
Abstract
In the presence of carbonate and uranine, the chemiluminescent intensity from the reaction of luminol with hydrogen peroxide was dramatically enhanced in a basic medium. Based on this fact and coupled with the technique of flow-injection analysis, a highly sensitive method was developed for the determination of carbonate with a wide linear range. The method provided the determination of carbonate with a wide linear range of 1.0 × 10−10–5.0 × 10−6 mol L−1 and a low detection limit (S/N = 3) of carbonate of 1.2 × 10−11 mol L−1. The average relative standard deviation for 1.0 × 10−9–9.0 × 10−7 mol L−1 of carbonate was 3.7% (n = 11). Combined with the wet oxidation of potassium persulfate, the method was applied to the simultaneous determination of total inorganic carbon (TIC) and total organic carbon (TOC) in water. The linear ranges for TIC and TOC were 1.2 × 10−6–6.0 × 10−2 mg L−1 and 0.08–30 mg L−1 carbon, respectively. Recoveries of 97.4–106.4% for TIC and 96.0–98.5% for TOC were obtained by adding 5 or 50 mg L−1 of carbon to the water samples. The relative standard deviations (RSDs) were 2.6–4.8% for TIC and 4.6–6.6% for TOC (n = 5). The mechanism of the chemiluminescent reaction was also explored and a reasonable explanation about chemical energy transfer from luminol to uranine was proposed.

Chemiluminescence profiles in batch system. 1, Injection of 100 μL of K2CO3 into 1.0 mL luminol-1.0 mL H2O2 solution; 2-3 and 4-5, Injection in sequence of 100 μL of K2CO3 and 100 μL of uranine into 1.0 ml luminol-1.0 mL H2O2 solution; Cluminol = 1.0 × 10−7 mol/L, CH2O2 = 1.0 × 10−5 mol/L, Curanine = 1.0 × 10−5 mol/L, CK2CO3 = 1.0 × 10−7 mol/L except for 4-5 where CK2CO3 = 1.0 × 10−4 mol/L
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Total inorganic carbon (TIC) and total organic carbon (TOC) are of significance in a variety of fields, particularly in research areas concerning the global carbon cycle, biological availability, chemical transport and interactions in aquatic systems, environmental quality, and pollution control etc. TIC and TOC have been recognized as parameters of “global indicators” [1, 2]. Furthermore, continuous findings have proved that organic matter is a precursor to carcinogenic and toxic disinfection byproducts (DBPs) such as trihalomethanes and haloacetic acids, which can be formed during drinking water disinfection or via a reaction of residual disinfectants with organic matter in natural water [3–5]. This leads to great interest in monitoring low levels of organic matter (TOC < 1 mg L−1 carbon). In addition, high technology industries such as semiconductor, computer, and laboratory applications also need purer water [6]. Therefore, a sensitive, convenient analytical method for the determination of TIC and TOC is needed.
The determination of TIC and TOC are mostly based on the detection of carbon dioxide. For TIC analysis the carbonate is decomposed in samples with acid and for TOC analysis the organic carbon is mineralized via wet or dry combustion, or chemical oxidation [3–8]. High-temperature catalytic combustion (HTC) is often employed and has advantages of greater oxidation efficiency and rapid determination. However, memory effects may arise because of carbon residues depositing on the wall of the HTC column, which may adversely affect the reproducibility and accuracy [3, 6, 7]. Moreover, the instrumentation is rather expensive. A chemical oxidation method is used because of its ease, cheapness, and far-ranging sample applicability despite its major limitation of relatively low oxidation efficiency [4, 5, 8]. Another interesting method developed by Vogl [9] for the determination of dissolved organic carbon (DOC) is inductively coupled plasma isotope dilution mass spectrometry (ICP-IDMS). It is rather expensive in the instrumentation, but capable of on-line measurements.
Various detection techniques have been used for the determination of carbon dioxide, including nondispersive infrared spectrometry (NDIR) [10, 11], thermal conductivity detector (TCD) [12], acid/base titration [13], gravimetry [14], membrane/conductivity [15], and potentiometry [16]. TCD and NDIR detector have high sensitivities. TCD may measure up to 1,000 g L−1 carbon, whereas NDIR detector has a detection limit of ca. 2.0 μg L−1 carbon [4]. However, TCD and NDIR require dedicated equipment. Moreover water, halides, SO2 or SO3, particulates, and other ionic solutions are interferences that can damage the detectors [1, 7]. Although membrane/conductivity and potentiometric techniques are commendable for their detection speed, the sensitivities reported vary largely and some of the techniques usually lack adequate selectivity and stability for the determination [7, 15, 16]. Other techniques, e.g., titration and gravimetry, are not sensitive enough for low level measurements.
The use of flow-injection chemiluminescence (CL) in analysis of organic and inorganic species at trace levels has received considerable attention mainly because of the high sensitivity, the wide dynamic range, the simplicity of the instrumentation, and the rapid and reproducible means of detection. Many environmental pollutants have been successfully determined with this technique [17]. However, to the best of our knowledge, only two works have been reported that are based on the use of flow-injection CL to determine chemical oxygen demand (COD) [18, 19], a parameter similar to TOC indicating the sum of organic compounds, and another to determine CO2 [20]. No CL system has been applied to the determination of TIC or TOC. In the works of Lan et al. [20, 21] and Xiao et al. [22], the enhancement effect of CO2 or \({\text{CO}}_3^{2 - }\) on the CL reaction of H2O2–luminol was investigated in the presence of heavy metal ions, thereby CL methods for the determination of CO2 and Co2+ were developed, respectively. The former method was applied to the determination of CO2 in atmospheric air and human breath. This allowed the determination range of 98–1,570 mg m−3, and a limit of detection of 2.9 mg m−3 [20].
In this paper the effects of both carbonate and uranine on the H2O2–luminol CL reaction in the absence of added heavy metal ions have been studied and it has been found that carbonate might play a crucial role in this chemiluminescence reaction, and uranine could dramatically enhance the reaction to emit more intensive light. Based on this finding and the technique of potassium persulfate oxidation [8], a new method for determining carbon fractions was established. The current method compared with the method developed by Lan et al. [20] for the determination of CO2 is more sensitive and more resistant to foreign interferences. The method has successfully been applied to the determination of TIC and TOC in water. In addition, the mechanism of the CL reaction was explored and a reasonable explanation was proposed.
Experimental
Apparatus
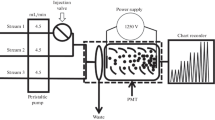
A versatile IFFS-A luminescence analyzer (Remex Electronic Science and Technology Co. Ltd., Xi’an, China) having both flow and static operational modes was employed in this work. The flow-injection manifold finally proposed for the chemiluminescence detection is schematically illustrated in Fig. 1. PTFE tubing (0.8-mm i.d.) was used as connection material in the flow system and a peristaltic pump (Chenghe, Suzhou, China) delivered the reagent and sample solutions. An eight-way injection valve equipped with a 105-μL sample loop was used as an injector. The CL reaction took place in a 3.5-turn spiral-shaped glass flow cell which was located directly facing the window of the photomultiplier tube. CL signals were recorded and processed by a personal computer. In addition, in order to understand the mechanism of the CL reaction, an RF-540 fluorescence spectrophotometer (Shimadzu, Japan) was utilized to obtain the CL emission spectra.

Schematic diagram of the flow-injection chemiluminescence manifold: a sample solution, b H2O2 solution, c uranine and luminol mixed solution, P peristaltic pump, V eight-way valve, FC flow-cell, W waste, PMT photomultiplier, PC personal computer
Reagents
All the chemicals were of analytical reagent grade and carbon-free water was used throughout. The carbon-free water was obtained by freshly redistilling the demineralized deionized water from acidic potassium permanganate prior to degassing further with ultrapure nitrogen (99.999%).
Luminol 0.01 mol L−1 was prepared by dissolving 0.18 g luminol (Sigma, USA) in 100 mL 0.1 mol L−1 sodium hydroxide solution and stored at 4 °C in a refrigerator to avoid exposure to light and air. Hydrogen peroxide 0.10 mol L−1 was prepared by dilution of 5.5 mL 30% (v/v) hydrogen peroxide (Beijing Chemical Reagent Company, Beijing, China) to 500 mL with water. A standard solutions of 0.01 mol L−1 potassium carbonate and 1,000 mg L−1 potassium hydrogen phthalate (KHP) were prepared respectively by dissolving 0.3455 g potassium carbonate (dried at 200 °C) and 0.5320 g KHP (dried at 120 °C, Beijing Chemical Reagent Company, Beijing, China) in 250 mL water. Other stock solutions, including 10−2 mol L−1 uranine (Aldrich, USA), 0.06 mol L−1 potassium persulfate (Beijing Chemical Reagent Company, Beijing, China), and 0.1 mol L−1 potassium hydroxide (Beijing Chemical Reagent Company, Beijing, China), were prepared respectively. The CO2 scrubbing solution consists of 0.1 mol L−1 potassium hydroxide containing 0.3% (v/v) 1-butanol solution. All working solutions were freshly prepared by appropriate dilution of the stock solutions.
Procedures
Sample preparation
The water samples were stored at 4 °C and were analyzed within 1 day after collection. The sample preparation procedure was based on the wet chemical oxidation method described by Fiedler et al. [8]. For the TIC determination, 10 mL of water sample was first acidified to ca. pH 2 using concentrated phosphoric acid, then purged with ultrapure nitrogen, and the CO2 released was absorbed by 50 mL scrubber solution. For the TOC determination, the TIC-free water sample was mixed with 10 mL 0.06 mol L−1 potassium persulfate and 5.0 mL 0.1 mol L−1 potassium hydroxide solution in a stoppered glass bottle. The mixture was boiled under pressure for 30 min. After the oxidation was carried out and the solution cooled down, and the purging and scrubbing procedures were repeated as described above. All the finally scrubbed solutions obtained were used for determination.
Sample detection
The peristaltic pump delivered the reaction solutions (H2O2, luminol–uranine, and sample) at a flow rate 2.8 mL min−1 (per tube). A 105-μL aliquot of the mixture solution of H2O2 and luminol–uranine was injected into the carrier stream (sample) by the injector and then reached the reaction flow cell. The emitted light was monitored by the photomultiplier tube (operated at −800 V). The quantitative determination was based on the net CL intensity \(\Delta I = I_{s} - I_{0} \), where I s is the CL intensity of carbonate sample and I 0 is the intensity of the blank signal.
Result and discussion
Characteristics of the CL reaction
The investigation of the characteristics of the CL reaction was performed in the static mode. A 10-mL glass vial was located directly on the window of the photomutiplier tube of the luminescence analyzer. The CL reagents were injected into the vial in various combinations. Some results of the CL reaction are shown in Fig. 2. It was found that no emission was observed for the injection of H2O2–luminol or H2O2 –uranine–K2CO3 and the combination of H2O2–luminol–uranine gave only weak CL intensity. However, when a small amount of K2CO3 was injected into the mixture of H2O2–luminol, an obviously stronger CL intensity occurred (Fig. 2, 1). Furthermore, if K2CO3 and uranine solution were injected in sequence into the H2O2–luminol solution, a relatively weak CL signal appeared first and then great CL intensity was emitted (Fig. 2, 2 and 3). It is interesting that the CL intensity was greatly dependent on the concentration of K2CO3. However, at a relative high concentration (e.g., 1.0 × 10−4 mol L−1) the CL intensity would be reduced dramatically and even disappeared (Fig. 2, 4 and 5). These results suggest that both K2CO3 and uranine enhance the CL reaction. The mechanism of the reaction will be explained later.

Chemiluminescence profiles in batch system: 1 injection of 100 μL K2CO3 into 1.0 mL luminol and 1.0 mL H2O2 mixed solution; 2,3 and 4,5 injection in sequence of 100 μL K2CO3 and 100 μL uranine into the mixed solution of 1.0 mL luminol and 1.0 mL H2O2. The concentrations of luminol, H2O2, and uranine are 1.0 × 10−7, 1.0 × 10−5, and 1.0 × 10−5 mol L−1, respectively. The concentrations of K2CO3 for peaks 1 to 3 are 1.0 × 10−7 mol L−1 and for peaks 4 and 5 are 1.0 × 10−4 mol L−1
Optimization of the CL system
Effect of enhancers
Five potential CL enhancers, namely, uranine, 2,7-dichlorofluorecein, Rhodamin 6G, Rhodamin B, and 8-hydroxyquinoline, were tested to investigate their effect on the H2O2–luminol–K2CO3 reaction. The results in Table 1 show that most of the compounds added could enhance the emission intensity to some extent except 8-hydroxyquinoline. Uranine itself showed the most significant enhancement effect on the CL reaction. The effect of uranine concentration, varying from 2.0 × 10−7 to 1.0 × 10−5 mol L−1, is shown in Fig. 3. It can be seen that 4.0 × 10−6 mol L−1 uranine provided the maximum CL intensity. Higher concentrations caused the signal to decrease due to its increasing inner filter effect. Hence, 4.0 × 10−6 mol L−1 uranine solution was used for further investigations.

Effect of concentration of uranine on the CL intensity. The concentrations of luminol, H2O2, and K2CO3 are 3.0 × 10−5, 9.0 × 10−7, and 1.0 × 10−8 mol L−1, respectively. The flow rate of each flow line is 2.4 mL min−1
pH of the medium
Since the luminol oxidation CL reaction is usually carried out under alkaline conditions, the effect of potassium hydroxide concentration in the range 1.0 × 10−4 –0.2 mol L−1 was examined and the result showed that 0.1 mol L−1 potassium hydroxide gave the strongest light intensity and the highest signal-to-noise ratio (S/N). Hence, a 0.1 mol L−1 potassium hydroxide solution was chosen.
Effect of luminol concentration
The effect of luminol concentration on CL intensity was investigated over the range 1.0 × 10−6–8.0 × 10−5 mol L−1. As shown in Fig. 4, the CL intensity increased with the increase of luminol concentration up to 4.0 × 10−5 mol L−1 and then decreased steadily with a continuous increase of the concentration. Hence, the 4.0 × 10−5 mol L−1 luminol solution was used throughout the research.

Effect of luminol concentration on the CL intensity. The concentration of uranine is 4.0 × 10−6 mol L−1. Other conditions are the same as in Fig. 3 except the luminol concentration
Effect of hydrogen peroxide concentration
The effect of hydrogen peroxide concentration on the CL intensity was investigated in the range 1.0 × 10−7–5.0 × 10−6 mol L−1. The results (Fig. 5) showed that at lower concentrations the CL intensity increased obviously with the increase of the hydrogen peroxide concentration, and the maximum CL intensity was obtained at 9.0 × 10−7 mol L−1. Above this concentration, the CL intensity decreased gradually. For the subsequent research, 1.0 × 10−6 mol L−1 hydrogen peroxide was chosen.

Effect of H2O2 concentration on the CL intensity. The concentration of luminol is 4.0 × 10−5 mol L−1. Other conditions are the same as in Fig. 4 except the concentration of H2O2
Parameters of the flow system
Both normal and reverse inject mode have been utilized in the investigation and the reverse one proved to be more effective for the CL reaction in addition to having the advantage of lower reagents consumption. Three key parameters, reagents flow rate, the distance from the injector to the flow cell, and the injection volume of the solution were examined. A 2.8 mL min−1 flow rate for each line, 75 mm for the injector–flow cell distance, and 105 μL for the injection volume were finally chosen as a compromise between the signal-to-noise ratio and the stability of the CL intensity.
Figures of merit
For the purpose of achieving a high determining accuracy, the concentration of CO3 2− (mol L−1) versus the relative CL intensity (mV) curve obtained under the selected optimum conditions was linearized in the range from 1.0 × 10−10 to 5.0 × 10−6 mol L−1, equivalent to TIC from 1.2 × 10−6 to 6.0 × 10−2 mg L−1 carbon. The limit of detection (LOD), based on a signal-to-noise ratio of 3, was calculated as 1.2 × 10−11 mol L−1, which is equivalent to 1.4 × 10−10 g L−1 carbon or 5.1 × 10−10 g L−1 CO2. The relative standard deviations (RSDs) at 1.0 × 10−9 and 9.0 × 10−7 mol L−1 CO3 2− for 11 determinations were 5.1% and 2.3%, respectively, and the average RSD was 3.7%. A typical CL signal versus c profile is shown in Fig. 6. Note that a perceptible blank signal was always observed in the determining procedure because utterly carbon-free water is unavailable. Therefore, as a corrective factor the blank value should be determined regularly.

CL signals with various concentrations of standard K2CO3 solution: 1 blank (scrubber solution), 2 9.0 × 10−7 mol L−1, 3 1.8 × 10−6 mol L−1, 4 2.7 × 10−6 mol L−1, 5 3.6 × 10−6 mol L−1. The concentrations of luminol, H2O2, and uranine are 4.0 × 10−5, 1.0 × 10−6, and 4.0 × 10−6 mol L−1, respectively. The flow rate of each flow line is 2.4 mL min−1
Compared with the method developed by Lan et al. [20], which provided a determination range of ca. 98–1,570 mg m−3 and a limit of detection of 2.9 mg m−3 for CO2, our method promises a wider linear range for the determination and is more sensitive.
Interference study
As is well known, all the CL reactions of the luminol–H2O2 type are greatly influenced by the presence of some heavy metal ions such as Fe2+, Co2+, Cu2+, and Cr3+. In this investigation, main interferences only were purgeable volatile chemicals assumed to be mainly inorganic compounds, because the sample was pre-treated with a purge and scrub process and metal ions were excluded from the scrubbing solution. So, the interferences of some common inorganic gases were examined in their corresponding ion forms in aqueous solution. The criterion for interference was a relative error of less than ±5% of the average CL signal taken for a K2CO3 concentration corresponding to 1.0 × 10−9 mol L−1. No interference was observed for 1.0 × 10−6 mol L−1 S2− (H2S), 5.0 × 10−6 mol L−1 SO3 2− (SO2), 8.0 × 10−5 mol L−1 NO2 − (NO), and for 0.5 mol L−1 SO4 2− (SO3), NH4 + (NH3), F− (HF), Cl− (HCl), NO3 − (NO2), respectively.
Determinations of TIC and TOC in water samples
Our method was applied to the determination of TIC and TOC in water. The sample preparation was described previously and the scrubbed solutions were determined directly (or after dilution). To validate the reliability of the method, sample recoveries for TIC and TOC were carried out by quantitatively adding standard solutions of potassium carbonate and KHP, respectively, to the sample. Because the determination of TOC was based on the assumption that all organic chemicals present in the water sample were oxidized to the same degree as that of KHP, the oxidization conditions must be examined and optimized. Using standard KHP solutions of 0.020–20.0 mg L−1 carbon as samples, it was found that the recoveries of KHP strongly depend on the concentrations of potassium persulfate and KHP (Fig. 7). The higher concentrations of potassium persulfate and KHP gave higher recoveries, whereas at a low concentration, KHP seemed difficult to be oxidized. When the concentration of potassium persulfate was 0.06 mol L−1, the recoveries for 0.08 and 20.0 mg L−1 carbon of KHP reached the maximum values of 94.5% and 99.3%, respectively. So, under the optimized conditions, a corrected calibration curve for the TOC determination was reconstructed with the standard KHP solutions as \(\Delta I = 222.8{\text{TOC}} + 238.7\) (TOC: ca. 0.08–30 mg L−1 carbon, r 2 = 0.9967). Note that here the calibration was carried out under the condition that the scrubbed solution was diluted 100 times so as to meet with the linear ranges of the CL.

Effect of K2S2O8 concentration on the oxidation of KHP: 10 mL KHP solution was mixed with 10 mL K2S2O8 and 5 mL 0.1 mol −1 KOH, and the mixture was boiled for 30 min
Table 2 shows the average values on the determinations of TIC and TOC for different water samples. It can be seen that good recoveries from 97.4 to 106.4% for TIC and from 96.0 to 98.5% for TOC were obtained. The relative standard deviations (RSDs) were in the range ca. 2.6–6.6% (n = 5). Hence, the proposed method was applicable to the determination of TIC and TOC in water.
Mechanism exploration
The mechanism of the luminol (LH2) CL reaction is complicated and its details are still poorly understood. It is usually considered to involve a radical reaction, and the emitting species is believed to be the excited intermediate 3-aminophthalic acid (AP2−*). Some heavy metal ions, peroxidases, and certain fluorescent dyes like fluorescein (FH2) are often used to accelerate the reaction for light emission in an alkaline buffer [17, 23]. Factors such as pH of the medium, dissolved oxygen [24], and carbon dioxide [20–22] also affect the reaction remarkably.
In order to explain the enhancement effect of carbon dioxide, Xiao et al. [22] suggested that it may be related to the formation of peroxycarbonate radicals (\( \cdot {\text{CO}}^{ - }_{4} \)), a more effective oxidant for the oxidation of luminol. Lan et al. [21] proposed that H2O2 formed from dissolved O2 may react with CO2 to produce carbonate radicals (\( \cdot {\text{CO}}^{ - }_{3} \)), which are responsible for the enhancing effect owing to their selective oxidation of luminol to afford luminol radical (\( \cdot {\text{L}}^{ - } \)). A similar mechanism was also suggested by Yeh et al. [25]. Mechanisms mentioned above all deal with carbon dioxide in the form of dissolved CO2 (g) rather than \({\text{CO}}^{{2 - }}_{3} \) or \({\text{HCO}}^{ - }_{3} \) which are evidently predominant in a strongly basic medium. Moreover, heavy metal ions are involved in the mechanisms and their effect should be taken into account. Thus, these mechanisms inevitably cannot guarantee a satisfactory interpretation of the CL systems used nor the results of our current investigation.
As the enhancement of luminol CL by fluorescein is concerned, although a mechanism of physical energy transfer was proposed [26, 27], Haas et al. [28] proved that simple physical energy transfer cannot occur between AP2−* (425 nm) and FH2 (535 nm), and FH2 interacts with the CL light-emitting processes likely via the formation of a fluorescein radical (·F−) species. As a result, a chemical energy transfer mechanism was assigned to CL the reaction.
In this work, characteristics of the effect of K2CO3 and the enhancement of uranine (F2−) on the LH2–H2O2 CL reaction have been elaborately carried out in the static experiments in the absence of heavy metal ions or peroxidases. Because no emission was detected from LH2–H2O2 or H2O2–F2−–K2CO3, whereas a relatively weak emission from LH2–H2O2 in the presence of an appropriate amount of K2CO3 and strong emissions were elicited in the presence of both K2CO3 and F2−, we can infer that the CL reaction may be triggered by interaction between \({\text{CO}}^{{2 - }}_{3} \) and H2O2 and then enhanced by F2−. In order to confirm this assumption, emission spectra of the CL reactions were studied further (Fig. 8). The maximum CL intensity of the LH2–H2O2–K2CO3 (Fig. 8, 1) was at a wavelength of 425 nm, which is obviously the emission of AP2−*. And for the LH2–H2O2– F2−–K2CO3 system (Fig. 8, 2 and 3), two emission maxima, one around 415 nm and another around 535 nm, appeared. This result was quite similar to that of the LH2–H2O2–F2−–horseradish peroxidases CL system [23] and suggested an energy transfer mechanism involving transfer of energy from LH2 (donor) to F2− (acceptor). The shift of the maximum emission wavelength of LH2 from 425 nm to 415 nm was the result of spectrum absorption of F2− due to the overlap between the spectra of the LH2 CL emission and the F2− adsorption [23]. Moreover, by comparing the spectrum of Fig. 8 parts 1 and 2, which were performed at the same concentration of K2CO3 (1.0 × 10−7 mol L−1) in the absence or presence of F2−, respectively, the CL intensity at 415 nm for the LH2–H2O2–F2−–K2CO3 become notably weaker than that at 425 nm for the LH2–H2O2–K2CO3 system. This result points again to the role of \({\text{CO}}^{{2 - }}_{3} \) in producing active species necessary for the CL reaction and a process of energy transfer from LH2 to F2−. Finally, we also found that at a relatively high concentration of K2CO3 above 5.0 × 10−6 mol L−1, the CL intensity decreased evidently with the increase of the concentration level, even near zero at 0.1 mol L−1. This can be ascribed to the increasing scavenging active of \({\text{CO}}^{{2 - }}_{3} \) at higher concentration level that quenches the CL reaction [21].

Spectra of the chemiluminescent emissions: 1 1.0 × 10−3 mol L−1 luminol+1.0 × 10−4 mol L−1 H2O2+1.0 × 10−7 mol L−1 K2CO3; 2 1.0 × 10−3 mol L−1 luminol+1.0 × 10−4 mol L−1 H2O2+5.0 × 10−3 mol L−1 uranine+1.0 × 10−7 mol L−1 K2CO3; 3 1.0 × 10−3 mol L−1 luminol + 1.0 × 10−4 mol L−1 H2O2 + 5.0 × 10−3 mol L−1 uranine + 4.0 × 10−7 mol L−1 K2CO3
By taking into consideration previous studies and the observations collected in this work, the following mechanism can be postulated:
-
i.
Formation of carbonate radicals\( \cdot {\text{CO}}^{ - }_{3} \):
$${\text{H}}_{{\text{2}}} {\text{O}}_{2} + {\text{OH}}^{ - } \to {\text{HO}}^{ - }_{2} + {\text{H}}_{2} {\text{O}}$$(1)$$ {\text{CO}}^{{2 - }}_{3} + {\text{H}}_{2} {\text{O}}\, \rightleftarrows {\text{HCO}}^{ - }_{3} + {\text{OH}}^{ - } $$(2)$${\text{HCO}}^{ - }_{3} + {\text{HO}}^{ - }_{2} \to {\text{HOO}} - {\text{COO}}^{ - } + {\text{OH}}^{ - } $$(3)$$2\,{\text{HOO}} - {\text{COO}}^{ - } \to ^{ - } {\text{OOC}} - {\text{OO}} - {\text{COO}}^{ - } \to 2\, \cdot {\text{CO}}^{{ - {\left[ {21} \right]}}}_{{\text{3}}} $$(4) -
ii.
If \({\text{CO}}^{{2 - }}_{3} \) is present but uranine is absent in the system:
$${\text{LH}}_{2} + 2\,{\text{OH}}^{ - } \to {\text{L}}^{{2 - }} + 2\,{\text{H}}_{2} {\text{O}}$$(5)$$ {\text{L}}^{{2 - }} + \cdot {\text{CO}}_{{\text{3}}} ^{ - } \to \cdot {\text{L}}^{ - } + {\text{CO}}_{{\text{3}}} ^{{2 - }} $$(6)$$ \cdot {\text{L}}^{ - } + 2\,{\text{H}}_{2} {\text{O}}_{2} \to {\text{L}} + \cdot {\text{O}}^{ - }_{2} + 2\,{\text{H}}_{2} {\text{O}}$$(7)$${\text{L}} + {\text{HO}}^{ - }_{2} + {\text{OH}}^{ - } \to {\text{LO}}^{{2 - }}_{2} + {\text{H}}_{2} {\text{O}}$$(8)$$ \cdot {\text{L}}^{ - } + \cdot {\text{O}}^{ - }_{2} \to {\text{LO}}^{{2 - }}_{2} $$(9)$${\text{LO}}^{{2 - }}_{2} \to {\text{AP}}^{{2 - * }} + {\text{N}}_{2} $$(10)$${\text{AP}}^{{2 - * }} \to {\text{AP}}^{{2 - }} + {\text{hv}}\,{\left( {425\,{\text{nm}}} \right)}$$(11) -
iii.
If both \({\text{CO}}^{{2 - }}_{3} \) and uranine are present in the system, in addition to the reactions of 1–11 described above a chemical energy transfer process occurs:
$$ {\text{F}}^{{2 - }} + \cdot {\text{CO}}^{ - }_{3} \to \cdot {\text{F}}^{ - } + {\text{CO}}^{{2 - }}_{3} $$(12)$${\text{LO}}^{{2 - }}_{2} + \cdot {\text{F}}^{ - } \to \cdot {\text{L}}^{ - } + \cdot {\text{FO}}^{{2 - }}_{2} $$(13)$${\text{FO}}^{{{\text{2}} - }}_{{\text{2}}} \to {\text{F}}^{{2 - * }} + {\text{O}}_{2} $$(14)$${\text{F}}^{{2 - * }} \to {\text{F}}^{{2 - }} + {\text{hv}}{\left( {536\,{\text{nm}}} \right)}$$(15)
where L is diazaquinone, (\({\text{LO}}^{{2 - }}_{2} \)) is α-hydroxyhydroperoxide, \({\text{FO}}^{{2 - }}_{2} \) is fluorescein endoperoxide anion, and the other symbols are as described in the text.
Conclusions
This study has demonstrated that \({\text{CO}}^{{2 - }}_{3} \), at a relatively low concentration level, plays a significant role in the CL reaction of the luminol–H2O2 system such that a highly sensitive approach for determining carbon fractions can be achieved with the strongly synergistic enhancement effect of uranine. The developed method promises a wide linear detection range spanning 4 orders of magnitude and a low limit of detection 1.2 × 10−11 mol L−1 for determination of \({\text{CO}}^{{2 - }}_{3} \). The method is precise, accurate, and resistant to foreign interferences. The successful application to the determination of TIC and TOC in water confirmed its usefulness.
Although the method has some other advantages like simplicity and high selectivity, it suffered from incomplete oxidation in the sample preparation procedure used when very low TOC concentration (<0.08 mg L−1 carbon) was determined. Hence, the method of the sample preparation, especially for more complex samples, should be improved further if lower TOC determination is needed.
Based on the previous investigations and the present exploration, a more reasonable mechanism was proposed for the CL reaction, which is probably triggered by the interaction of H2O2 and K2CO3 and then enhanced by uranine via chemical energy transfer from luminol to uranine.
To sum up, the potential of the CL system is apparent and the established method is a promising alternative approach for the determination of CO2 in gases or total carbon (TC) in waters.
References
Bisutti I, Hilke I, Raessler M (2004) Trends Anal Chem 23:716–726
Visco G, Campanella L, Nobili V (2005) Microchem J 79:185–191
Aiken G, Kaplan LA, Weishaar J (2002) J Environ Monit 4:70–74
Wallace B, Purcell M, Furlong J (2002) J Environ Monit 4:35–42
Urbansky ET (2001) J Environ Monit 3:102–112
Fung YS, Wu Z, Dao KL (1996) Anal Chem 68:2186–2190
Qian J, Mopper K (1996) Anal Chem 68:3090–3097
Fiedler U, Hansen EH, Ruzicka J (1975) Anal Chim Acta 74:423–435
Vogl J, Heumann KG (1998) Anal Chem 70:2038–2043
Paniz JNG, Flores EMM, Dressler VL, Martins AF (2001) Anal Chim Acta 445:139–144
Peter BA, Pardue HL (1992) Anal Chem 64:200–204
Liu SJ, Tubino M (1998) Talanta 47:711–717
Neal RH, Younglove T (1993) Commun Soil Sci Plant Anal 24:2733–2746
Rabenhorst MC (1988) Soil Sci Soc Am J 52:965–969
Kuck LR, Godec RD, Kosenka PP, Birks JW (1998) Anal Chem 70:4678–4682
Choi YS, Lvova L, Shin JH, Oh SH, Lee CS, Kim BH, Cha GS, Nam H (2002) Anal Chem 74:2435–2440
Lin JM (2004) Chemiluminescence-basic principle and applications. Chemical Industry Press, Beijing, pp 33–54
Hu Y, Yang Z (2004) Talanta 63:521–526
Li B, Zhang Z, Wang J, Xu C (2003) Talanta 61:651–658
Lan ZH, Aottola HA (1996) Anal Chim Acta 329:305–310
Lan ZH, Aottola HA (1996) Analyst 121:211–218
Xiao C, Palmer DA, Wesolowski DJ, Lovitz SB, King WD (2002) Anal Chem 74:2210–2216
Diaz AN, Garcia JAG, Lovillo J (1997) J Biolumin Chemilumin 12:199–205
Rose AL, Waite TD (2001) Anal Chem 73:5909–5920
Yeh HC, Lin WY (2003) Talanta 59:1029–1038
Hadjianestis J, Nikokavouras J (1993) J Photochem Photobiol A 69:337–343
Voicescu M, Vasilescu M, Meghea A (2000) J Fluoresc 10:229–236
Haas Y, Wurzberg E (1979) J Phys Chem 83:2692–2696
Acknowledgements
The authors gratefully acknowledge financial support of the National Natural Science Foundation of China (Nos. 20437020, 20575008) and Major Research Program of Chinese Academy of Sciences (KZCX3-SW-432).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Fan, SL., Qu, F., Zhao, L. et al. Flow-injection analysis for the determination of total inorganic carbon and total organic carbon in water using the H2O2–luminol–uranine chemiluminescent reaction. Anal Bioanal Chem 386, 2175–2182 (2006). https://doi.org/10.1007/s00216-006-0853-z
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00216-006-0853-z