
Abstract
This study reports the first application of coupled-column liquid chromatography–photochemically induced fluorimetry–fluorescence detection (LC-LC-PIF-FD), demonstrating its potential for the quantitative and selective detection of seven pyrethroids in vegetable samples such as cucumber, green bean, tomato and aubergine. An internal surface reversed-phase (ISRP) column coupled to a C18 column for analyte clean-up and determination were used, respectively. In comparison with a C18 column, the ISRP substantially improved the separation between analytes and interferences from the vegetable matrix. The limits of detection ranged from 0.01 to 0.22 μg kg−1 in the vegetable samples (equivalent to 0.01 and 0.13 μg L−1 in the extract injected), and limits of determination ranged from 0.56 to 8.33 μg kg−1 in the vegetable samples (equivalent to 0.34 and 5.00 μg L−1 in the extract injected). Samples were extracted into dichloromethane to yield mean recoveries at two levels of concentration between 72.8 and 110.0% in all cases. Relative standard deviations were lower than 11%.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Pesticide residues from crops and environmental samples can be handled and analyzed by using a number of techniques. Current analytical methodology usually consists on off-line SPE of sample extract for the clean-up and instrumental analysis of the extracts by GC with ECD and NPD detection and/or HPLC with UV plus MS detection for additional confirmation in order to avoid false positives [1]. However, MS instruments are expensive and matrix effects on MS detection, such as signal suppression [2], have been observed.
Fluorimetric detection has been closely bound to the important development of HPLC instrumentation as it is generally more sensitive than the classical UV absorption detection. In addition, fluorescence detectors are very selective, overcoming matrix interference [3]. However, although few compounds are fluorescent, some of them possess the necessary degree of aromaticity and may be converted to fluorescent species by using derivatization methods.
Among the numerous existing derivatization procedures, photochemically induced fluorimetry (PIF) has found extensive application [3]. This methodology is based on the conversion of non-fluorescent analytes into strongly fluorescent photoproducts by UV irradiation. PIF is more recent than chemical derivatization and has been much less applied to quantitative pesticide analysis. This method has several advantages compared to other fluorogenic derivatization techniques such as simplicity, shorter analysis time, and enhanced sensitivity and selectivity. In addition, no post-column pumps or other devices are required. PIF is adaptable to various experimental conditions such as TLC [4], stationary liquid solutions [5], and flow systems, including HPLC [6, 7] and FIA [8, 9]. Advantages of the post-column approach include separation of analytes in their original form with no necessity for complete derivatization reaction, assuming reproducibility.
Pyrethroids are non-systemic insecticides and acaricides with contact and stomach action which are used for the control of a wide range of pests in cereals, ornamentals, vegetables, cotton, and other crops. Their neurotoxic effect caused by a prolonged opening of the sodium channel, which provokes a repetitive nerve action associated with hyperactivity, tremor, ataxia, convulsions, and possible paralysis [10].
The pyrethroid insecticides have no native fluorescence, but they can be photolysed into strongly fluorescent photoproducts. The photodegradation mechanism of pyrethroid insecticides has been widely investigated [11–14]. The identification of a number of photoproducts during the photolysis of fenvalerate [14] and deltamethrin [11, 12] under various experimental conditions indicates the complexity of the photolysis mechanisms.
Multidimensional HPLC is a powerful tool for the analysis of samples with a high degree of complexity. Coupled-column liquid chromatography (LC-LC) techniques have been used for pesticide quantification in different matrices with UV [15, 16], fluorescence [17], and MS [18] detection but there are no reports in the literature coupling LC-LC to post-column PIF and fluorescence detection (FD). Usually, LC-LC has been applied to the determination of polar analytes in water [18, 19] or aqueous samples [20, 21]. In these cases, problems may be encountered when highly polar analytes have to be determined, because retention is low even on highly hydrophobic phases and, consequently, trace enrichment becomes difficult as analytes start to elute during injection. Moreover, clean-up becomes less efficient, because the possibility of separating the analytes of interest from the early-eluting interferences will be rather limited. However, coupled-columns using direct large-volume injections may well prove to be an elegant monitoring method for most of the medium-polarity pesticides in use today [19]. It may even be very useful to analyze low polar pesticides (such as pyrethroids, which will remain highly retained on hydrophobic phases) in vegetable extracts obtained in solvents with high elutropic strength.
We have developed a new and efficient LC-LC-PIF-FD method for the determination of pyrethroids in vegetable samples from Almería (Spain). Its main advantages are that the coupling LC-LC performs an efficient clean-up of extracts and PIF-FD allows an improvement in sensitivity and selectivity
Experimental
Chemicals and solvents
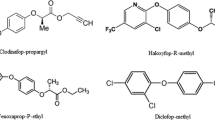
Analytical standards (pestanal quality) of fenpropathrin (FENP), λ-cyhalothrin (λ-CYH), deltamethrin (DELT), fenvalerate (FENV), permethrin (PERM), τ-fluvalinate (τ-FLUV), and bifenthrin (BIFE) were obtained from Riedel-de Haën (Seelze, Germany). Chemical structures of the compounds are presented in Fig.1.

Chemical structures of the pyrethroids used in this study
HPLC-grade acetonitrile (ACN), methanol (MeOH), and ethanol (EtOH) were obtained from Merck (Darmstadt, Germany). Dichloromethane and n-hexane for analysis of pesticide residue quality were obtained from Riedel-de Haën. Anhydrous sodium sulfate for residue analysis was obtained from Panreac (Barcelona, Spain).
HPLC-grade water was obtained by purifying demineralized water in a Milli-Q system (Millipore, Bedford, MA, USA). Mobile phases were filtered through a 0.45-μm cellulose acetate (water) or Teflon (organic solvents) and degassed with helium prior and during use. All standards and samples were filtered through Millipore membrane Teflon filters (0.45-μm particle size) before injection into the chromatographic system.
Instrumentation
The system consists of an isocratic Model 510 LC pump (P-1) and gradient Model 600 LC pump (P-2) from Waters (Milford, MA, USA), a Rheodyne six-port injection valve (Model 7725) with a 1,000-μL sample loop, a type 7000 high-pressure column-switching valve (V) from Rheodyne (Berkeley, CA, USA), and a Model 474 variable-wavelength scanning fluorescence detector (D) from Waters.
LC-LC separations were performed with a 50×4.6-mm-i.d. first separation column (C-1) packed with 5-μm GFF-II (ISRP, Pinkerton) from Regis and a 250×4.6-mm-i.d. second separation column (C-2) packed with 3.5-μm Symmetry C18 (Waters).
The photochemical reaction was carried out in a post-column photochemical reactor (Softron GmbH, Gynkotek HPLC, Germering, Germany) fitted with a knitted open tube reactor coil (5 m × 1.6-mm e.d. and 0.3-mm i.d.) PTFE and a 4-W Xenon lamp. Recording of chromatograms and quantitative measurements of peak areas were performed with Millennium32 Software from Waters (Milford, MA, USA).
A Model PT 2100 polytron (Kinematica AG, Luzern, Switzerland) was used to blend vegetable samples. A Model VV2000 LIF rotary vacuum evaporator (Heidolpf) thermostated by water circulation with a N-010 KN-18 vacuum pump (Telstar) was used to evaporate the extracts.
Preparation of standards and spiked samples
Standard solutions of pesticides (200 mg L−1) were prepared by exactly weighing and dissolving the corresponding compounds in n-hexane. These standard solutions were stable for a period of at least 3 months. Dilutions in ACN:water (70:30 v/v) were freshly prepared for the working solutions. These solutions were obtained by evaporating to dryness aliquots of the standard solutions in n-hexane under a gentle N2 stream and redissolving in AcN:water (70:30 v/v). All solutions were protected against light and were stored in a refrigerator at 4°C.
For recovery determinations, 15-g samples of finely chopped vegetable were spiked at concentration levels of 16.7 and 167 μg kg−1 for each of the pesticides except for bifenthrin which was at 6.25 and 62.5 μg kg−1. The spiked samples were allowed to stand for a few minutes before extraction to allow the spiked solution to penetrate the test material.
Extraction procedure
A 15-g sample of vegetable was placed in a container glass and mixed with 30 mL of dichloromethane for 2 min with the polytron. Then, 25 g of sodium sulfate was added and the mixture was homogenized for 1 min. The extract was filtered through a porous plate and washed with two successive 15-mL portions of dichloromethane. The combined extraction fractions were collected in a 250-mL spherical flask and evaporated to nearly dryness in a rotating vacuum evaporator with a water bath at 40±1°C, and the remaining solvent was allowed to evaporate under a slight N2 stream.
The residue obtained from the extract was redissolved in 5 mL ACN, and 1 mL of this sample extract, equivalent to 3 g vegetable sample, was redissolved in 5 mL ACN:water (70:30, v/v) and then filtered through 0.45-μm Teflon filter. Finally, 1 mL of the preceding disolution was injected into the LC-LC-PIF-FD.
LC-LC-PIF-FD analysis
ACN:water (70:30 v/v) was used as first mobile phase (M-1); the second (M-2) comprised an ACN:water gradient as follows: initially 18 min isocratic with ACN:water (80:20 v/v) and 1 min linear gradient to ACN:water (100:0 v/v); then 8 min isocratic with ACN:water (100:0 v/v) followed by an additional period of 1 min linear gradient to the initial conditions, and finally 3 min under the initial conditions was sufficient time before subsequent analysis runs. Mobile phase flows were set at 1 mL min−1 for both M-1 and C-2. The fluorimetric detection was performed at an excitation wavelength (λex) of 283 nm and at an emission wavelength (λem) of 330 nm for all pesticides.
The schematic diagram of the dual-column system used in the present study is well known [16, 22]. The analytical process was as follows: a 1-mL aliquot of vegetable extract was injected into the sample loop and was transferred by M-1 to the pre-separation column C-1 at 1 mL min−1. At 1.3 min from injection, the switching valve was turned to the alternate position so that the first and main separation columns were connected. The mobile phase used to carry analytes from the first column to the main column was ACN:water (80:20 v/v).
After 4.4 min, the switching valve was turned back to the initial position to disconnect the first and the main separation columns to re-equilibrate the former with mobile phase in the pre-separation. The analytes were eluted from the main column with the mobile phase M-2.
Results and discussion
Fluorescence detection after photolysis
In PIF, two parameters affect the sensitivity of the method, namely the UV irradiation time and the type of solvent used [5, 23]. According to some previous works pyrethroids have a good affinity for protic solvents giving high PIF intensity [24]. In our case, batch studies showed that the strongest fluorescence signal for the target pesticides occurred in binary aqueous mixtures with organic solvents, such as MeOH, ethanol, and ACN, in agreement with results previously obtained by other authors for deltamethrin and fenvalerate [5].
The maxima of excitation and emission wavelengths of photoproduct from each pyrethroid were determined from their excitation and emission spectra, which were located between 280–287 and 319–338 nm, respectively. All pyrethroids under study show closely located excitation maxima (and emission maxima), so we selected a λex and λem of 283 and 330 nm, respectively, as the better compromise wavelengths. Samples were injected in ACN:water 70:30 (v/v) because under these conditions the higher signal was obtained for all pesticides.
In the chromatographic system, the analytical response increased when the flow rate decreased, because the residence time of the analytes in the photoreactor and the flow rate are inversely proportional. As no well-defined maxima were reached, a flow rate was selected as a compromise between the residence time in the reactor and the band broadening on the chromatogram.
Switching parameters
The Pinkerton ISRP column [25] can be used in the coupled-column or column-switching mode. In this technique a short ISRP (5-μm, GFF-II) column serves as the pre-column or initial column and the analytes are transferred to a secondary column after matrix elution by a column-switching valve, giving a better separation and a cleaner chromatogram, which contributes to the enhancement of selectivity and sensitivity.
The degree of retention of these pyrethroids on the ISRP column is enough to carry out a good clean-up step. Clean-up and transfer volumes were estimated when connecting C-1 with the photoreactor and fluorescence detector. Experiments indicated that a mobile phase of ACN:water (70:30 v/v) at a flow rate of 1 mL min−1 for M-1 was a good compromise between clean-up and preventing elution of the first-eluting analyte. A mobile phase containing ACN:water (80:20 v/v), also at 1 mL min−1, was adequate for transfer of the fraction containing analytes from C-1 to C-2 and to attain peak compression. By using the above mobile phases, a clean-up volume of 2.3 mL (sample injection included) and a transfer volume of 2.1 mL were attained. Figure 2 shows that under the optimized LC-LC-PIF-FD conditions most of the interfering compounds were removed to provide chromatograms with reduced background.

Chromatograms of 1-mL sample injections on the C-1 column illustrating the selection of the column-switching conditions: a injection of blank green bean sample; b injection of the pyrethroid pesticides sample containing 200 μg L−1 in ACN:water (70:30 v/v)
On account of the high separating capacity of the RP18 material, a 3.5-μm C18 column (250×4.6-mm i.d.) was selected as the second column (C-2).
Binary mixtures of aqueous solutions with ACN or MeOH in several ratios were tested to obtain maximum responses and selectivity with minimum broadening on the chromatograms in C-2. Retention times of pyrethroids were too long with MeOH:H2O.
The highest fluorescence responses and the best separation with shortest time were achieved using the AcN:water gradient program described in “LC-LC-PIF-FD analysis” section. Thus, M-2 combines the process of transferring analytes from C-1 to C-2 (ACN:water 80:20 v/v) and their separation into C-2.
The volume of injection is usually optimized by connecting C-1 to the detector. This study was performed in this way by injecting 80 ng of each compound onto C-1 with ACN:water (70:30 v/v) as mobile phase. The volumes tested ranged from 20 to 2,000 μL, showing the possibility of performing LVI with good peak shape (width and symmetry) even for a 2.0-mL injection.
When injection volume is large, the sensitivity and the analysis time are higher. A sample volume of 1 mL was selected as a compromise between the required sensitivity and speed of analysis. Figure 3 shows a chromatogram of a standard solution spiked at a level of 50 μg L−1 using the method optimized above. As can be seen, one chromatographic peak was obtained for each compound, except for permethrin, which yields two peaks (peak numbers 5 and 7), corresponding to the cis and trans isomers, according to the solid standard used. In all cases, the peak area was used as analytical signal for quantitation. For permethrin, the total area of the two peaks was used [26].

LC-LC-PIF-FD chromatogram of a standard solution containing 50 μg L−1 of analytes registered at λex=283 nm and λem=330 nm. Volume of sample injected 1 mL. 1 fenpropathrin, 2 λ-cyhalothrin, 3 deltamethrin, 4 fenvalerate, 5 permethrin, 6 τ-fluvalinate, 7 bifenthrin
Extraction and analysis
In a previous paper, we reported that dichloromethane is a satisfactory extractant for the studied pyrethroids [26]. The extraction procedure described in the “Experimental” section was applied to spiked cucumber, green bean, tomato, and aubergine samples. The LC-LC-PIF-FD analysis was carried out on-line by means of LVI (1 mL), which provided sufficient sensitivity. We found very clean chromatograms for all vegetables assayed. Figure 4 shows the LC-LC-PIF-FD chromatogram of a blank green bean extract and the chromatogram corresponding to a extract containing all the analytes at a spiking level of 8.33 μg kg−1 (corresponding to 5.00 μg L−1 in the extract injected), using the final selected chromatographic conditions in both cases. The peaks of pyrethroids were well resolved and showed no interferences with the vegetable matrix.

LC-LC-PIF-FD chromatogram at λex=283 nm and λem=330 nm of a green bean sample blank extract (dashed line) and a green bean extract spiked at 8.33 μg kg−1 of each pyrethroid and analyzed by the proposed method (solid line). Volume finally injected 1 mL. 1 fenpropathrin, 2 λ-cyhalothrin, 3 deltamethrin, 4 fenvalerate, 5 permethrin, 6 τ-fluvalinate, 7 bifenthrin
In our previous paper [26], an analytical HPLC-PIF-FD method for the determination of seven pyrethroids in vegetables was developed. In this procedure, samples were extracted with dichloromethane and further cleaned up by SPE, which involves a great consumption of time, solvents, and disposable cartridges. In the present work, the use of LC-LC-PIF-FD offers a much more powerful technique for the qualitative and quantitative analysis of the target compounds, since the LVI combines with the sensitivity and selectivity of PIF-FD yield, low cost, and decrease in time.
Validation
Limits of detection (LODs) and limits of quantification (LOQs) were calculated statistically [27] as 3.84 and ten times, respectively, the standard deviation of the signals, corresponding to 10 blanks divided by the slope of the calibration curve. The LOQs were also calculated, according to the EURACHEM guidance [28], as the lowest concentration for which the relative standard deviation is equal to or less than a fixed percentage (10% in our case).
Linear range was established for each pesticide, the lower limit being the LOQ calculated according to the last criterion and the upper limit the concentration for which the signal deviates from the linearity by 3–5% [29]. Retention time windows (RTWs) were calculated as retention time (tR) averages ± 3 standard deviation of retention time.
Analytical figures of merit, obtained by using an external standard method and pure solvent standards for calibration are given in Table 1. Wide linear ranges between 0.23 and 0.72 μg L−1 according to the compound, until between 200 and 600 μg L−1 according to the compound, which characterize generally fluorimetric methods, and determination coefficients higher than 0.999 were obtained in all cases.
The method was also checked for condition of uniform variance (homoscedasticy) over the linear range [29]. For calibration experiments, this means that the precision of the measurements is independent of the concentration. Each calibration graph was obtained in triplicate and the comparison of standard deviations showed that the dispersion of the measurements was independent of the analyte concentration.
The presence of constant systematic errors due to additive signals from matrices was checked by comparing the intercepts of calibration graphs built using solvent-based and matrix-matched standards [29]. In this study, no significant differences were found. Relative systematic errors due to matrix effect or matrix interferences may affect the slope of calibration graphs. They were tested by comparing the slopes of solvent-based and matrix-matched calibration graphs [29]. No matrix effect was observed for the analytes in the target matrices, except for permethrin and τ-fluvalinate in green beans. Low responses for these two pesticides in matrix extract (Fig. 5) can be attributed to incomplete photoderivatization of pesticides in the coil or to quenching processes produced by coeluting bulk plant co-extracts (e.g., by pigments and cuticular waxes) which do not constitute interferent peaks because they are not fluorescent species in the working conditions. For this reason, matrix-matched standards were used for quantification of permethrin and τ-fluvalinate and solvent-based standard were used in all other cases.

Calibration curves of a permethrin in CH3CN:H2O (70:30 v/v) solvent (diamonds) and green bean matrix (triangles); b τ-fluvalinate in CH3CN:H2O (70:30 v/v) solvent (diamonds) and green bean matrix (triangles)
Retention time windows obtained using spiked blank extract matrix were not different from those obtained using solvent. LODs and LOQs obtained using blank extract of matrices (Table 2) are higher than those obtained using solvent for both criteria. As values were obtained using blank extract matrix were more realistic, they were chosen as characteristic parameters of the method. On the other hand, LOQs obtained using the criterion proposed by the EURACHEM guidance estimated more realistic values. In any case, these values are lower than the maximum residue levels (MRLs) established by Spanish and European [[31, 32] legislation and are also in compliance with the criteria established by the Directorate General Health and Consumer Protection [30].
The LOQs obtained using LC-LC-PIF-FD (0.56–8.33 μg kg−1) are lower than reported values obtained by HPLC-PIF (0.80–9.00 μg kg−1) [26]. These values are also lower than those reported in the literature for the determination of the same insecticides by other techniques, such as GC-ECD [32], TLC [33], HPLC-UV [34], or PIF[35, 36]. Besides, the imprecision associated with those measurements (Table 1) is usually about double.
Evaluation of method selectivity
Thirty-two pesticides, belonging to different pesticide families, which can be used simultaneously to pyrethroids were checked for the possibility of giving chromatographic interferences [26]. Pesticides giving no response to the fluorescence detector do not affect to analytical signals of target analytes. Pesticides showing native fluorescence or UV-induced fluorescence give no response at the working wavelength.
Evaluation of precision and accuracy
The extraction procedure described above was applied to cucumber, green bean, tomato, and aubergine samples spiked with the analytes at levels of 16.7 and 166.7 μg kg−1 for each pesticide except for bifenthrin, which, owing to its higher sensitivity, was at levels of 6.25 and 62.5 μg kg−1 (equivalent to 10 and 100 μg L−1, and 3.75 and 37.5 μg L−1, respectively, in the final extract). The procedure is efficient for extracting pesticide residues from vegetable samples, as the analytes were determined at two concentration levels (low and intermediate) and using six replicates with recoveries ranging between 78.0 and 96.4% in cucumber samples, 73.5 and 95.6% in green bean samples, 72.8 and 105.1% in tomato samples, and 73.9 and 110.0% in aubergine samples (Table 3). The RSD ranged between 1.5–11.1 and 1.1–7.3% for the low and high concentrations, respectively. These recoveries are between 70 and 110% and RSDs (%) are lower than 20%, as establish the Directorate General Health and Consumer Protection for routine residue analysis in post-registration monitoring and control [30].
The efficiency of the analytical column was not affected by the vegetable injections onto the first column during all injections that were necessary to perform this study.
Conclusions
In order to determine selected pyrethroids in vegetable samples, we have developed an efficient and sensitive LC-LC-PIF-FD method by combining on-line clean-up with selective column switching using ISRP, and conventional separation of analytes with C18 column. This methodology requires minimal manipulation of the vegetable samples before injection. The automation of the system is subject to a minimum of human errors and contamination. Reductions in costs for sample pre-treatment (solvent and solid phases for clean-up) and method development times are also achieved.
The availability of an easy-to-operate LC-LC-PIF-FD system offering high sensitivity and selectivity makes it a suitable technique for the trace analysis of pesticides in vegetable samples. LOD and LOQ are lower than MRLs established by the European Community (EU) for vegetables. Precision and accuracy of the method are also convenient for routine applications.
References
Hogendoorn EA, Westhuis K, Dijkman E, Heusinkveld HAG, den Boer AC, Evers EAIM, Baumann RA (1999) J Chromatogr A 858:45–54
Hogenboom AC, Hoffman MP, Jolly DA, Niessen WMA, Brinkman UATh (2000) J Chromatogr A 885:377–388
Aaron JJ (1993) In: Schulman SG (ed) Molecular luminescence spectroscopy: methods and applications. Wiley, New York, p 85
Brown KK, Tomboulian P, Walters SM (1988) J Res Natl Bur Stand USA 93:301–305
Coly A, Aaron JJ (1994) Analyst 119:1205–1209
Muñoz de la Peña A, Mahedero MC, Bautista Sánchez A (2002) J Chromatogr A 950:287–291
Martínez Galera M, López López T, Gil García MD, Martínez Vidal JL, Parrilla Vázquez P (2001) J Chromatogr A 918:79–85
Coly A, Aaron JJ (1999) Anal Chim Acta 392:255–264
Vilchez JL, Valencia MC, Navalón A, Molinero-Morales B, Capitán-Vallvey LF (2001) Anal Chim Acta 439:299–305
Narahashi T (1992) Nerve membrane Na channels as targets of insecticides. TIPS 13:236–241
Ruzo LO, Holmstead RL, Casida JE (1976) Tetrahedron Lett 35:3045–3048
Ruzo LO, Holmstead RL, Casida JE (1977) J Agric Food Chem 25:1385–1394
Ruzo LO, Casida JE (1981) J Agric Food Chem 29:702–706
Mikami N, Takahashi N, Hayashi K, Miyamoto J (1980) J Pest Sci 5:225–236
Parrilla P, Kaim P, Hogendoorn EA, Baumann RA (1999) Fresenius J Anal Chem 363:77–82
Parrilla Vázquez P, Martínez Vidal JL, Martínez Fernández J (2000) Analyst 125:1549–1553
Hogendoorn EA, Ossendrijver FM, Dijkman E, Baumann RA (1999) J Chromatogr A, 833:67–73
van der Heeft E, Dijkman E, Baumann RA, Hogendoorn EA (2000) J Chromatogr A, 879:39–50
Hogendoorn A, Brinkman UATh, Van Zoonen P (1993) J Chromatogr A 644:307–314
Oertel R, Richter K, Gramatté T, Kirch W (1998) J Chromatogr A 797:203–209
Sancho JV, Puzo OJ, Hernández F (2000) Rapid Commun Mass Spectrom 14:1485–1490
Hidalgo C, Sancho JV, Hernández F (1997) Anal Chim Acta 338:223–229
Patel BM, Moye HA, Weinberger R (1991) Talanta 38:913–922
Coly A, Aaron JJ (1998) Talanta 46:815–843
Hagestam IH, Pinkerton TC (1985) Anal Chem 57:1757–1763
López-López T, Gil García MD, Martínez Vidal JL, Martínez Galera M (2001) Anal Chim Acta 447:101–111
Currie LA (1999) Anal Chim Acta 391:105–126
EURACHEM Guidance Document No 1 WELAC Guidance Document No WGD 2 “Accreditation for Chemical Laboratories: Guidance on the interpretation of the EN 45000 series of Standards and ISO/IEC Guide 25” (1993). Available from the EURACHEM Secretariat, PO Box 46, Teddington, Middlesex, TW11 ONH, UK
Massart DL, Vandeginste BGM, Buydens LMC, De Jong S, Lewi PJ, Smeyers-Verbeke J (1997) Handbook of chemometrics and qualimetrics, Part 2. Elsevier Science, Amsterdam (The Netherlands)
Guidance document on residue analytical methods SANCO/825/00 Rev.6. 20/06/00
Legislación Internacional de Residuos de Plaguicidas en Productos Vegetales, Secretaria General de Comercio Exterior de España, NIPO: 101-99-108-9, 1999
Mukherjee I, Gopal M (1992) Pesticide Sci 36:175–179
Corbini G, Biondi C, Proietti D, Dreassi E, Corti P (1993) Analyst 118:183–187
Pang GF, Chao YZ, Fan CL, Zhang JJ, Li XM, Zhao TS (1995) J AOAC Int 78:1481–1488
Aaron JJ, Coly A (1996) Analyst 121:1545–1549
Coly A, Aaron JJ (1998) Anal Chim Acta 360:129–141
Acknowledgments
The authors are grateful to the INIA (Project CAL-00–063) and FIAPA for financial support for this work.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Parrilla Vázquez, P., Gil García, M.D., Barranco Martínez, D. et al. Application of coupled-column liquid chromatography combined with post-column photochemically induced fluorimetry derivatization and fluorescence detection to the determination of pyrethroid insecticides in vegetable samples. Anal Bioanal Chem 381, 1217–1225 (2005). https://doi.org/10.1007/s00216-004-3043-x
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00216-004-3043-x