactions must be taken into account—compounds with a lower ionization potentials are preferred during ionization. In comparison to β-radiation, UV ionization permits quantitative analyses over a larger concentration range. The linearity is not limited by the restricted number of reactant ions available.
Ionization by using different kinds of discharges is more complicated, because of the defragmentation of the molecules and the number of different analyte ions formed is usually higher than in the other methods. However, discharges produce both positive and negative ions and their use bypasses the need to handle radioactive materials. For further details on partial discharge IMS, refer to [14–16, 24, 48].
We now review the application of IMS to detecting trace contaminations in gas phases.
Applications of IMS
Gas insulated substations
Although gas insulated switchgear and substations (GIS) are amongst the most reliable components of electric power networks (and usually operate for years without complications), failures still occur. These failures are unpredictable and typically cause severe damage and economic loss. Such failures can be attributed to sparks or arcs inside high-voltage switches filled with SF6, an electrical insulating gas. Over a long period, undetected partial discharges may degrade the quality of the insulating gas, ultimately leading to a single catastrophic failure. Such a degradation in gas quality is associated with increases in corrosive and toxic byproducts (which also leads to secondary industrial hygiene concerns). An in situ IMS analyzser placed inside each switch can provide, on demand, measurements of gas quality while the switches are operating. It is possible to apply this technique to each switch due to the small size, low cost and high reliability of mobility drift tubes. In principle, switches equipped with in situ analyzers for continuous or regular diagnostic measurements can be used to anticipate switch failure, preventing failures in power grids. The chemistry of the switch gas was independent of the producer of the switch, and so suitable for such diagnostics. Other mature advanced methods, such as gas chromatography/mass spectrometry (GC/MS) or infrared spectroscopy (IR), can provide considerable information on the chemical compositions of samples. However, these are too slow, too expensive or are not suitable for monitoring individual gas switches in situ. Portable methods such as reagent detector tubes could be used at a switching station, but cannot be localized to minor components, which govern the quality of the sulfur hexafluoride. Ion mobility spectrometry is a good compromise, providing sufficient information at moderate cost; the analyzer can be placed directly inside the gas insulated switch and measurements taken automatically [14, 16, 39, 49].
One immediate complication that arises when adapting IMS to monitor SF6 is the ionization source; commonly used sources such as radioactive sources or UV lamps are unacceptable. The possible occurrence of corrosive byproducts rules out the use of radioactive materials, and the very strong UV absorption of the SF6 matrix, as well as the ionization potentials of the expected compounds (which are higher than 11.8 eV) make it impossible to use photoionisation. Hence, a partial discharge source was configured and developed as an ionization source for an IMS. A second innovation in this application was the use of the sample gas, SF6, as both the supporting atmosphere for the source and the drift gas. Thus, no additional gases were needed to operate the analyzer, which is a major advantage, especially for measurements taken in remote locales [48].
Under these conditions, the ions created in the SF6 include those of the original sulfur hexafluoride and byproducts from manufacture or in situ decomposition processes. Thus, any chemical changes in the SF6 will be seen in the ion mobility spectra and peak positions can be compared in order to evaluate the level of electrical stress induced by partial discharges in the SF6.
Typical ion mobility spectra used to characterize SF6 are shown in Fig. 2. A single spectrum of pure SF6 and another of used SF6 are plotted together. Shifts in the peak maximum provide sensitive and specific information regarding the quality of the gas and any electrochemical events occurring inside the gas insulating switch. For example, peak shifts are caused by ions with different mobilities, while peak broadening arises when the number of different ions increases (which may be due to the production of impurities during discharges). As shown in Fig. 2, large shifts in peak positions suggest high levels of decomposition products within the gas. Calibration procedures, including reference FT–IR measurements, and a training procedure for partial discharge IMS are described in the literature. Figure 3 shows the operation of an IMS in a gas insulated substation on a circuit breaker line.

Spectra of pure (left spectrum) and used (right spectrum) SF6, showing the peak shift used as as a measure of the quality of the gas inside a gas insulated high voltage switch. The spectrum on the right is from a sample obtained from a highly contaminated gas insulated circuit breaker (>4.000 ppmv, maximum limit 2.000 ppmv)

Making ion mobility spectrometer measurements at a German gas insulated substation
The conclusion from this measurement was that significant levels of decomposition products were present in the gas of this specific circuit breaker, despite the presence of molecular sieves in the gas switches. The extent of the peak shift can be correlated with the mean concentration of decomposition products [49] and critical levels that precede switch failure have been identified. The exact identities of these impurities are still being investigated, although this has not stopped the productive use of IMS for in situ monitoring. While deriving the exact nature of the decomposition products (considering species like SF4, SOF2, SOF4, SO2F2, S2F10) using Gaussian curve analysis is one target, in practice it is probably more convenient for a technician to see a green, yellow or red lamp depending on the status of the switch. Therefore, the concept of automated monitoring of SF6-insulated substations by IMS has been industrially realized. Slight changes in the quality of the filling gas can be detected and preventative maintenance undertaken (Fig. 4).

Gaussian curve analysis of spectra from pure and used SF6; the rectangles indicate the differences in the concentrations of the single analytes
Water contamination
Some of the organic substances used in the industrial sector can degrade the aquatic environment, especially when they are released into groundwater and surface water in operating accidents. Such compounds are often toxic at low concentrations and can accumulate in sediments and organisms living in the water. It is therefore important to identify the contaminant as quickly as possible after an environmental accident, or in conjunction with remediation measures. This is necessary to control the extent of contamination and to avoid dangerous dumping.
Most analytical methods officially recommended for water pollution control require time-consuming sample preparation processes, including the use of organic solvents. This means that well-equipped analytical laboratories need to be available. However, the combination of a well established preconcentration procedure with fast preseparation coupled to IMS can be used for efficient on-site detection of organic water contaminants to acceptable sensitivities, as we will now see.
To transfer the water pollutants into the gas phase (necessary for IMS), solid phase microextraction is applied as an intermediate step. This method is based on enriching the analyte molecules on chemically modified fused silica fibers, and it is well known from its common application to gas chromatography. The solvent-free SPME technique has turned out to be a particularly effective technique for isolating contaminants from water matrices on timescales of minutes, allowing preconcentration and clean-up in a single step. Headspace SPME is used instead of methods like purge and trap or liquid–liquid and solid phase extraction. The transfer of the adsorbed compounds from the microfiber into the gas phase can easily be accomplished by thermodesorption.
The following procedure was realized successfully. The groundwater was sampled directly from special boreholes which were in hydraulic equilibrium with the aquifer. Using a peristaltic pump, the water was transferred into glass bottles until the conductivity and pH were constant. For HS–SPME, the water sample under study (10 mL) is pipetted into a 25 mL glass vessel, which is then closed by a septum. The fused silica fiber is pushed through the septum with a syringe and the tip is adjusted to be 1 cm above the surface of the stirred (magnetic stirrer) water sample. By choosing suitable fiber coatings it is possible to separate trace substances from matrix components. In the present case, a polydimethylsiloxane (PDMS) fiber was used to extract nonpolar substances, whereas the polar substances were collected on a polyacrylate (PA) fibre. After 5 min (volatiles) or 15 min (semi-volatiles) of exposure, the fiber is removed and mounted in a custom-designed thermodesorption unit (Fig. 5). To achieve complete desorption, the fiber must be exposed for 1–5 min at a temperature just above the highest boiling point of the substances under investigation. The completeness of the desorption is regularly tested by a subsequent second desorption. The flow rate of the carrier gas (nitrogen) through the thermodesorption unit is 1 L/min. The desorbed compounds are transferred with a carrier gas (nitrogen) at 1 L/min to the inlet of a multicapillary column (MC-1, HT, Alltech GmbH, Unterhaching) and, after separation at ambient temperature, to the ionization chamber (10.6 eV low pressure gas discharge lamp) of the attached UV–IMS (see Fig. 5). To validate the developed headspace SPME/MCC/UV–IMS method, the groundwater samples were also analyzed using standard analytical methods. Quantification of the key substances benzene and naphthalene in the groundwater at the gasworks site was carried out by GC/FID analysis according to DIN 38407 part 9-2.

Set-up for SPME/MCC/UV–IMS; from [52]
Thus, the developed method makes use of the advantages of HS–SPME/thermodesorption, fast preseparation using MCCs, and IMS used as a mass selective detector with ms response time. Since the IMS works at ambient pressure and comes in a wide range of sizes and operating conditions, it has found widespread use in on-site analysis. Accordingly, the set-up presented here is well suited for field measurements of volatile and semi-volatile contaminants in water.
Figure 6 shows a 2D-MCC/UV–IMS-chromatogram recorded after performing HS–SPME of contaminants from groundwater at a former gasworks site. Two MCC/photoionization detection chromatograms (total ion current) are also presented in the figure. One was obtained with the same groundwater sample, and the other one with a standard solution of 10 mg/L benzene and 1 mg/L naphthalene in water. Figure 6 shows that benzene and naphthalene are separated on the MCC in 250 s, which should be compared with the >40 min required for the GC/FID standard analytical method. Whereas the latter only delivers the retention time for substance identification, the new technique additionally yields an orthogonal substance-selective parameter, namely the drift time in the IMS. This significantly improves the reliability of the analytical results.

MCC/UV–IMS chromatograms obtained after HS–SPME of the contaminants in groundwater from a former gasworks site; from [52]
The developed method yields results with acceptable reproducibility. The statistical parameters are of the same order of magnitude as those obtained using standardized analytical methods performed in accredited environmental analytical laboratories. The relative standard deviation and the recovery were calculated to be 9.4 and 98.8% for the detection of o-xylene (c=2.5 mg/L; 20 repetitive measurements). The same experiments carried out with 1,4-dichlorobenzene (c=2.5 mg/L) revealed a relative standard deviation of 7.3% and a recovery of 99.9% [50].
The combination of headspace solid phase microextraction (HS–SPME) with fast chromatographic preseparation via MCC coupled to an IMS with a 10.6 eV photoionisation source (UV–IMS) appears to be an instrument suitable for on-site monitoring of ground and surface water contaminants. Comparisons of the presented method with two standardized methods have shown that the HS–SPME/MCC/UV–IMS is as good quantitativelybut better qualitatively because it provides two substance selective parameters, namely the chromatographic retention time and the drift time in the IMS. This feature considerably improves the certainty of the analytical information. This tool can be used for emergency field investigations because it can be operated on-site at relatively small expenditure and provides results within a short time [17, 32, 50–52].
Odoration of natural gas
Gasodor S Free (trademark of Symrise, Holzminden, Germany) is the first DVGW (German Technical and Scientific Association for Gas and Water)-certified sulfur-free odorant, containing the principal ingredients methylacrylate and ethylacrylate. It combines environmental compatibility with the highest levels of safety and economy, and it is intended to replace other odorants such as tetrahydrothiophene (THT).
When changing the odoration of natural gas from sulfur-containing to sulfur-free, a fast and sensitive detection method plays a key role. Using a hand-held IMS, the odoration content of natural gas can be derived in less than 2 min total analysis time. The concentrations of methylacrylate and ethylacrylate (which can be between 0.2 and 23 mg/m3) can be monitored using IMS, and the THT content (still commonly used) can also be found using this method.
Gas chromatography (Varian, Agilent and others) or olfactometric (Axel–Semrau) procedures are used to detect the odorants within natural gas. Dräger is developing detection techniques for small pipes based on color changes. These methods have different detection limits, total analysis times, and prices. Often, the full procedure takes several minutes. Some methods give a measurement error of about 30%; for details refer to [53].
When applying IMS to detect the major odorants methylacrylate and ethylacrylate in the hydrocarbon matrix of natural gas, the goal is to perform the ionization using the low-energy electrons provided by a β-radiation source (63 Ni). The acrylates are ionized and monomer and dimer ions are formed via reactant ions and ion-molecule reactions that occur within the ionization region of the IMS. The concentrations of the analytes can be determined without preseparation using gas chromatography columns. By carefully selecting the ionization conditions, the interactions of the different ions can be neglected, as can other ions potentially formed within the rather complex matrix.
The spectra of methylacrylate and ethylacrylate (used as single analytes) are shown in Fig. 7. The reaction ion peak (RIP) that always occurs in air is visible at 2.98 ms. Another two peaks from methylacrylate (3.2 and 3.8 ms) and ethylacrylate (3.4 and 4.2 ms) can also be distinguished. If the acrylates are absent, only the reaction ions arise in air, so that only the reaction ion peak becomes visible. Monomer peaks are formed at low concentrations of the analytes. At higher concentrations of the monomers, solely monomeric ions don’t suffice any more; dimer ions are formed in addition to the monomer ions.

Ion mobility spectra of the major sulfur-free odorants in natural gas, methyl- and ethylacrylate (RIP reactant ion peak); from [53]
In Fig. 8, peak height diagrams are shown for different concentrations of odorant Gasodor S-Free in natural gas. The time (number of the spectrum collected) refers to the time that the measurement was taken. Small monomer peaks occur at concentrations of 200 and 500 μg/m3. At 2 and 5 mg/m3, the dimer ion peak becomes visible. At even higher concentrations of 11 and 19 mg/m3, no monomer ions are visible and the dimer peaks reach maximum intensity values. All charges are transferred from the reactant ions to the analytes. Therefore, the reaction ion peak is no longer visible. No further analyte ions can be formed. However, this is not important in this case since the experimental parameters chosen here ensure that the complete concentration range of interest is covered, between 0 and 23 mg/m3. It is apparent that the odorants can be characterized by using either the position of the peak on the drift time scale or by analyzing the peak intensity as function of drift time and running time of the experiment, which is related to the time needed for the analyte to enter the IMS and the time taken for the analytes to be washed out (to make the IMS clean again). It is clear that applying suitable pattern recognition methods would pick out differences in the concentrations levels of the odorants [53]. The question of potential interactions between the acrylates themselves should also be considered.

Peak height diagrams of the odorants methyl- and ethylacrylate, present at different concentrations in natural gas, as a function of the experimental time; from [53]
Breath analysis and metabolites of bacteria
Volatile metabolites that occur in air exhaled by humans can be directly correlated with different kinds of diseases. Some metabolites are biomarkers: acetone is related to diabetes, nitric acid to asthma and ammonia to hepatitis, while others arise from bacteria. An IMS was coupled to a MCC preseparation unit and this sytem was used to identify and quantify volatile metabolites occurring in human breath. As examples, here we discuss spectra obtained from patients suffering from chronic obstructive pulmonary disease (COPD) and pneumonia in detail, as well as IMS chromatograms of metabolites of bacteria.
The aim of the study was to investigate whether selected diseases can be rapidly diagnosed by detecting the presence of certain metabolites in human breath using IMS, and whether IMS can be used to monitor the effectiveness of drug delivery [54].
Using a chromatographic column with IMS enables multidimensional data analysis to be performed; peaks can be identified using chromatographic data (retention times) and specific ion mobility data (arrival times at the Faraday plate). The retention times of the compounds separated by the MCC and the drift time values of the analytes are plotted in “IMS chromatograms”. The MCC reduces the number of interactions arising from the formation of clusters in the ionization region of the IMS and avoids concurrent charge discharge transfer by causing the analytes to enter the region at different times. Thus, it is possible to ensure the effective ionization of almost all analytes in a given sample by proper selection of the gas chromatography conditions (including by ensuring effective temperature programming of the chromatographic column or by keeping the temperature constant as in the present case) [55–57].
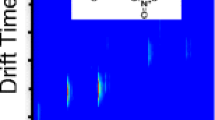
Figure 9 shows the results for the exhaled air from a healthy human, which contains major peaks from ammonia, acetone, ethanol and the reactant ion peak (RIP). Smaller peaks will not be considered here. In addition, the mobilities of the ions, calculated from the drift times of the ion peaks, are included in the inset (in brackets), showing a single spectrum at a fixed retention time. It is also clear from Fig. 9 that the humidity can reach very high values initially. In the first 20 s rather high moisture levels enter the ionization region of the IMS. In general, the MCC will reduce the influence of the humidity. The main regions of interest are between 17 and 29 ms and 20 and 400 s, as shown in the following figures, and these are not affected by the initial high humidity.

IMS chromatogram of human exhaled air (inset: single spectrum at a fixed retention time of 3 s showing signals from the major components ammonia, ethanol and acetone); from [56]
In co-operation with a German lung hospital, on-site measurements were carried out on the exhaled air of 40 subjects, including 22 patients suffering from different pulmonary lung infections. The exhaled air from 18 different healthy persons were analyzed on-line using MCC–63Ni-IMS. To reduce the risk of cross-contamination from other processes, subjects were not allowed to drink, eat or smoke 2 h before the breath measurements. The analyses were always performed in the same room, where the air in the room was determined before each of the breath measurements. Figure 10 shows an IMS chromatogram from the exhaled air of a patient suffering from a bacterial lung infection. The peaks are labelled with their mobility values, K 0, calculated using the individual experimental parameters [55–57].
As mentioned above, the formation of reactant (arising from the carrier gas) and product ions (formed from the analytes) in IMS can be influenced by moisture. Therefore, separation out water molecules is of high importance when working with breath samples. Figure 10 shows that the breath moisture (mobility value K 01=2.11 cm2/V s) was separated out effectively using the MCC. The signals correlated with moisture are always located right at the beginning of the IMS chromatogram. Thus, moisture has no influence on peaks with longer retention times. Acetone was identified in the topographic plot of the exhaled air from a healthy person based on its reduced mobility value (K 04=1.78 cm2/V s). Acetone has a similar retention time to water, but it possesses a stronger proton affinity, enough for acetone to be ionized via ion-molecule reactions [55–57].

IMS chromatogram of the breath of a patient suffering from a lung infection (K0 refers to the mobility values calculated for each major peak); from [56]
Upon comparing the two IMS chromatograms in Figs. 9 and 10, the differences are conspicuous. The 2-D plot in Fig. 10 exhibits additional peaks. Two major peaks occur at retention times of 28 s (K 02=1.95 cm2/V s) and 36 s (K 03=1.77 cm2/V s). Both are probably from degradation products of antibiotics or other drugs, because they were also found in the exhaled air from several people who had taken similar antibiotics. The peak at 50 s and K 05=1.42 cm2/V s was also seen in the exhaled air from other patients suffering from pneumonia. Two other larger analytes with lower mobility values (K 06=1.39 cm2/V s and K 07=1.28 cm2/V s) and longer retention times (t ret6=244 s, t ret7=330 s) were often detected in the exhaled air from patients with bacterial infections and airway inflammations [55–57].
Figure 11 shows the exhaled air from a patient with chronic obstructive pulmonary disease (COPD) is shown. Two major peaks can be seen. The peak at the lower retention time (P-Co01-01) is identical to a peak found for Staphylococcus aureus cell cultures (B-Sa-01). The second peak at higher retention and drift times, indicating a higher molecular substance, is not correlated to Staphylococcus aureus or any other bacteria. However, we need to account for metabolites from bacteria that occur in the lung. Therefore, the emissions from these bacteria were also investigated. Figure 12 shows an IMS chromatogram of the volatile emissions obtained from Staphylococcus aurens. A single peak (B-Sa-01) at an ion mobility of 1.86 cm2/V s and a retention time of 33 s (B-Sa-01) is clearly visible [55–57].

IMS chromatogram of a patient suffering from chronic obstructive pulmonary disease (COPD), and two typical corresponding single spectra (inset); from [56]

IMS chromatogram of Staphylococcus aureus and the corresponding spectrum (inset); from [56]
To demonstrate the rather complex situation in cases where different diseases and bacteria are involved, Fig. 13 shows the IMS chromatogram of a COPD patient with different infections. Five clearly separated major peaks are apparent. One of them, called P-Co02-04, is identical to the peak B-Ec-05 that arises from Escherichia coli [55–57].

IMS chromatogram and related single spectra (inset) of a patient suffering from infections and COPD; from [56]
Summarizing, by coupling an IMS to a MCC for preseparation, it was possible to directly detect volatile metabolites in exhaled air from humans. Total analysis times were less than 10 min. The system is sensitive enough to directly detect metabolites, which are identified using characteristic IMS chromatograms. Results from investigating patients suffering from pneumonia show different peaks in comparison to healthy persons [55–57].
Further investigations using mass spectrometric methods should permit the identification of all of the metabolites found by ion mobility spectrometry. The ability to perform on-site and rapid analysis using air at ambient pressure as the carrier gas are the most important benefits of the technique developed.
Using MCC–IMS directly for clinical diagnosis allows results to be obtained in minutes, and it provides additional information that can be used to determine the therapeutic strategy. Databases of MCC–IMS data can be constructed for several diseases. Thus, these studies could lead to the analysis, via ion mobility spectrometry, of volatile metabolites in human breath as a way to obtain early diagnoses of selected diseases [55–57].
Conclusions
Ion mobility spectrometry offers a range of applications and possibilities for use in field and process analytical applications. Generally, IMS is not very useful for identifying unknown compounds. However, very low detection limits are achievable in a controlled environment, especially when using air as the analyte carrier gas. It has recently been applied to more complex matrices, like natural gas or biological applications like investigations of human breath, where more than 200 different analytes are reported. The complexities of ion-molecule reactions can be managed and IMS should be considered whenever size, weight, power and cost are important factors for analytes analysis in a particular process. A comparison with other analytical methods shows that IMS has a relatively large information density with a comparatively low weight, power and size burden, although there are techniques that give a greater information density (such as mass spectrometry) and there are others which are smaller and more economical in terms of power (such as surface acoustic wave or electrochemical sensors). Detection limits are comparable or better than other technologies. Costs should be comparatively low in theory, although IMS actually costs the same as gas chromatographs configured for the field. IMS detectors are less expensive than mass spectrometers and more expensive than chemical sensors. The specificity of IMS is dependent upon ion source and sample concentration and the linear range can be high due to a combination of drift time and ionization properties (dependent upon the ion source). When unfavourable conditions exist, hyphenated GC–IMS is preferable. In terms of consumption, IMS is superior to mass spectrometry and gas chromatography, since it does not consume gas, does not require a vacuum, and draws relatively little power (batteries are often sufficient). Compared to FT–IR, it has a better detection limit, and it bypasses optical interference and optical windows. The main disadvantage of the surface acoustic wave detector is the high level of cross-sensitivity. In summary, IMS gives a good balance between usable information and convenience, reliability and portability.
References
Baumbach JI, Eiceman GA (1999) Appl Spectrosc 53:338A–355A
Eiceman GA, Karpas Z (1994) Ion mobility spectrometry. CRC, Boca Raton, FL, pp 1–228
Hill HH Jr, Siems WF, St Louis RH, McMinn DG (1990) Anal Chem 62:1201A–1209A
Roehl RE (1991) Appl Spectrosc Rev 26:1–57
Baumbach JI, Schmidt H, Sielemann S, Pilzecker PJ (2004) Process Anal Chem 8:1–20
Vautz W, Sielemann S, Baumbach JI (2004) Int J Ion Mobility Spectrom 7:18–23
Vautz W, Sielemann S, Uhde E, Baumbach JI (2004) Int J Ion Mobility Spectrom 7:30–34
Vautz W, Sielemann S, Baumbach JI (2003) Int J Ion Mobility Spectrom 6:21–29
Ruzsanyi V, Baumbach JI, Eiceman GA (2003) Int J Ion Mobility Spectrom 6:53–57
Li F, Eiceman GA, Ruzsanyi V, Baumbach JI (2003) Int J Ion Mobility Spectrom 6:64–66
Li F, Xie Y, Schmidt H, Sielemann S, Baumbach JI (2002) Spectrosc Spectral Anal (China) 372:606–610
Ruzsanyi V, Sielemann S, Baumbach JI (2002) Int J Ion Mobility Spectrom 5:45–48
Xie Z, Sielemann S, Schmidt H, Li F, Baumbach JI (2002) Anal Bioanal Chem 372:606–610
Soppart O, Pilzecker P, Baumbach JI, Klockow D, Trindade E (2000) IEEE T Dielect El In 7:229–233
Soppart O, Baumbach JI, Trindade E, Alberti SM, Moraes e Silva JM, Klockow D, Peier D (2000) Eur T Electr Power 10:179–184
Baumbach JI, Pilzecker P, Trindade E (1999) Int J Ion Mobility Spectrom 2:35–39
Xie Z, Sielemann S, Schmidt H, Baumbach JI (2001) Int J Ion Mobility Spectrom 4:77–83
Lubman DM (1988) Ion mobility spectrometry with laser-produced ions. In: Report ARO–2244416–CH. Gov Rep Announce Index (U.S.) 88(24) Abstr No 860641
Spangler GE (2000) Field Anal Chem Tech 4:255–267
Wu C, Hill HH Jr, Gamerdinger AP (1998) Field Anal Chem Technol 2:155–161
Spangler EG (2000) Field Anal Chem Technol 4:255–267
Bell SE, Ewing RG, Eiceman GA, Karpas ZJ (1994) Am Soc Mass Spectrom 5:177–185
Wu C, Hill HH, Rasulev UK, Nazarov EG (1999) Anal Chem 71:273–278
Schmidt H, Baumbach JI, Pilzecker P, Klockow D (2000) Int J Ion Mobility Spectrom 3:8–14
Tabrizchi M, Khayamian T, Taj N (2000) Rev Sci Inst 71:2321–2328
Borsdorf H, Rudolph M (2000) Int J Ion Mobility Spectrom 3:1–7
Borsdorf H, Schelhorn H, Rudolph M, Flachowsky J, Stach J (1999) Int J Ion Mobility Spectrom 2:40–44
Teepe M, Kang WJ, Neyer A, Baumbach JI, Schmidt H (2001) Int J Ion Mobility Spectrom 4(2):173–176
Leonhardt JW, Rohrbeck W, Bensch H (2000) Int J Ion Mobility Spectrom 3:43–49
Leonhardt JWJ (1995) Radioanal Nucl Chem 206:333–339
Leonhardt JW, Rohrbeck W, Bensch H (1995) In: Proc Fourth Int Workshop on Ion Mobility Spectrometry, 6–9 August 1995, Cambridge, UK
Sielemann S, Baumbach JI, Pilzecker P, Walendzik G (1999) Int J Ion Mobility Spectrom 2:15–21
Sielemann S, Baumbach JI, Schmidt H, Pilzecker P (2001) Anal Chim Acta 431:293–301
Kang WJ, Teepe M, Neyer A, Baumbach JI, Schmidt H, Sielemann S (2001) Int J Mobility Spectrom 4(2):108–111
Sielemann S, Li F, Schmidt H, Baumbach JI (2001) Int J Ion Mobility Spectrom 4(2):81–84
Vautz W, Ruzsanyi V, Sielemann S, Baumbach JI (2004) Int J Ion Mobility Spectrom 7:3–8
Leasure CS, Fleischer ME, Anderson GK, Eiceman GA (1986) Anal Chem 58:2142–2147
Shumate CB, Hill HH Jr (1989) Anal Chem 61:601–606
Baumbach JI, Pilzecker P, Trindade E (1999) Int J Ion Mobility Spectrom 2:35–39
Borsdorf H, Schelhorn H, Flachowski J, Döring HR, Stach J (2000) Anal Chem Acta 403:235–242
Xu J, Whitten WB, Lewis TA, Ramsey JM (2001) Int J Ion Mobility Spectrom 4(1):3–6
Borsdorf H, Schelhorn H, Flachowsky J, Döring H, Stach J (1999) Int J Ion Mobility Spectrom 2:9–14
Tabrizchi M, Khayamian T (2001) Int J Ion Mobility Spectrom 4:52–56
Tabrizchi M, Abedi A (2001) Int J Ion Mobility Spectrom 4(2):129–131
Tabrizchi M, Abedi A (2002) Int J Mass Spectrom 218:75–85
Hill CA, Thomas CLP (2003) Analyst 128:55–60
Hill C, Thomas P (2002) Int J Ion Mobility Spectrom 5:155–160
Baumbach JI, Soppart O, Pilzecker P, Trindade E, Klockow D (1999) Elektrizitätswirtschaft 3:37–42
Pilzecker P, Baumbach JI, Kurte R, Heise HM (2002) Int J Ion Mobility Spectrom 5:132–137
Walendzik G (2004) Dissertation. Universität Dortmund, Germany
Jauzein M, Baumbach JI, Gutierrez JI, Sardin M (1996) Measurement and modelling of VOC mobility in soils and groundwaters for environmental risk assessment. In: Barton J, Biletewski B, Büsing J, Jackson DV, Jager J, Jauzein M, Kresja P, Ujma KH (eds) Recycling technologies, treatment of waste and contaminated sites (DGXII/D-1). European Commission, Brussels, 15:361–371
Walendzik G, Baumbach JI, Sielemann S, Klockow D (1998) Proc Sixth Int Workshop on Ion Mobility Spectrometry, 10–14 August 1997, Bastei, Germany, p 326
Sielemann S, Teepe M, Vautz W, Baumbach JI (2004) GWF—Gas—Erdgas 145:642–646
Baumbach JI (2001) Uni-Report Universität Dortmund 33:44–48
Baumbach JI, Vautz W, Ruzsanyi V, Freitag L (2005) Metabolites in human breath: ion mobility spectrometers as diagnostic tools for lung diseases. In: Amann A, Smith D (eds) Breath gas analysis for medical diagnostics. World Scientific, Singapore
Baumbach JI, Vautz W, Ruzsanyi V, Freitag L (2005) Metabolites in human breath: ion mobility spectrometers as diagnostic tools for lung diseases. In: Knäblein J (ed) Modern biopharmaceuticals. Wiley–VCH, Weinheim
Ruzsanyi V (2005) Dissertation, Universität Dortmund, Germany
Acknowledgements
Major contributions to common experimental and laboratory work were made by Dr. Li Fang, Peter Pilzecker, Dr. Vera Ruzsanyi, Dr. Stefanie Sielemann, Dr. Gudrun Walendzik, Dr. Wolfgang Vautz and should be mentioned in gratitude. Some of the results discussed above were published partly in the PhD thesis of Dr. Gudrun Walendzik (water contamination) and Dr. Vera Ruzsanyi (breath analysis and metabolites of bacteria), and so they deserve hearty thanks. The co-operation and common experimental work done with G.A.S. Gesellschaft für analytische Sensorsysteme mbH, Dortmund, Germany, in the fields of applications of IMS to gas insulated substations and supervision of the odoration level of natural gas are gratefully acknowledged. Lucia Seifert, Stefanie Güssgen and the mechanical department of the ISAS headed by Hans–Georg Krebs made essential contributions to the results obtained. The co-operation, helpful discussions and support obtained at the Hemer lung hospital, especially from Dr. Lutz Freitag, Dr. Michael Westphal and Dr. Patrick Litterst, should also be mentioned. Additionally, detailed discussions on fundamental questions and application problems with Prof. Dr. Gary Eiceman, New Mexico State University, are gratefully acknowledged. The financial support of the Bundesministerium für Bildung und Forschung and the Ministerium für Wissenschaft und Forschung des Landes Nordrhein-Westfalen and the G.A.S. Gesellschaft für analytische Sensorsysteme mbH, Dortmund, is also gratefully acknowledged.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Baumbach, J.I. Process analysis using ion mobility spectrometry. Anal Bioanal Chem 384, 1059–1070 (2006). https://doi.org/10.1007/s00216-005-3397-8
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00216-005-3397-8