
Abstract
A luminol chemiluminescence (CL) detection/flow injection analysis technique coupled with ion chromatography (IC) has been employed for the determination of low levels of Cu(II) and Co(II) in drinking water samples. The detection system was the CL of luminol/perborate or luminol/percarbonate in alkaline medium catalyzed by these transition metals. Oxalic acid in a solution of KOH and N(CH3)4OH was used as an eluent in the IC to improve the column selectivity (Dionex CS5A). Concentration and pH of the eluent affected simultaneously the CL intensity and the retention times (t R). Under the elution conditions used here, the retention times of both metal ions were much greater when the concentration of oxalic acid was decreased. Thus, R t(Cu) = 2.15 min and t R(Co) = 4.50 min were measured at 80 mM oxalic acid concentration, while t R raised to 4.12 and 18 min for Cu(II) and Co(II), respectively, using a 10-mM concentration, but on the other hand, the CL signals showed substantially higher values when the concentration of oxalic acid was lesser in the eluent. An optimum oxalic acid concentration of 20 mM and an eluent pH = 4.7 were selected in order to have reproducible signals with a total analysis time of 10 min. The optimum flow rate for the mobile phase was 1.5 mL min−1. The concentration and pH of the postcolumn reagents also affected the CL signal, obtaining optimum concentrations of 5 mM for both oxidants (perborate or percarbonate) and luminol, this last dissolved in a 0.1-M borate buffer at pH 12. The optimum flow rate for the postcolumn reagents was 1 mL min−1. Linear calibrations for both transition metal ions were established, with calculated detection limits of 0.15 ng mL−1 for Co(II) and 0.20 μg mL−1 for Cu(II). Others ions commonly present in natural waters showed little or no interference. The method was successfully applied to water samples spiked with Cu(II) and Co(II), obtaining recoveries in the range of 85–128%, depending on the metal concentrations.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
The determination of trace metals in the environment is of interest in many fields, including the environmental health, biochemical and geochemical studies (Dulaquais et al. 2014). Cobalt, for instance, is required as nutrient for many algae including diatoms, chrysophytes and dinoflagellates and in growth enhancement of some terrestrial plants at low concentrations. However, at higher concentrations, cobalt is toxic to terrestrial and aquatic animals and plants; in humans, the incidence of goiter is higher in regions containing increased levels of Co in water and soils (Yamane et al. 1998). On the other hand, the determination of the cationic copper is a good tool for environmental monitoring studies, and the removal of this metal from aqueous medium such as effluents is of great interest for health purposes (Lemos et al. 2006).
Many analytical procedures are available for the determination of Cu and Co at trace or ultra-trace levels, for example, the atomic absorption spectrometry, the inductively coupled plasma, the atomic emission spectroscopy and the amperometric techniques. However, sophisticated and high-cost equipments are required, limiting their use in most laboratories, making necessary the development of simple, sensitive, accurate and selective techniques, which do not use expensive devices (Ahmed and Uddin 2007).
Flow injection analysis with chemiluminescence detection has been successfully employed for the analysis of metals in several matrices, achieving very low detection limits without preconcentration steps (Issam et al. 2015; Li et al. 2006). Luminol chemiluminescence reaction with hydrogen peroxide is the most used, owing to its ease of use, high sensitivity, simplicity of the instrumentation and because the CL emission is strongly enhanced by transition metal cations by a mechanism of catalysis, which lies with the activation of the step where the HO· radicals are derived from the hydrogen peroxide (Issam et al. 2015; Roda and Guardigli 2012; Correa and Vitorino 2002); these radicals are highly reactive accelerating the production of the excited intermediary.
The luminol CL has the advantage of the use of simple and low-cost equipments and reagents, but it lacks of selectivity. This limitation can be overcome by the coupling of the ionic chromatography to the CL detection (Murillo et al. 2013; Gammelgaard et al. 1997). With this system, Chen et al. (2012) were able to carry out the speciation of ferrous and ferric iron in water.
In its most simple form, the IC consists of a chromatographic column connected to the CL detector, and through an injection valve in a pneumatic system, the sample is introduced into the column head and the mixture of eluent–sample is moved along the column by using a pump. This low-cost system requires few inputs and is relatively free of maintenance.
In this work, the influences of different experimental conditions relative to the chromatographic separation and the CL emission were studied for the simultaneous determination of divalent copper and cobalt ions in environmental samples (water, soil and plants), mainly the effects of the oxalic acid concentration and pH on the chromatographic separation and CL signal. The luminol pH and the use of sodium perborate or sodium percarbonate as alternative oxidants to the usual hydrogen peroxide were also evaluated, because scarce information exists in the literature about the use of these compounds as oxidants in the develop of analytical methods based on the luminol chemiluminescence.
Materials and methods
Reagents
All chemicals were of analytical grade and used without further purification. Water was obtained from a Milli-Q purification system. Copper nitrate and cobalt nitrate were obtained from Merck. Luminol was obtained from Fluka. Sodium perborate, sodium percarbonate and sodium oxalate were obtained from Sigma-Aldrich. Nitric acid and hydrochloric acid were supplied by Panreac. Stock solutions of 250 μg mL−1 Cu(II) and Co(II) were prepared by dissolving their nitrates in water. These stock solutions were diluted in the ultra-pure water to prepare working standard solutions containing both ions. Luminol (5 mM) was dissolved in a 0.1-M borate buffer solution at pH 11 and stored for 24 h before use. Aqueous solutions of sodium perborate or sodium percarbonate were daily prepared.
Samples
River and sea water samples were collected at Alicante coast, Spain. The samples were filtered and acidified with 0.1 mol L−1 nitric acid and stored in dark bottles. The drinking water sample was obtained from the local market.
The certified reference materials Montana Soil NIST 2711 (National Institute of Standards, USA), CRM-Soil 020-050 (RT Corp, USA) and GBW07602 “bush branches and leaves” (National Research Center for Certified Reference Materials of China) were used to validate the analytical method.
Equipment
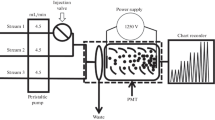
The IC-CL system (Fig. 1) consisted of a peristaltic pump (Gilson Minipuls 3) for postcolumn reagents (luminol in borate buffer solution at pH 12, and perborate or percarbonate), which were carried at a rate of 1.0 mL min−1, and a simplified ionic chromatograph system, which consisted in polyethylene bottle which contained the eluent under N2 pressure at 0.6 bar. The eluent flows to the pump, and then, it is pumped to the injection valve, where a sample is loaded in a fixed volume loop (25 μL) and injected into the chromatography stream. The system is provided with a Dionex Ion Pac CG5A guard column (50 mm × 4 mm i.d., Dionex, Sunnyvale, CA, USA) and an Ion Pac CS5A analytical column (250 mm × 4 mm i.d., 9-μm bead diameter, ethylvinylbenzene functionalized with both quaternary ammonium and sulfonate functional groups, Dionex, Sunnyvale, CA, USA). The CL detector was a Camspec Chemiluminescence Detector CL-2 (photosensor module Hamamatsu 45773-20 spectral response from 300 to 900 nm; spiral-type flow cell, volume 120 μL; Sawston, Cambridge). The detector was connected to a computer by a digital analogical converter; data were acquired using the Clarity version 2.4.1.77 (Data s) software to integrate chromatographic peaks in order to have the peak area under the CL response signal.

Maniford of the IC-CL coupling for the CL evaluation of Cu(II) and Co(II)
Four eluent solutions with oxalic acid concentrations between 10 and 80 mM were prepared in order to evaluate ligand effect in terms of retention time and peak area. Solutions that contained simultaneously copper and cobalt were injected in the chromatographic system, by peak integration areas that were calculated along with peak width and retention times. Once the optimum H2C2O4 concentration was determined, the maximum column efficiency was established by calculating the number of theoretical plates (N) and the equivalent height of a theoretical plate (HETP) at the following mobile-phase velocities: 1.2, 1.5, 1.8 and 2.1 mL min−1.
Optimization of CL emission
The pH of the luminol solution has been identified as a key factor in the CL reaction; for that reason, 5 mM luminol solutions buffered at pH 10.5, 11.0, 12.0 and 12.5 were prepared and mixed with 5 mM solutions of the oxidants. Both reagents have to be mixed before entering the detector; therefore, the rate of mixing is another factor that influences the intensity of the chemiluminescence emission. In this experiment, mixing rates of 0.6, 0.7, 1.0, 1.2 and 1.4 mL min−1 were tested.
Effect of foreign ions
A study of potential interferences in the determination of copper and cobalt was performed. Solutions containing Cu 2+ (1.2 μg mL−1) and Co2+ (0.12 μg mL−1) and other ions such as Na+, Ca2+, Fe3+, Mg2+, CO3 2−, NO3 −, SO4 2−, Cl− and PO4 3− were tested under the optimized conditions for the IC-CL system, and the maximum tolerance allowed in the peak area in the presence of the interferences was ±3σ of the peak area of the pure Cu or Co solutions.
Determination of Cu2+ and Co2+ in environmental samples
Water samples were previously analyzed to determine their actual concentration of both metals. Then, in order to evaluate the recoveries, the samples were spiked with known amounts of Cu(II) and Co(II) from their nitrates.
For the dissolution of the Cu(II) and Co(II) ions in the soil and plant certified materials, digestion procedures in closed systems (Parr bombs) with a microwave heating were applied. Briefly, 100 ± 1 mg of each soil or 250 mg of the plant material was weighed into the microwave transparent body of the Parr 4782 bomb. For the mineralization of the plant sample, 1 mL of the (1:1) HNO3–H2O2 was added, while for the soil samples, 1.0 mL of aqua regia was employed. The system was closed tightly by hand and after placed in a microwave oven (Samsung, Japan) to be irradiated for 20 s at full power (800 W). Once the bomb was cool, the vessel was open and 10 mL of water was added. Finally, the resulting solution was filtered through a Whatman No. 1 filter paper into a 50-mL volumetric flask and made up to volume with Milli-Q water. Blank and standard solutions containing the Cu(II) and Co(II) ions were digested in the same way to obtain the calibration curves.
The flame atomic absorption spectrometry was also applied for the determination of Cu2+ in water as an alternate method to evaluate the proposed IC-CL procedure. A Varian spectrophotometer model Spectra 300 provided with a Cu hollow cathode lamp was employed.
Results and discussion
Operational conditions
Effect of oxalic acid concentration and pH on CL signal of luminol oxidation by sodium perborate catalyzed by Cu(II) and Co(II)
Figure 2 shows that the increasing oxalic acid concentration leads to a decrease in the CL emission for both metals, owing to the lesser concentration of the free ions in the aqueous solution, because oxalic acid tends to form chelates with these ions, resulting in an inhibition of their catalytic effect on the luminol oxidation. Similar results have been reported for other ligands such as tartrate, citrate, salicylate and phthalate Lu et al. (2003). Also, Kim et al. (2013) found that the complexes of Cu(II) with chelating reagents lost their catalytic activity due to the chelating reagents acting as masking agents.

Effect of pH and concentration of oxalic acid on the luminol chemiluminescence
On the other hand, the resulting pH after the mixing of reagents depends on the pH of the buffered luminol solution as well as the acidity of the oxalic acid solution in which the metallic cations are dissolved. Badocco et al. (2007) have found that the optimum pH to produce the maximum CL emission is around 10. When the luminol pH is buffered at 11.5 and the pH of the oxalic acid solution was fixed at 5.0, the maximum CL intensity could be obtained (Fig. 2), because at these conditions, the resulting pH after the mixing was 10.2, very close to the optimum.
Effect of perborate or percarbonate concentration on CL signal
Luminol was oxidized by perborate and percarbonate producing the excited phthalate intermediate responsible for the CL emission. Results showed no differences between both oxidants with a maximum intensity at 10 mM. This concentration was employed in further experiments.
Optimization of IC separation of Cu2+ and Co2+: oxalic acid concentration
For the simultaneous determination of metallic cations, the eluent solution must contain complexing agents to improve the column selectivity. The concentration of oxalic acid in the eluent affected the retention times as well as the CL response in terms of peak area. At an oxalic acid concentration of 80 mmol L−1, the solute migration was faster, with retention times of 1.6 and 3.06 min for Cu2+ and Co2+, respectively. The presence of oxalate ions in the eluent favors the formation of soluble complexes, reducing, at the same time, the adsorption of the analytes by the stationary phase. Although this resulted in shorter run times, the CL emission also diminishes with negative effects on the sensibility of the proposed method. The maximum peak areas were produced when the chromatographic separation was carried out using the most diluted eluent (10 mmol L−1) and, in this case, the free metallic cations could be more retained by the column, which explains the increase in the retention times.
The optimum concentration of the ligand was selected as a compromise between sensibility and time needed to complete the analysis, in such way a concentration of 20 mM was selected as the most convenient, with a run time of 12 min.
Mobile-phase velocity
Increasing the flow rate of the mobile phase, a reduction in the retention time was observed for both cations. While copper emerges from the column within 1.8 min with a flow of 1.2 mL min−1, only 1.2 min are needed using a flow of 2.1 mL min−1. The effect was more evident for the cobalt ions; the retention times experienced a significant reduction from 5.3 min to 3.3 at the highest flow rate.
The column efficiency, expressed as the equivalent height of a theoretical plate (HETP), was determined using the Van Deemter equation. Retention times and peaks width were used to obtain the HETP values, which were then plotted against the mobile-phase velocity giving the minimum value for HETP around 1.8 mL min−1, corresponding to the optimum velocity for Cu and Co separation.
Optimization of the CL emission: effect of the pH on the luminol-oxidant reaction
Under the conditions described above, the solutions of luminol and perborate o percarbonate have to be mixed with the emerging mobile phase buffered at pH 5.0 and final pH in the mixture cannot be easily predicted. For that reason, solutions of luminol buffered at several pH values were prepared using a borate buffer to adjust the final pH as closer as possible to the optimum of 10.
Experimental results showed that the maximum peak areas for the Cu2+ or Co2+ catalysis occur in the solutions buffered at pH 12. Also, the CL emission seems not to be dependent on the type of oxidant employed, because chromatographic peaks obtained using either perborate or percarbonate are similar, indicating that the oxidation mechanism would be the same for both oxidants.
Luminol-oxidant flow rate
The rate of luminol and perborate mixing also affected the CL emission. The maximum peak area was obtained with a rate of 1 mL min−1. At lesser velocities, the reaction may start before reaching the cell inside the detector device. At higher velocities, the time of residence would not be enough to achieve the maximum intensity. It has been reported that several factors, including the way in which reagents are mixed, may modify the detector response, as a consequence of kinetic factors that modify the concentrations of the species involved in the intensity and duration of the chemiluminescence (Xiao et al. 2000, Burguera et al. 1981).
Calibration curves and analytical parameters
The optimum operational parameters for the coupling IC-CL are given in Table 1. In such conditions, well-resolved peaks were obtained (Fig. 3), which by integration of the areas give linear responses for both ions in the concentrations intervals of 0.25–2.5 ng mL−1 for Co2+ and 0.20–1.4 µg mL−1 for Cu2+, indicating that cobalt ions have a major catalytic effect on luminal decomposition (Gao et al. 2015).

Typical ionic chromatogram for the separation of Cu(II) and Co(II) at the optimum operative conditions
Linear equation regression of the type A = (b ± S b ) [Cation] + (a ± S a ) was found (Table 2). In this general equation, A represents the peak area, a and b are the slope and intercept of the calibration plot, while S a and S b are the respective standard deviations. The calibration features given in Table 2 suggested that these equations are adequate for the quantification of both metals under the optimized conditions.
The detection and quantification limits were calculated by the root mean square error (RMSE) method. According to Corley (2002) in dynamic systems such as chromatography, if a linear relationship exists between the detector response and the analyte concentration, RMSE is easier to measure and more reliable than the methods based on the standard deviation of blanks.
The optimized method can detect between 0.15 and 0.20 ng mL−1 for Co2+ or 0.15–0.18 µg mL−1 for Cu2+ using perborate and percarbonate as the oxidants. Khorrami et al. (2006) obtained a detection limit for Co of 0.08 ng mL−1 using a sophisticated and expensive system, which included inductively coupled plasma and preconcentration procedures. The method proposed here is more simple and does not require any preconcentration step, with quantification limits suitable for the quantification of traces of this metal in drinking water, where the maximum recommended level for cobalt ions is 1 μg mL−1 or for drinking water for humans where the upper limits are even lower (Neal et al. 2000).
Interferences
The selectivity of this method is evidenced by the absence of effects associated with the foreign ions in the IC-CL determination. Alkali and alkali earth ions or mono- and bivalent anions did not interfere at maximum concentrations of 500 μg mL−1, which are much greater than the concentration usually found in natural waters.
Determination of copper and cobalt in the environmental samples
Results of the metals determination in water samples are summarized in Tables 3 and 4; in all cases, the recoveries were between 85 and 128%, depending on the amount of metals spiked. No differences were observed in the use of perborate or percarbonate for the determination of copper or cobalt. It was observed good agreements between the results obtained for Cu2+ by the IC-CL method and by using the atomic absorption spectrometry as an alternate method. Therefore, the chemiluminescent method could be used instead of the AAS, with the advantage of the simultaneous determination of cobalt, while the AAS requires the replacement of lamps for the analysis of each single ion.
Also a good agreement was found between the certified values for cooper and cobalt in soils and plant certified materials (Table 5), with recoveries between 93 and 105%, showing that the IC coupled with CL detection allowed a selective detection of Cu(II) and Co(II) without the interference of other ions which are also dissolved by the digestion with aqua regia or with the mixture of hydrogen peroxide and nitric acid.
Conclusion
The alkaline luminol/perborate or luminol–percarbonate-based CL detection of Cu(II) and Co(II) coupled with the chromatographic separation is a simple, fast, accurate and free of interferences procedure for the simultaneous analysis of these metals in different environmental samples, such as waters, soils and plants. This method employs a very simple system requiring short times to complete an entire analysis making it attractive for the determination of these metals in a large number of samples in environmental monitoring studies. Due to the low cost, simplicity and low detection limits, it becomes as an alternative for those methods that require more sophisticated devices.
References
Ahmed MJ, Uddin MN (2007) A simple spectrophotometric method for the determination of cobalt in industrial, environmental, biological and soil samples using bis(salicylaldehyde)orthophenylenediamine. Chemosphere 67(10):2020–2027
Badocco D, Pastore P, Favaro G, Macca C (2007) Effect of eluent composition and pH and chemiluminescent reagent pH on ion chromatographic selectivity and luminol-based chemiluminescent detection of Co2+, Mn2+ and Fe2+ at trace levels. Talanta 72:249–255
Burguera JL, Burguera M, Towsend A (1981) Determination of zinc and cadmium by flow injection analysis and chemiluminescence. Anal Chim Acta 127:199–201
Chen YC, Jian YL, Chiu KH, Yak HK (2012) Simultaneous speciation of Iron (II) and Iron(III) by ion chromatography with chemiluminescence detection. Anal Sci 28:795–799
Corley J (2002) Best practices in establishing detection and quantification limits for pesticide residues in foods. In: Lee P (ed) Handbook of residue analytical methods for agrochemicals. Wiley, West Sussex
Correa E, Vitorino A (2002) Chemiluminescence as an analytical tool: from the mechanism to applications of the reaction of luminol in kinetic based methods. Quimica Nova 25(6):1003–1011
Dulaquais G, Boye M, Middag R, Owens S, Puigcorbe V, BuesselerK Masques P, de Baar H, Carton X (2014) Contrasting biogeochemical cycles of cobalt in the surface western Atlantic Ocean. Global Biogeochem Cycles 28(12):1387–1412
Gammelgaard B, Liao Y, Jons O (1997) Improvement on simultaneous determination of chromium species in aqueous solution by ion chromatography and chemiluminescence detection. Anal Chim Acta 354(1):107–113
Gao W, Qi W, Jianping L, Qi L, Saadat M, Guobao X (2015) Thiourea dioxide as a unique eco-friendly coreactant for luminol chemiluminescence in the sensitive detection of luminol, thiourea dioxide and cobalt ions. Chem Commun 51:1620–1623
Issam M, Shakir A, Zaineb F, Hassan A (2015) Chemiluminometric-CFIA for the determination of Cobalt(ll) ion in commercial cobalt–molybdenum catalyst (K F124-3E) used in desulphurization processes of petroleum products via multi gel beads reactor. Iraqi J Sci 56(1):38–52
Khorrami AR, Hashempur T, Mahmoudi A, Karimi AR (2006) Determination of ultra trace amounts of cobalt and nickel in water samples by inductively coupled plasma-optical emission spectrometry after preconcentration on modified C18-silica extraction disks. Microchem J 84:75–79
Kim KM, Kim YH, Oh S, Lee SH (2013) A chelate complex-enhanced luminol system for selective determination of Co(II), Fe(II) and Cr(III). Luminescence 28:372–377
Lemos V, Santos JS, Baliza PX (2006) Me-BTABr reagent in cloud point extraction for spectrometric determination of copper in water samples. J Braz Chem Soc 17(1):30–35
Li B, Wang D, Lv J, Zhang Z (2006) Flow-injection chemiluminescence simultaneous determination of Cobalt(II) and Copper(II) using partial least squares calibration. Talanta 69:160–165
Lu C, Lin C, Huie W, Yamada M (2003) Simultaneous determination of Copper(II) and Cobalt(II) by ion chromatography coupled with chemiluminescent detection. Anal Sci 19:551–557
Murillo JA, García L, Carrasquero A (2013) Fast simultaneous determination of traces of Cu(II) and Co(II) in soils and sediments with the luminol/perborate chemiluminescent system. Environ Monit Assess 158(1):573–577
Neal C, Jarvie HP, Whitton BA, Gemmell J (2000) A simple spectrophotometric method for the determination of cobalt in industrial, environmental, biological and soil samples using bis(salicylaldehyde)orthophenylenediamine. Sci Total Environ 251(252):153–172
Roda A, Guardigli M (2012) Analytical chemiluminescence and bioluminescence: latest achievements and new horizons. Anal Bioanal Chem 402:69–76
Xiao CB, King DW, Palmer DA, Wesolowski DJ (2000) Study of enhancement effects in the chemiluminescence method for Cr(III) in the ng l−1 range. Anal Chim Acta 415(1–2):209–219
Yamane T, Watanabe K, Mottola A (1998) Continuous-flow system for the determination of cobalt in sea and river water: in-line preconcentration/separation coupled with catalytic determination. Anal Chim Acta 207:331–336
Author information
Authors and Affiliations
Corresponding author
Additional information
Editorial responsibility: M. Abbaspour.
Rights and permissions
About this article

Cite this article
Murillo Pulgarín, J.A., García Bermejo, L.F. & Carrasquero Durán, A. An easy and fast IC-CL procedure for the monitoring of Cu(II) and Co(II) in environmental samples. Int. J. Environ. Sci. Technol. 14, 233–240 (2017). https://doi.org/10.1007/s13762-016-1154-5
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13762-016-1154-5