
Abstract
The ligands at the surface of a gold nanoparticle (GNP) have a significant influence on the optical and physical properties, that may render different functionalities to the GNP. Therefore, there is a need in understanding the nature of the interaction at atomic resolution in order to allow rational design of GNPs with desired physico-chemical properties. The interaction between Au\(_{79}\) and a series of small organic molecules has been systematically studied at the quantum mechanical level : methane, methanol, formic acid, hydrogen sulfide, benzene, and ammonia. The reactivity of Au\(_{79}\) has been first analyzed by performing the condensed Fukui analysis to emphasize that the surface of Au\(_{79}\) is dominated by electrophilic sites, with higher reactivity at the corner and edge atoms. The net charge transfer flowing from the organic molecules toward Au\(_{79}\) comes from the electrophilic behavior of the GNP. Furthermore, the shape of the frontier molecular orbitals of Au\(_{79}\) and of the incoming organic molecules has been found to dictate the preferred orientation of the adsorption. Several quantum chemical topological analyses of the electron density have been performed to further classify the interactions to weak dispersive or van der Waals interactions in methane and stronger non-covalent interactions in ammonia, benzene, hydrogen sulfide, methanol, and formic acid. The analysis of the electron localization function (ELF), on the other hand, provides more insight about the charge transfer, as the population of the basins of the organic molecules has decreased after interacting with Au\(_{79}\).
Similar content being viewed by others
Avoid common mistakes on your manuscript.
1 Introduction
Gold nanoparticles (GNPs), due to their versatility and unique optical properties, have largely been applied in many different fields that include catalysis, drug delivery, imaging, molecular recognition, sensors, and biomedical applications. All of these applications require the gold nanoparticles to be surrounded by ligands of an increasing complexity. The tunability of the size and geometrical shape of GNP, along with the variation in surface charge, further adds into the richness of the gold nanoparticles systems. However, we also could not exclude the role of solvent packing at the interface of gold nanoparticles, which have been shown to increase the radiosensitizing properties of gold nanoparticles [1,2,3]. Therefore, there is an interest in understanding the interaction between GNP and ligands in the presence of solvent at atomic level to allow design of optimal ligands for GNP with desired properties. The pursuit of such understanding has been undertaken by the researchers worldwide. With the development of surface-sensitive spectroscopy techniques, such as Surface plasmon resonance (SPR), attenuated total reflection Fourier transform infrared (ATR-FTIR), Sum Frequency Generation (SFG), Polarization Modulation Infrared Reflectance Absorption Spectroscopy (PM-IRRAS), Time-of-flight Secondary Ion Mass Spectrometry (TOF-SIMS), Surface Enhanced Raman Spectroscopy (SERS), Circular Dichroism (CD), and X-ray Absorption Spectroscopy (XAS), it is possible to probe into the interactions at the interface [4,5,6,7,8,9,10,11,12,13]. Computational modeling approaches have also been employed to provide atomistic insight into the nature of the interactions between nanoparticles and biomolecules [14,15,16,17,18,19,20,21,22,23,24,25,26,27,28]. Yao et al, has reported a combined experimental and theoretical study on the adsorption behavior of l-cystein on gold nanoparticles. The experimentally obtained SERS spectra have been qualitatively compared to calculated spectra for a series of cysteine geometries interacting with gold nanoclusters. They conclude that the S and O sites of cysteine are the binding sites with the nanoclusters [29]. Shao et al performed a thorough investigation on the interaction of amino acids with gold nanoparticles with classical molecular dynamics simulation. The variables considered during their simulation were the size of GNPs, the preference of either backbone or sidechain, and the influence of the solvation shell, which demonstrated the complexity of the interaction [30]. Yet, atomic investigation into the interaction with gold nanoparticles is still limited. The precise information on the interaction will present the opportunity to design optimal ligands for desired properties or to predict the bonding orientation of biomolecules.
Therefore, the goal of the current article is to decipher the nature of the interaction between Au\(_{79}\) and several small organic molecules, whose functional groups are commonly present in amino acids. The Density Functional Theory (DFT) framework was used and followed by thorough quantum chemical topological analysis to characterize the interaction. The study starts by computing the electronic property and reactivity index of Au\(_{79}\). Then, representative organic molecules, such as methane, ammonia, methanol, hydrogen sulfide, formic acid, and benzene, are adsorbed to the surface of the GNP. The interaction is investigated by electronic structure calculations and quantum chemical topological analysis of the electron density combining several theoretical tools.
2 Methodology
Systems of interest Au\(_{79}\) (1.15 nm of diameter) has been selected as the model GNP throughout the article, and representative organic molecules have been selected with commonly available functional groups: methane, ammonia, formic acid, hydrogen sulfide, benzene, and methanol (see Fig. 1). The structure of Au\(_{79}\) has been taken from the dataset for silver nanoparticles and re-optimized with gold atoms at the DFT level [31].

Left: schematic representation of Au\(_{79}\) interacting with methane, ammonia, formic acid, hydrogen sulfide, benzene, and methanol. Right: different interaction sites on Au\(_{79}\)
Fukui analysis The reactivity of Au\(_{79}\) is analyzed by computing the Fukui function f(r) to identify the electrophilic and nucleophilic sites. Investigations on the Au clusters [32], AuNPs [33], and Au [111] surface [34] based on the Fukui’s frontier molecular orbitals theory have demonstrated the effectiveness of the analysis on the prediction of their reactivity. Fukui function analysis at the DFT level is defined as follows:
with \(\mu \) as the chemical potential, \(\upsilon (r)\) as the external potential, \(\rho (r)\) as the electron density and N as the total number of electrons. Based on equation (1), Fukui function can be defined as the sensitivity of the chemical potential to a change in the external potential and as the change in the electronic density as the number of electrons change. Therefore, the electrophilic (\(f^{-}\)) and nucleophilic attacks (\(f^{+}\)) can be evaluated using a finite different approach:
Yang and Mortier proposed a method that allows evaluating the Fukui function at the atomic center [35], which is also known as condensed Fukui function, in terms of the charges, as follows:
with k indices representing the atomic centers. With the introduction of dual descriptors, the simultaneous nucleophilic and electrophilic behaviors of the system at point r have been made possible. This dual descriptor, called \(\Delta f\), is defined as the difference between \(f^{+}\) and \(f^{-}\). When \(\Delta f > 0\) the atom behaves as electrophilic species and when \(\Delta f < 0\), the atom behaves as nucleophilic species [36, 37]. In this study, the analytic Fukui function calculation has been performed as implemented in deMon2k [38, 39], which uses auxiliary density perturbation theory (ADPT). ADPT is based on the variational fitting of the coulombic potential and the associated approximation of exchange-correlation energy.
DFT calculations All DFT calculations have been performed with deMon2k code [40], which generates auxiliary density that are variationally fitted against the Kohn–Sham density, resulting in reduced computational cost that depends on the number of auxiliary functions, instead of the size of the basis set. The electronic properties, such as frontier molecular orbitals were initially calculated for Au\(_{79}\), with unrestricted Kohn–Sham method (convergence criteria: for SCF (self-consistency field convergence) tolerance of \(1.0.10^{-9}\) a.u., for CDF (auxiliary density convergence) tolerance of \(5.0.10^{-7}\) a.u.). Initially, the organic molecules were placed at the three different positions as shown in Fig. 1. Calculations were performed using the PBE functional, with empirical dispersion added [41]. DZVP Gaussian-type basis sets were used for all the organic molecules and relativistic large-core effective core potential (11 valence electrons treated explicitly) with the associated basis set for Au [42]. Local geometry optimizations were performed with the criteria of \(1.0.10^{-4}\) a.u. and with \(1.0.10^{-7}\) a.u. and \(1.0.10^{-5}\) a.u. for SCF and CDF tolerance, respectively. Due to the small electronic gap and degenerated orbitals between the frontier molecular orbitals (MOs), the spin multiplicity was set to 2 at the beginning of the calculation and a smearing process was activated to allow fractional occupation of orbitals with energy difference of 0.01 a.u.. The interaction energy, \(E_\mathrm{INT}\), was obtained via:
with the \(E_\mathrm{TOT}\) the energy of the system, \(E_\mathrm{GNP}\) the electronic energy of Au\(_{79}\), and \(E_\mathrm{ORG}\) the electronic energy of the respective organic molecule at the geometry of the full system. Basis set superposition error (BSSE) was estimated for several complexes but frequently, the open-shell nature of the systems with different basis sets results to SCF convergence issues. Consequently, BSSE corrections were not included in the interaction energies to simplify the calculations. The provided interaction energies are thus qualitative but the underlying trends are preserved. The charge transfer (CT) was obtained by subtracting the total charge of Au\(_{79}\) before and after interacting with organic molecules using different population schemes.
Quantum chemical topology analysis All quantum chemical topological analyses [43, 44] were performed with the Multiwfn code [45], by analyzing the electron density generated from the deMon2k code with tight SCF and CDF criteria of \(1.0.10^{-9}\) a.u. and \(5.0.10^{-7}\) a.u. respectively. Calculations of bond critical points in the quantum theory of atoms in molecule (QTAIM) framework were performed [46, 47], associated to the computation of several properties, such as the electron density \(\rho (r)\), the energy density H(r), and the electron density Laplacian descriptor to characterize the bonding [48]. Then, the electron localization function (ELF) basins of the system were calculated [49], to observe the change in the population of the basins as the molecule interact with Au\(_{79}\). Lastly, the analysis of Non-Covalent Interactions(NCI) were calculated based on the reduced density gradient (RDG) [50].
3 Results and discussion
3.1 Electronic properties of Au\(_{79}\)
We initially calculated the electronic properties of Au\(_{79}\), and the five highest occupied and five lowest unoccupied molecular orbitals (HOMO and LUMO, respectively) are reported in Figure S1. Considering the degenerated molecular orbitals, the fractional occupation of the orbitals has been allowed during the calculation. Therefore, the five HOMOs could act as both electron donor and electron acceptor, while the LUMO can only act as the electron acceptor. The reactivity of Au\(_{79}\) was then analyzed by performing both condensed Fukui analysis and the electrostatic potential (ESP) calculation to pinpoint the nucleophilic and electrophilic sites. On the left of Fig. 2, the plot of \(\Delta f\) function is shown, with the blue lobes (positive isosurface) corresponding to the electrophilic sites and red lobes (negative isosurface) to the nucleophilic sites (the isosurfaces of \(f^{+}\), \(f^{-}\) and the average between the two are available in Figure S2, numerical values are provided in Table S1). In this representation, the surface of Au\(_{79}\) is more susceptible to the attacks by nucleophile species, while the area sensitive to the electrophile attack is within the core of the GNP. Furthermore, the size of the electrophilic lobes at the corner and edges positions are larger than that of the face positions, which implies an increased reactivity at these sites. [33] As a result, nucleophiles are more likely to adsorb on the corner and edges atoms rather than the face atoms. The electrostatic potential of Au\(_{79}\) is shown in Fig. 2 right, where the blue isosurface represents the positive potentials (electrophilic sites) and the red isosurface represents the negative potentials (nucleophilic sites). In this analysis, similarly, the surface of GNP has been dominated by the electrophilic sites, while the nucleophilic sites are rather localized at the top and face positions of GNP. Therefore, the ESP also predicts that GNP behaves more as an electrophile and that nucleophile species prefer interacting with the corner and edges atoms rather than the atoms at the surface.

Left: representation of the condensed Fukui analysis of Au\(_{79}\) (isovalue of 0.00025): the blue lobes represent the electrophilic sites and the red lobes the nucleophilic sites. Right: electrostatic potential of Au\(_{79}\) with the blue isosurface (+0.005) represents positive potentials and red isosurface (−0.001) represents negative potentials
3.2 Interaction with small organic molecules
To validate the prediction, geometry optimizations of Au\(_{79}\) interacting with the representative organic molecules were performed. The molecules were initially placed at the three different sites (Fig. 1). The optimized geometries for different binding sites along with the interaction energy and charge transfer are shown in Figure S3. From the optimization step, several observations can be made. Firstly, the preferred binding sites vary with the ligand, but in most cases they prefer either the corner or edge sites to the face sites, which is consistent with the Fukui analysis. Secondly, the interaction is considered as weak with an interaction energy range between −0.18 and −0.72 eV. The order of interaction strength, considering the most favorable binding site for each system, is found to follow this trend: methane, formic acid, methanol, hydrogen sulfide, benzene, and ammonia, as shown in Fig. 3. Methane, considered as weak nucleophile and electrophile, interacts very weakly with Au\(_{79}\), while the other organic molecules form stronger interaction through their functional groups. The availability of lone pairs on these moieties could render them as the anchoring points on the surface of GNP.

Trend in interaction energy (ev) and charge transfer (e) using the ADCH [51] population scheme across the different organic molecules
Thirdly, CT was calculated using different population schemes, i.e. Mulliken, Hirshfeld, Atomic Dipole Corrected Hirshfeld (ADCH), Voronoi, Becke, and Bader analyses, as the change in the total charge of Au\(_{79}\) before and after interacting with the ligand. In most cases, GNP receives electrons which implies that it is acting as electrophile, except in the case of the interaction with methane computed using Hirshfeld and ADCH population schemes. This behavior is consistent with the Fukui analysis. Depending on the population scheme, the calculated CT does not exactly follow the trend for the interaction energy (see Figure S4). Among them, the ADCH scheme provides a good correlation as shown in Fig. 3. However, due to the arbitrary nature of the population analysis, and the different reactivity of the molecules investigated, it is less straightforward to draw a direct comparison between the charge transfer and interaction energy. Benzene, owing to the relatively richer \(\pi \)-electron, is more likely to donate more electrons, which has been shown by the different population schemes (Figure S4). Methanol and formic acid, despite having two lone pairs, do not donate more electron than ammonia, with one lone pair on the nitrogen atom. Therefore, further analysis on the interaction would be necessary in order to probe the possibility of other contributing factors to the interaction energy, in addition to the charge transfer.
Lastly, the possible role of the shape of MOs in the orientation of the organic molecules at the surface is investigated. Considering that organic molecules behave as electron donors, the HOMO is expected to interact with empty MOs of GNP (Figure S1). It turns out that the shape of MOs does influence the orientation of the organic molecules (Fig. 4). In the example of ammonia and benzene molecules, the HOMO approaches the GNP and re-orientates itself to maximize the overlap with the MOs of Au\(_{79}\). In all of the geometries for ammonia and benzene interacting with GNP (Figure S3), ammonia always adopts the same orientation where the hydrogen atoms are pointing away from the surface while nitrogen atom is pointing toward the surface. Meanwhile, the benzene always adopts a flat configuration and the perpendicular configuration is not stable (back to the flat orientation). The same behavior is expected for the other organic molecules as well. Therefore, the MOs of the two species are important in determining the orientation of the ligand, which will be helpful in prediction of possible interaction between GNP of any size with other organic molecules.

Approaching HOMO of ammonia (left) and benzene (right) to Au\(_{79}\)
3.3 Quantum chemical topological analysis
To further complement the analysis of the interaction, a thorough quantum chemical topological analysis on each system is presented to gain more insight into the nature of the interaction.
3.3.1 BCP analysis
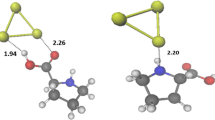
In the QTAIM framework developed by Bader and co-workers [52], we performed a search for the bond critical points (BCP) and bond paths (BP) with the focus on the interaction sites. BCPs and BPs are present for every system that was analyzed, validating the presence of bonds for the interaction. To characterize the nature of these bonds, several local properties were calculated, i.e. electron density, the Laplacian of the electron density and the energy density, at these BCPs based on the classification proposed by Bianchi and coworkers [48]. The BCPs and properties of the gold-molecule interaction regions are shown in Table 1, in Fig. 5 and Figure S5.

BCP analysis and the respective RDG and NCI analysis on methane (top) and formic acid (bottom)
The electron density at each BCP is rather small, in contrast to covalent bonds (whose electron density is greater than 0.1 a.u.), which confirms weak interaction. For some of the ligands, i.e. methanol, formic acid, and benzene, there are two BCPs, which indicate the presence of another weak interaction site. Subsequently, the Laplacian for all of the systems is greater than 0, which further clarifies that the interaction is not covalent. The discriminating factor will then be the energy density, whose sign is a good indicator of the stability of the interaction. According to the classification [48], the interaction formed with methane is classified as van der Waals interactions (H(r)\(>0\)). Meanwhile, the other organic molecules form at least one interaction, which is classified as dative bonding and characteristic of ligand to metal interactions (H(r)\(<0\)). In the case of methanol, two bond critical points are found, one is dative interaction and the other is van der Waals interaction (which forms between the methyl group and the surface). In the case of formic acid, we also found two BCPs between the oxygen of carbonyl group and –OH group with the H pointing to the surface of Au\(_{79}\), respectively. The interaction between the oxygen of carbonyl group is supposedly stronger than that of the –OH, as seen from the electron density. However, it is clear that this interaction contributes to the overall interaction energy. The quantum chemical topological analysis of formic acid oriented differently (see configuration 1 in Figure S6) at the surface reveals that the second interaction point is not favorable, which result in a lower value of the interaction energy. Therefore, the orientation of the ligand at the surface plays a significant role in determining the interaction energy of a system. Lastly, two bond critical points were found for benzene, with the formation of two dative bonds.
3.3.2 ELF analysis
Then, the ELF analysis was performed. As expected for such weak interactions, we do not observe the presence of a valence basin that signifies covalent interaction between the two interacting species. Nevertheless, the population of the detected basins that belong to the ligands was investigated (Table 2). Interestingly, a decrease in the overall population of the ligands interacting with Au\(_{79}\), as compared to the isolated molecule, is highlighted. For CH\(_{4}\), we observe a very small change in the total population of the basins, which supports the fact that methane forms a very weak interaction with GNP. For ammonia, the population of the lone pair of the nitrogen atom decreases from 2.109 (isolated molecule) to 1.871 upon interaction with GNP. As for H\(_{2}\)S, the population of the lone pairs also decreases and rearranges from 2.127 each to 1.840 and 2.186, respectively. Similarly, the population of the lone pairs of methanol also changes from 2.412 and 2.368 to 2.728 and 1.898. However for formic acid, the population of the lone pairs of both oxygen atoms slightly increases from 4.311 and 5.117 for –OH and C=O respectively, to 4.231 and 5.261, which could probably due to the formation of favorable interaction between Au\(\cdots \)H–O. Lastly, the total population of the benzene changes slightly, from 29.293 to 29.228. This analysis demonstrates that the organic ligands form interaction with Au\(_{79}\) that involves intermolecular charge transfer. ELF results are consistent with the CT population analysis in section 3.2 and the electrophilic character of GNP.
To check the possibility of back donation from Au\(_{79}\) to the ligands, the contribution of gold atoms to the basins of organic ligands was calculated. The results are shown in the last column of Table 2. Though the contribution is small, but it is still non-negligible, and it demonstrates the possibilities of the overlapping of the LUMO of ligands and HOMO of Au\(_{79}\), which results in stronger interactions. [53, 54]
3.3.3 Non-covalent interaction analysis
The NCI analysis, developed by Johnson and coworkers [50], allows the detection and the visualization of weak interactions, such as steric effects, hydrogen bonds, and dispersion interactions. Here, NCI analysis was performed to identify the weak interaction between the Au\(_{79}\) and the organic molecules. The plots of the reduced density gradient with respect to the electron density multiplied by the sign of the second eigenvalues of the Hessian matrix [55] restricted to the intermolecular regions are shown in the middle parts of Fig. 5 and of Figure S5. Dative interactions are found to overlap with metal-metal interaction, while the dispersion/van der Waals interactions are seen at a very low electron density. The color-coded NCI basins representation [50, 55] on the right parts of Fig. 5 clearly reveal non-covalent interactions. The green-colored basins between the interacting species represents the weak interactions, i.e. dispersive interactions, as in the case of methane. The color gradually changes to blue as the interaction shifts to the stronger dative interactions as in the case of ammonia. There is a consistent trend observed between this analysis and AIM results in the previous section, where multiple interaction basins have been observed on some of the molecules. There are two interaction points for methanol, with one strong dative interaction and one weak dispersive interaction (NCI basis and RDG plots). Similarly, the formic acid forms one strong dative interaction with one relatively weaker dative interaction. For benzene, two strong dative interactions are identified. Again, this analysis further demonstrates the multiple cooperating weak interactions that eventually contribute to the overall interaction energy.
4 Conclusion
To summarize, we have performed Fukui function and electrostatic potential analysis to identify the reactivity of Au\(_{79}\), based on the electrophilic and nucleophilic sites. These reactivity descriptors predicted that the Au\(_{79}\) is more likely to behave as an electrophilic species, with the corner and edges atom identified as the more reactive sites as compared to the surface sites. This prediction has been fulfilled as the geometry optimizated GNP-organic molecules complex indeed demonstrated stronger interaction energy at the corner and edge sites of Au\(_{79}\) as compared to the face sites (0.1 to 0.2 eV). Furthermore, the electron rich moieties (O, S, C=O, \(\pi \) electrons) of the organic molecules act as anchor points on the surface of Au\(_{79}\). The subsequent calculation of charge transfer shows that Au\(_{79}\) behaves as an electrophile as it accepts electrons from the incoming organic molecules. Among several population analyses, only the dipole-corrected Hirshfeld analysis provides charge transfer which follows the interaction energy trend. Possibly, charge transfer is not the unique contributing factor. Meanwhile, the role of the shapes of frontier molecular orbitals has been proposed to be important in determining the orientation of organic molecules on the surface. Therefore, various quantum chemical topological analyses have been performed to gain deeper insight into the interaction.
The analysis of bond critical points and bond paths in the AIM framework has classified the bond forming between methane to be weak van der Waals interaction, while the other organic molecules form at least one dative interaction. Moreover, the analysis on the different geometries of formic acid has demonstrated the importance of structural orientation in the overall interaction energy. The ELF analysis has helped in following the rearrangement of the electron density, which supports the charge flow from the ligand to GNP. Lastly, NCI analysis highlights weak interactions, which are consistent with the calculation of BCPs in QTAIM. This global quantum chemical topological analysis allows identification of the multiple interaction points for organic molecules, whose contribution to the overall interaction energy could be important.
All in all, this study has investigated thoroughly the interaction between Au\(_{79}\) with small organic molecules. The conclusion from this study is a first step in predicting the way larger molecules, with multiple functional groups, would interact with GNPs.
References
Sicard-Roselli C, Brun E, Gilles M, Baldacchino G, Kelsey C, McQuaid H, Polin C, Wardlow N, Currell F (2014) Small 10(16):3338. https://doi.org/10.1002/smll.201400110
Baldacchino G, Brun E, Denden I, Bouhadoun S, Roux R, Khodja H, Sicard-Roselli C (2019) Cancer Nanotechnol 10(1):3. https://doi.org/10.1186/s12645-019-0047-y
Gilles M, Brun E, Sicard-Roselli C (2018) J Colloid Interface Sci 525:31. https://doi.org/10.1016/j.jcis.2018.04.017
Iosin M, Toderas F, Baldeck PL, Astilean S (2009) J Mol Struct 924–926:196. https://doi.org/10.1016/j.molstruc.2009.02.004
Schwenk N, Mizaikoff B, Cárdenas S, Lopez-Lorente AI (2018) Analyst 143:5103. https://doi.org/10.1039/C8AN00804C
Carnovale C, Bryant G, Shukla R, Bansal V (2018) Phys Chem Chem Phys 20(46):29558. https://doi.org/10.1039/C8CP05938A
Brancolini G, Kokh DB, Calzolai L, Wade RC, Corni S (2012) ACS Nano 6(11):9863. https://doi.org/10.1021/nn303444b
Novelli F, BernalLopez M, Schwaab G, RoldanCuenya B, Havenith M (2019). J Phys Chem B. https://doi.org/10.1021/acs.jpcb.9b02358
Shen YR, Ostroverkhov V (2006) Chem Rev 106(4):1140. https://doi.org/10.1021/cr040377d
Velasco-Velez JJ, Wu CH, Pascal TA, Wan LF, Guo J, Prendergast D, Salmeron M (2014) Science 346(6211):831. https://doi.org/10.1126/science.1259437
Hoarau M, Badieyan S, Marsh ENG (2017) Org Biomol Chem 15(45):9539. https://doi.org/10.1039/C7OB01880K
Gao J, Hu Y, Li S, Zhang Y, Chen X (2013) Spectrochim Acta A Mol Biomol Spectrosc 104:41. https://doi.org/10.1016/j.saa.2012.11.103
Petit T, Yuzawa H, Nagasaka M, Yamanoi R, Osawa E, Kosugi N, Aziz EF (2015) J Phys Chem Lett 6(15):2909. https://doi.org/10.1021/acs.jpclett.5b00820
Tang M, Gandhi NS, Burrage K, Gu Y (2019) Langmuir 35(13):4435. https://doi.org/10.1021/acs.langmuir.8b03680
Rosa M, DiFelice R, Corni S (2018) Langmuir 34(49):14749. https://doi.org/10.1021/acs.langmuir.8b00065
Le JB, Cheng J (2020) Curr Opin Electrochem 19:129. https://doi.org/10.1016/j.coelec.2019.11.008
Ramezani F, Amanlou M, Rafii-Tabar H (2014) J Nanoparticle Res 16(7):2512. https://doi.org/10.1007/s11051-014-2512-1
Hong G, Heinz H, Naik RR, Farmer BL, Pachter R, Appl ACS (2009) Mater Interfaces 1(2):388. https://doi.org/10.1021/am800099z
Lee SS, Kim B, Lee S (2014) J Phys Chem C 118(36):20840. https://doi.org/10.1021/jp412438f
Hoefling M, Iori F, Corni S, Gottschalk KE (2010) Langmuir 26(11):8347. https://doi.org/10.1021/la904765u
Wang JG, Selloni A (2009) J Phys Chem C 113(20):8895. https://doi.org/10.1021/jp901842p
El-Mageed HRA, Taha M (2019) Liq J Mol 296:111903. https://doi.org/10.1016/j.molliq.2019.111903
Xu Z, Yuan SL, Yan H, Liu CB (2011) Colloid Surf A Physicochem Eng Asp 380(1):135. https://doi.org/10.1016/j.colsurfa.2011.02.046
Feng J, Slocik JM, Sarikaya M, Naik RR, Farmer BL, Heinz H (2012) Small 8(7):1049. https://doi.org/10.1002/smll.201102066
Feng J, Pandey RB, Berry RJ, Farmer BL, Naik RR, Heinz H (2011) Soft Matter 7(5):2113. https://doi.org/10.1039/c0sm01118e
Hoefling M, Iori F, Corni S, Gottschalk KE (2010) ChemPhysChem 11(8):1763. https://doi.org/10.1002/cphc.200900990
Domínguez-Castro A, Hernández D, Guzmán F (2017) Theor Chem Acc 136:7. https://doi.org/10.1007/s00214-017-2118-7
DarvishGanji M, TavassoliLarijani H, Alamol-hoda R, Mehdizadeh M (2018) Sci. Rep. 8(1):11400. https://doi.org/10.1038/s41598-018-29887-5
Yao G, Huang Q (2018) J Phys Chem C 122(27):15241. https://doi.org/10.1021/acs.jpcc.8b00949
Shao Q, Hall CK (2016) Langmuir 32(31):7888. https://doi.org/10.1021/acs.langmuir.6b01693
Barnard A, Sun B, MotevalliSoumehsaraei B, Opletal G (2017) https://data.csiro.au/collections/#collection/CIcsiro:23472v3/DItrue. Silver nanoparticle data set
SekharDe H, Krishnamurty S, Pal S (2010) J Phys Chem C 114(14):6690. https://doi.org/10.1021/jp1004852
Allison TC, Tong YJ (2011) Phys Chem Chem Phys 13:12858. https://doi.org/10.1039/C1CP20376B
Chrétien S, Gordon MS, Metiu H (2004) J Chem Phys 121(8):3756. https://doi.org/10.1063/1.1769366
Yang W, Mortier WJ (1986) J Am Chem Soc 108(19):5708. https://doi.org/10.1021/ja00279a008
Martínez J (2009) Chem Phys Lett 478(4):310
Morell C, Grand A, Toro-Labbé A (2006) Chem Phys Lett 425(4):342
Flores-Moreno R, Melin J, Ortiz JV, Merino G (2008) J Chem Phys 129(22):224105. https://doi.org/10.1063/1.3036926
Flores-Moreno R (2010) J Chem Theory Comput 6(1):48. https://doi.org/10.1021/ct9002527
Geudtner G, Calaminici P, Carmona-Espíndola J, Campo JMD, Domínguez-Soria VD, Moreno RF, Gamboa GU, Goursot A, Köster AM, Reveles JU, Mineva T, Vásquez-Pérez JM, Vela A, Zúñinga-Gutierrez B, Salahub DR (2012) WIREs Comput Mol Sci 2(4):548. https://doi.org/10.1002/wcms.98
Goursot A, Mineva T, Kevorkyants R, Talbi D (2007) J Chem Theory Comput 3(3):755. https://doi.org/10.1021/ct600373f
Ross RB, Powers JM, Atashroo T, Ermler WC, LaJohn LA, Christiansen PA (1990) J Chem Phys 93(9):6654. https://doi.org/10.1063/1.458934
Popelier PLA, Aicken FM (2003) ChemPhysChem 4(8):824. https://doi.org/10.1002/cphc.200300737
Popelier PLA (2014) in The Chemical Bond (Wiley-VCH Verlag GmbH & Co. KGaA 271–308. https://doi.org/10.1002/9783527664696.ch8
Lu T, Chen F (2012) J Comput Chem 33(5):580. https://doi.org/10.1002/jcc.22885
Bader RFW, MacDougall PJ, Lau CDH (1984) J Am Chem Soc 106(6):1594. https://doi.org/10.1021/ja00318a009
Bader RFW (1998) J Phys Chem A 102(37):7314. https://doi.org/10.1021/jp981794v
Bianchi R, Gervasio G, Marabello D (2000) Inorg Chem 39(11):2360. https://doi.org/10.1021/ic991316e
Becke AD, Edgecombe KE (1990) J Chem Phys 92(9):5397. https://doi.org/10.1063/1.458517
Johnson ER, Keinan S, Mori-Sánchez P, Contreras-García J, Cohen AJ, Yang W (2010) J Am Chem Soc 132(18):6498. https://doi.org/10.1021/ja100936w
Lu T, Chen F (2012) J Theor Comput Chem 11(01):163. https://doi.org/10.1142/s0219633612500113
Bader RFW (1985) Acc Chem Res 18(1):9. https://doi.org/10.1021/ar00109a003
Puyo M, Lebon E, Vendier L, Kahn ML, Fau P, Fajerwerg K, Lepetit C (2020) Inorg Chem 59(7):4328. https://doi.org/10.1021/acs.inorgchem.9b03166
Bulteau Y, Lepetit C, Lacaze-Dufaure C (2020) Inorg Chem 59(24):17916. https://doi.org/10.1021/acs.inorgchem.0c01972
Laplaza R, Peccati F, Boto RA, Quan C, Carbone A, Piquemal JP, Maday Y, Contreras-García J (2020) WIREs Comput Mol Sci 11:2. https://doi.org/10.1002/wcms.1497
Acknowledgements
The authors thank Dr. D. Lauvergnat and Dr. J.P. Dognon for a careful reading of the manuscript and helpful discussions. This work was performed using HPC resources from the GENCI (CINES/IDRIS, Grant No. 2020-A0080806830).
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Published as part of the special collection of articles “20th deMon Developers Workshop”.
Supplementary Information
Valence molecular orbitals of Au79, condensed Fukui analysis, geometries, interaction energies and charge transfers for the different interaction sites, charge transfer from various population analyses, and quantum chemical topological data are provided in Supplementary Information. A set of coordinates for the different complexes is also provided. Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Tandiana, R., Van-Oanh, NT. & Clavaguéra, C. Interaction between organic molecules and a gold nanoparticle: a quantum chemical topological analysis. Theor Chem Acc 140, 118 (2021). https://doi.org/10.1007/s00214-021-02821-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00214-021-02821-1