
Abstract
A method for the analysis of several macrolide and ionophore antibiotics as well as tiamulin in liquid manure was developed. Reversed-phase liquid chromatography and atmospheric pressure chemical ionisation (APCI) tandem mass spectrometry was used for detection.
High-performance liquid chromatographic (HPLC) separation of the antibiotics was achieved in 35 min. The analytes were extracted with ethyl acetate and the extracts were cleaned up by solid-phase extraction on a diol SPE cartridge.
Recovery experiments with spiked liquid manure concentrations varying from 6 to 2,000 µg kg−1 gave constant recovery rates. The recovery rates for the macrolides erythromycin, roxithromycin and oleandomycin were 75–94%, that for the ionophore salinomycin was 119%, while that for the pleuromutilin tiamulin was 123%, when using a macrolide internal standard. The relative standard deviation was found to be 15–36% and the limits of detection were 0.4–11.0 µg kg−1.
The maximum concentrations found in manure samples were 43 µg kg−1 for tiamulin and 11 µg kg−1 for salinomycin.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Most of the 2,900 pharmaceuticals registered in Germany were used in animal husbandry as well as in human medical applications [1]. Some of those have been detected in the environment [2, 3, 4]. Toxic effects on fauna have been observed as well [5]. First results about resistance of bacteria to the majority of existing antibiotics were reported by Neu [6].
Over 10,000 t of antibiotics were applied in Europe in 1997 as antibacterial agents. About 50% of this was used in human medicine while the other half was applied in large-scale animal husbandry [7]. Three fields of application for animal husbandry were significant: growth promoter (salinomycin and monensin and formerly the tetracyclines), treatment of infections in livestock (pleuromutilins, sulfonamides, ionophores and macrolides) and prevention of infections especially if pigs from different breeders are brought together (pleuromutilins, sulfonamides, ionophores and macrolides). Growth promotion with sodium-monensin, sodium-salinomycin, flavophospholipol and avilamycin will be phased out in the EU on 1 January 2006 [8]. Fifty to ninety percent of the administered pharmaceutical dose is excreted rapidly after the treatment [9]. The respective parent compounds as well as their primary metabolites are prevalent in excretions. Thus large quantities of these pharmaceuticals, applied in animal husbandry, are transferred together with liquid manure to manure tanks. The final homogenate is dispersed on the fields after varying time periods in Germany.
Little is known about the behaviour and the degradability of antibiotics in soil. Pharmaceuticals may accumulate in soil [10] and influence soil organisms. On the other hand a very hydrophilic drug may be mobile in soil and can contaminate the ground water.
Several methods for the analysis of macrolides, ionophores and tiamulin in animal tissues, milk and plasma have been described using liquid chromatography/ultra violet detection (LC/UV), liquid chromatography/mass spectrometry (LC/MS) and liquid chromatography/tandem mass spectrometry (LC/MS/MS) [11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23]. Methods for the analysis of sulfonamides in manure [24] and tetracyclines in soil fertilised with manure by using LC/MS/MS have also been described [25, 26]. A review of several analytical strategies for the screening of veterinary drugs was presented by Aerts et al. [27].
Because manure is a complex matrix, an efficient clean-up procedure is necessary to remove interfering matrix. The aim of the present work was to develop a reproducible and sensitive multiresidue method to investigate the commonly used macrolides, erythromycin, roxithromycin, oleandomycin, tylosin and ivermectin, ionophores, salinomycin and monensin as well as the pleuromutilin derivate tiamulin in liquid swine manure as a source of soil contamination. The structural formulae of some of the analytes and the internal standard are shown in Fig. 1.

Structural formulae of a erythromycin, b oleandomycin, c roxithromycin, d internal standard, e salinomycin, f tiamulin
Experimental
Materials
Acetonitrile (HPLC-S gradient grade) was purchased from Biosolv (Valkensward, Netherlands). Water (HPLC grade) was obtained from Mallinckrodt Baker (Griesheim, Germany). Isooctane, methanol (suprasolv grade), acetone and ethyl acetate (analytical grade) were obtained from Merck (Darmstadt, Germany).
Ammonium acetate, dipotassium hydrogen phosphate, potassium dihydrogen phosphate, urea, disodium ethylenediaminetetraacetate, sodium sulfate and calcium carbonate were of analytical grade and were purchased from Merck. Erythromycin, ivermectin, roxithromycin, tylosin tartrate and O-methylhydroxylamine hydrochloride were provided by Sigma–Aldrich (Seelze, Germany). Oleandomycin phosphate dihydrate, monensin sodium salt, salinomycin SV sodium salt 2.5-hydrate, tiamulin fumarate (Vetranal) and sodium chloride (p.a.) were obtained from Riedel–de Haën (Seelze, Germany).
Internal standard
The synthesis of (E)-9-[O-(2-methyloxime)]-erythromycin was similar to the procedure described by Gasc et al. [28].
Calcium carbonate (1,052 mg) and 415 mg of O-methylhydroxylamine hydrochloride were added to a solution of 707 mg erythromycin in 25 mL dry methanol and the mixture was stirred at room temperature for 96 h. This solution was poured into 50 mL of a 5% ammonium hydroxide solution and the resulting mixture was cooled in an ice–water bath. The mixture was extracted thrice with 30 mL methylene chloride.
The crude product was purified by chromatography on silica gel 60 (Merck, Darmstadt, Germany) with a chloroform/triethylamine mixture (9:1, v/v). The HPLC–MS separation of the derivative revealed a purity of 92% at mass 763.5. MS conditions were full scan from 150 to 1,000 amu, device parameters are described below. No erythromycin was detected. The 1H NMR signals of the modification (300 MHz, CDCl3) reveal δ 3.83 ppm (s, N-OCH3) and δ 3.33 ppm (s, 4″-OCH3). This is in agreement with the data from the literature [28]. This new macrolide was used as internal standard.
HPLC
Separations were performed using a Phenosphere–Next RP18 column (2-mm i.d., length 150 mm, particle size 3 µm) and a SecurityGuard (Phenomenex, Torrance, CA, USA) at 25 °C. The flow rate was 0.2 mL min−1. The HPLC gradient was produced by using two mobile phases: phase A, 0.1 M aqueous ammonium acetate solution and phase B, pure acetonitrile. Chromatographic separation was achieved with the following gradient: 0–1 min 10% B, 1 min→14 min 10%→100% B, 14–29 min 100% B, 29 min→30 min 100%→10% B, 30–35 min 10% B. Ten µL of each sample were injected.
The HPLC system consisted of a GINA 50 autosampler, a P 580A HPG HPLC pump, a degasser unit DEGASYS DG-1210 and a column oven STG 585 (all from Dionex, Idstein, Germany). The dead time of the HPLC system was 1.8 min. After HPLC separation, the analytes were determined by atmospheric pressure chemical ionisation/tandem mass spectrometry (APCI–MS/MS) in positive ion mode and single reaction monitoring (SRM).
Mass spectrometry
The triple quadrupole mass spectrometer (TSQ 7000, Finnigan-MAT, Bremen, Germany) was equipped with an APCI 2 source and operated under the following conditions: capillary temperature, 180 °C; sheath gas, 40 psi; corona current, 5 µA; vaporiser temperature, 450 °C; auxiliary gas, off; q0 offset, −4.4 V; collision cell pressure, 2.0 mTorr; collision gas, argon; multiplier, 1,900 V (1,600 V in full scan mode). The potential difference between the capillary and the tube lens was held at 70 V. The fused silica capillary of the APCI 2 source was replaced by a steel capillary in order to reduce tailing of antibiotics adsorbing on the silica surface [29]. APCI was preferred because this ionisation is less vulnerable to matrix effects than ESI [24].
A post-column Valco divert valve was used to direct most of the non-significant LC flow of a sample to waste. Diverting the flow minimized contamination of the MS source: 0–8 min divert to waste, 8–28 min flow to mass spectrometer, 28–35 min divert to waste. An additional flow of 50 µL min−1 water/acetonitrile (3:7, v/v) pumped by an LC-10 AT HPLC (Shimadzu, Duisburg, Germany) compensated the missing flow from the HPLC during waste positing operation. Automatic data acquisition was triggered using a short contact closure signal of the autosampler.
Selected reaction monitoring (SRM) was chosen to gain higher selectivity. The optimal collision energy was determined by means of a software procedure controlling the automatic switching between the different voltages with a step size of 1 eV scan−1 and a range from −5 to −70 eV. A pre-scan voltage setting time of 2 ms and a cycle time (9 transitions) of 1.0 s were used for SRM. Key parameter settings for SRM are given in Fig. 2. The data obtained was processed by using Xcalibur 1.2 software.

APCI SRM traces of selected macrolides, ionophores and tiamulin for quantification in spiked manure (100 µL stock solution)
Sample preparation
Manure samples were stored at 4 °C. The manure was homogenized for 5 min at 25,000 rpm using an ultra Turrax homogeniser (VF2/IKA, Staufen, Germany). Homogenised manure (15 g) was transferred into 75-mL centrifuge glass tubes with a screw cap (Schott, Mainz, Germany) and 5 g urea was added. The samples were buffered to pH 8.0 by the addition of 6 mL phosphate buffer (33.5 g K2HPO4, 1.1 g KH2PO4 in 1 L water).
Liquid–liquid extraction
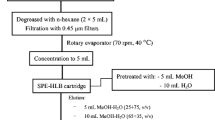
The buffered manure was extracted with 40 mL ethyl acetate by shaking for 20 min on a horizontal shaker (Kottermann, type 4020, Haenigsen, Germany) at 150 min−1. After shaking 25 µL of internal standard (IS) (10 mg (E)-9-[O-(2-methyloxime)]-erythromycin in 100 mL acetonitrile) was added to the mixture and the centrifuge glass was shaken by hand for 1 min. The phases were separated by centrifugation at 800 g for 20 min (BeckmannCoulter, Avanti J25, Unterschleissheim, Germany). The organic phase was removed and stored. The aqueous phase was mixed with 6 mL EDTA solution (37.3 g disodium ethylenediaminetetraacetic acid in 1 L water) and the mixture was extracted again with 40 mL ethyl acetate, with shaking (20 min) and centrifugation (800 g for 20 min). The organic phases from the 1st and the 2nd extraction were combined and the sample volume was reduced to 5 mL at 60 °C and 320 hPa on the rotary evaporator. The residue was dissolved in 20 mL isooctane and the volume was reduced again to 10 mL at 60 °C and 170 hPa. Figure 3 shows the procedure for analysis of antibiotics in manure.

Sample preparation scheme
SPE clean-up
Samples were cleaned up by a modification of the method developed by Delépine et al. [19]. Diol solid-phase extraction cartridges from UCT (2,000 mg, Bristol/PA, USA) were conditioned with 10 mL isooctane. A solid-phase extraction manifold (IST, Grenzach–Wyhlen, Germany), with PTFE stopcock and outlet, was used. The manure extract (10 mL) was passed through the cartridge at a speed of 5 mL min−1 (vacuum). The cartridge was washed with 10 mL isooctane to remove lipids and dried for 20 min by sucking air through the column followed by a wash step with 10 mL water to remove salt. The analytes were eluted twice from the cartridge with 4 mL of an acetonitrile/0.1 M aqueous ammonium acetate (3:2, v/v) mixture. An aliquot of 0.8 mL of the eluate was transferred to a 1.5-mL autosampler vial for HPLC/MS/MS analysis.
Calibration and validation
The calibration was performed as an internal standard calibration in the presence of manure matrix to avoid matrix effects [24, 30]. A liquid manure sample, from a pig farm, with a very high dissolved organic carbon (8.4 mg mL−1) content and a relative high dry weight (11%) was selected to simulate a worst-case scenario. This antibiotic-free manure had a pH of 7.7. The cleaned-up extracts of this manure were used for preparation of the standards in the presence of manure matrix for LC/MS/MS determination.
A stock solution was produced by dissolving 10 mg of the macrolides, ionophores and tiamulin in 100 mL acetonitrile. This standard solution was stored at 4 °C in the dark and was stable for at least 3 months. Calibration standards (5, 10, 50, 100, 500, 1,000 and 5,000 ng mL−1) were made by serial dilution of the stock solution. The IS was added to the calibration standards in a concentration of 5 µL mL−1. The calibration standard solution (0.5 mL) was filled in 1.5-mL HPLC vials and 0.5 mL manure matrix was added. The manure matrix solution was produced by the established method described above. The calibration curves were calculated using a weighted (1/X) linear regression model.
Results and discussion
All analytes were completely separated by HPLC. The selected APCI SRM traces for quantification are shown in Fig. 2.
The calibration graphs are linear in the range from the limit of quantification (LOQ) up to 5,000 ng mL−1 with correlation coefficients (r 2) better than 0.98 (Table 1).
Validation of the method
The method was validated by spiking 15 g of homogenised antibiotic-free manure aliquots, as described above, with 0.3–300 µL of the stock solution (2, 6, 20, 200 and 2,000 µg kg−1 manure) and shaking manually for 1 min. The following sample preparation, extraction and clean-up was identical to the procedures described above.
Recovery experiments for the macrolides, ionophores and tiamulin were carried out at five concentration levels in triplicate.
The recoveries are given in Fig. 4. Since there was no significant concentration (2, 6, 20, 200 and 2,000 µg kg−1) dependency of recoveries, all experiments were averaged (Table 2).

Recovery rates for roxithromycin at five concentration levels (2, 6, 20, 200 and 2,000 µg kg−1 manure) The standard deviation (SD) for three replicates is indicated by an error bar. b Recovery rates for tiamulin at five concentration levels (2, 6, 20, 200 and 2,000 µg kg−1 manure) The standard deviation (SD) for three replicates is indicated by an error bar
Mean recoveries of 75% (RSD 21%) to 94% (RSD 36%) were obtained for the macrolides; the recoveries of salinomycin and tiamulin were 119% (RSD 26%) and 123% (RSD 15%), respectively. The limit of detection (LOD) was taken as a signal-to-noise ratio of 3:1 and the limit of quantification (LOQ) was defined as a signal-to-noise ratio of 10:1 (Table 2). This method was also applied to ivermectin, monensin and tylosin but did not give constant recovery rates for these three compounds. The method was tested for several samples in order to investigate the persistence of antibiotics in different manure samples. One of the four samples investigated contained tiamulin (43 µg kg−1) and salinomycin (11 µg kg−1): Fig. 5 shows the SRM trace of this sample. Additionally the respective farmer gave the information that both compounds had been applied two months before sampling, together with information on dosage. The manure was stored several months before it was homogenised in the manure tanks and successively sampled. This manure had a dry weight of 5% and the total organic carbon was 29 mg mL−1. The concentrations of tiamulin and salinomycin were two powers of ten lower than the expected concentrations of 2,000 µg kg−1 manure [31, 32]. This expected concentration is based on the assumption that the administered dosage of 2 kg antibiotic is excreted completely by the 800 pigs and the whole liquid manure was deposited in the 1,000-m3-manure tank.

APCI SRM traces of a manure sample from a farm that applied tiamulin and salinomycin. Tiamulin (43 µg kg−1) and salinomycin (11 µg kg−1) were measured
These antibiotics are probably not very stable in manure. Time and temperature-dependent degradation experiments are necessary to obtain more information about the long-term stability of these compounds in manure.
Conclusions
A precise and rapid multimethod with low LOQ has been developed to analyse macrolides, ionophores and tiamulin in manure. Liquid–liquid extraction followed by a diol SPE clean-up step resulted in sufficient clean extracts, which were analysed by APCI LC/MS/MS. Recoveries for the macrolides were75–94%; for the ionophore salinomycin the recovery rate was 119%, while the pleuromutilin tiamulin has a recovery rate of 123%, that is salinomycin and tiamulin are not significantly higher than 100%. Recoveries were not dependent on the concentration level. No blank problems were detected during the method validation and the applications. The limits of detection were 0.4–3.3 µg kg−1, and LOQs were 1.4–11.0 µg kg−1. In the tested samples tiamulin was found at concentrations of 43 µg kg−1 manure and salinomycin at concentrations of 11 µg kg−1. This method is more sensitive that of Hamscher et al. [26] who investigated tetracycline antibiotics in manure.
References
Rote Liste Service GmbH (2002) (eds) Rote Liste, ECV Editio Cantor Verlag für Medizin und Naturwissenschaften GmbH
Hirsch R, Ternes TA, Haberer K, Mehlich A, Ballwanz F, Kratz KL (1998) J Chromatogr A 815:213–223
Jørgensen SE, Halling-Sørensen B (2000) Chemosphere 40:691–699
Stuer-Lauridsen F, Birkved M, Hansen LP, Holten Lützhøft HC, Halling-Sørensen B (2000) Chemosphere 40:783–793
Wollenberger L, Halling-Sørensen B, Kusk KO (2000) Chemosphere 40:723–730
Neu HC (1992) Science 257:1064–1073
Winkler C, Grafe A (2000) Umweltbundesamt-Bericht 297 33 911
European Commission (2002) 153 final 2002/0073 (COD) Official Journal C 203 E:1–41
Kroker R (1983) Wissenschaft und Umwelt 4:305–308
Norwara A, Burhenne J, Spiteller M (1997) J Agric Food Chem 45:1459–1463
Kruzik P, Weiser M, Damoser J, Helsberg I (1990) Wien Tieraerztl Monat 77:141–146
Harris JA, Russell CAL, Wilkins JPG (1998) Analyst 123:2625–2628
Chan W, Gerhardt GC, Salisbury CDC (1994) J AOAC Int 77:331–333
Juhel-Gaugain M, Anger B, Laurentie M (1999) J AOAC Int 82:1046–1053
Liguoro MD, Montesissa C, Anfossi P, Angeletti R (1998) Analyst 123:1279–1282
Dreassi E, Corti P, Bezzini F, Furlanetto S (2000) Analyst 125:1077–1081
Markus JR, Sherma J (1993) J AOAC Int 76:447–448
Dubois M, Fluchard D, Sior E, Delahaut P (2001) J Chromatogr B 753:189–202
Delépine B, Hurtaud-Pessel D, Sanders P (1996) J AOAC Int 79:396–404
Lim JH, Jang BS, Lee RK, Park SC, Yun HI (2000) J Chromatogr B 746:219–225
Ali MS, Sun T, McLeroy G E, Phillippo ET (2000) J AOAC Int 83:39–52
Scarano G, Esposito M, Grasso L, Soprano V, Oliviero G (1998) Analyst 123:2541–2553
Hormazabal V, Yndestad M (2000) J Liq Chrom Rel Technol 23:1585–1598
Pfeifer T, Tuerk J, Bester K, Spiteller M (2002) Rapid Commun Mass Spectrom 16:663–669
Hamscher G, Sczesny S, Abu-Qare A, Höper H, Nau H (2000) Deut tieraerztl Woch 107:332–334
Hamscher G, Sczesny S, Höper H, Nau H (2002) Anal Chem 74:1509–1518
Aerts MML, Hogenboom AC, Brinkman UATh (1995) J Chromatogr B 667:1–40
Gasc JC, D'Ambrieres SG, Lutz A, Chantot JF (1991) J Antibiot 44:313–330
Pfeifer T, Spiteller M (2000) Rapid Commun Mass Spectrom 15:2206–2207
Bester K, Bordin G, Rodriguez A, Schimmel H, Pauwels J, VanVyncht G (2001) Fresen J Anal Chem 371:501–555
Committee for veterinary medicinal products (1997) EMEA/CVP/055/96-Final:1–41
Montforts MHMM, Kalf DF, van Vlaardingen PLA, Linders JBHJ (1999) Sci Total Environ 225:119–133
Acknowledgements
The authors are indebted to the two farmers in North-Rhine-Westphalia who cooperated in the manure sampling and provided data on the application of antibiotics during conventional farm management. The authors are grateful to Thomas Pfeifer who made a substantial contribution during the method development on the mass spectrometer. This study was supported by the Ministry for Education, Science and Research (MSWF) of North-Rhine-Westphalia (NRW), Germany.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Schlüsener, M.P., Bester, K. & Spiteller, M. Determination of antibiotics such as macrolides, ionophores and tiamulin in liquid manure by HPLC–MS/MS. Anal Bioanal Chem 375, 942–947 (2003). https://doi.org/10.1007/s00216-003-1838-9
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00216-003-1838-9