
Abstract
The geometrical parameters, the electronic structures and the nature of the chemical bonding for complexes of the type (NqMes)M[Cl(CO)2] (NqMes = naphtoquinone-annulated NHC ligand and M = V, Mn, Re, Co, Rh, Cu) which possess even electron counts have been investigated at the BP86 and B3LYP levels of theory using orbital molecular and energy decomposition analyses. The coordination of the six-membered ring is strongly disfavoured (structures A), while the structures B1 and B2 which differ by the orientation of the M[Cl(CO)2] unit are in competition with regard to the metal’s nature and the spin state. The carbene–metal bonds are governed chiefly by electrostatic attractions contributing 65–75% of the bonding attractive interactions. The carbene–metal bonds are strong in electron-moderately rich Re element and weak in electron-rich Cu metal. In the most cases, orbital interactions are more important in symmetrical species than in the unsymmetrical ones, which are mainly described by σ-bonding with the π-backbonding part less than 20%.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
1 Introduction
The N-heterocyclic carbenes (NHCs) [1, 2] ligands have always played a central role in the development of organometallic chemistry and have found remarkable usefulness for modulating the activities of transition metals [3,4,5]. The first reports on the isolation of the complexes containing N-heterocyclic carbenes (NHCs) are known since more than four decades [6,7,8,9]. This breakthrough has opened an outstanding route facilitating its development due to their significant activity in homogeneous catalysis, where a new class of catalysts has lately been developed with N-heterocyclic carbenes rather than organophosphine ligands [3]. A wide variety of NHC–metal complexes with early and late transition metals exist, and many methods to generate them are known [10,11,12]. The understanding of the nature governing the interaction between metals and NHCs has been intensively investigated. It is well known that N-heterocyclic carbenes (NHCs) are strong σ-donor ligands that coordinate to transition metals; however, the π-backdonation in NHC-transition metal complexes was revealed to be negligible as elucidated in previous experimental and theoretical works [13, 14]. Recently, many efforts have been devoted in both theoretical and experimental aspects to clarify the importance of the steric and electronic contributions to the NHC–metal bonding, where several calculations pointed out the existence of both σ- and π-type interactions between the carbene and the metal ions [15,16,17,18,19,20,21].
The DFT study has been achieved on a series of neutral complexes of the type (NqMes)M[Cl(CO)2] (NqMes = naphtoquinone-annulated NHC “Fig. 1” and M = V, Mn, Re, Co, Rh, Cu) to establish a stability order between different conformations for same transition metal in function of the spin state and to provide an in-depth look into the nature of the interactions governing the NHC–Metal bonding depending on the nature of the metal ion and the orientation of the MCl(CO)2 fragment. The BP86 and B3LYP functionals have shown their utility in determining the electronic, structural and interaction types within the monometallic complexes of various molecular structures [22,23,24,25,26,27,28,29,30,31,32].

Atom numbering (left) and BP86-optimized bond distances in Å (right) of the NHC ligand (NqMes = naphtoquinone-annulated NHC) of C2v symmetry
2 Computational details
All calculations were carried out using the 2016.01 version of the Amsterdam Density Functional (ADF) program [33] developed by Baerends and co-workers by means of the density functional theory (DFT) [34,35,36,37,38]. Electron correlation was treated within the local density approximation (LDA) in the Vosko–Wilk–Nusair parametrization [39]. The generalized gradient approximation BP86 [40,41,42,43] and the hybrid-type B3LYP functionals [44, 45] have been used for calculations. The atom electronic configurations were described by a triple-\( \varsigma \)Slater-type orbital (STO) basis set for H 1s, C 2s and 2p, N 2S and 2p, O 2s and 2p augmented with a 3d single-\( \varsigma \) polarization for C, N and O atoms and with a 2p single-\( \varsigma \) polarization for H atoms. A triple-\( \varsigma \)Slater-type orbital basis set for Cl 3s and 3p augmented with a 3d single-\( \varsigma \) polarization for P and Cl atoms. A triple-\( \varsigma \) STO basis set was used for the first-row transition metals 3d and 4s augmented with a single-\( \varsigma \) 4p polarization function, for the Rh 4d and 5s augmented with a single-\( \varsigma \) 5p polarization function for Rh, and a triple-\( \varsigma \) STO basis set was used for the for Re 5d and 6s augmented with a single-\( \varsigma \) 6p polarization function for Re. The scalar relativistic zero-order regular approximation (ZORA) was used for the heavy Rh and Re atoms, with the associated optimized valence basis set [46,47,48]. The BP86 calculations were performed assuming the frozen–core approximation up to 1s for C, 2p for Cl, 3p for the first-row metals, 4p for Rh and 4f for Re [34,35,36,37,38]. Full geometry optimizations were carried out using the analytical gradient method implemented by Versluis and Ziegler [49]. Vibrational frequency calculations were performed on all the optimized geometries to verify that these structures are true minima on the potential energy surface [50, 51]. All the energy values reported in the supplementary information include zero-point energy correction. Representations of the orbitals and of the molecular structures were done by using the ADF-GUI [33] programs. The natural population-based (NPAI) [52] were obtained by the NBO 6.0 program [53, 54].
3 Results and discussion
In order to understand the bonding type, the electronic structures and the electronic transfers into the series of (NHC)M[Cl(CO)2] complexes, full geometry optimizations and the energy decomposition analysis have been achieved. Indeed, two different sites of coordination of the ligand have been envisaged to establish the stability order between isomers depending on the metal nature and the spin state. The first site of coordination is relative to the external six-membered ring (configuration A), while the second one (configuration B) to two conformations: one symmetrical B1 (Cs symmetry) and other unsymmetrical B2 (C1 symmetry) which are obtained from each other by rotation as illustrated in Scheme 1.

Possible conformations arising from the coordination between the NHC ligand and the M[Cl(CO)2] fragment
4 Vanadium complexes
Several previous works giving rise to vanadium cation with different oxidation states have been reported [10]. In the frame of this work, optimized structures of the V(I) complexes showed that the V[Cl(CO)2] fragment prefers to be bound to the carbene site rather than the six-membered ring as gathered in Table 1 and illustrated in Fig. 2. The unsymmetrical V–B2(T) isomer in its triplet state corresponds to the global minimum obtained by both BP86 and B3LYP adopting a pseudo-tetrahedral geometry with short V–C(1) and V–Cl bond distances of 2.094 and 2.258 Å, respectively, putting emphasis on strong interactions between the metal and the carbon of the NHC ligand. However, the V–B2(S) isomer of singlet state lies 8.7 (BP86) or 7.6 kcal/mol (B3LYP) above its homolog V–B2(T) as global minimum, adopts a trigonal pyramidal geometry around the metal ion with slightly short V–C and V–Cl bond distances (Table 1) and exhibits a small HOMO–LUMO gap of 0.20 eV (BP86).

BP86-optimized structures for (NHC)V[Cl(CO)2] complexes. S and T designated the singlet and triplet spin states, respectively. Relative energies between isomers ΔE1 (BP86) and ΔE2 (B3LYP) are given in kcal/mol
Both symmetrical V–B1(S) and V–B1(T) complexes adopt a trigonal pyramidal geometry around the vanadium cation V(I) showing resemblances with open OC–V–CO angles of about 180° and lying above their homologous of the unsymmetrical structures. The V–B1(S) structure displays a small HOMO–LUMO gap of 0.15 (BP86) or 0.68 eV (B3LYP). For both V–B1(T) and V–B2(T), the two unpaired electrons are mainly localized on the vanadium ion as evidenced by the spin density values of 2.02 and 1.91 as given in Table 1.
The electronic configuration of the metal in all vanadium cases shows significant donation (depopulation of the 3d AO) and backdonation into the 4s AO (population of the 4s AO) as given in Table 1.
The elongation of N–C bond lengths from 1.378 in free ligand to 1.383 Å in V–B2(S) is consistent with the π-backbonding from the filled orbitals of the metallic fragment into the vacant π * C–N ones of the carbene fragment.
It is important to note that the energy barriers of the rotation of the V[Cl(CO)2] fragment for this kind of compounds are in the range of 17–33 kcal/mol; thus, the rotational movement changes the stability order between the isomers favouring the unsymmetrical structures.
Despite its full η6-coordination mode, the V–A(S) and V–A(T) species are found high in energy by both BP86 and B3LYP used methods exhibiting a small HOMO–LUMO gap of 0.26 (BP86), but large one of 1.74 eV (B3LYP) is obtained very high in energy than the global minimum V–B2(T) of triplet spin state.
5 Manganese and rhenium complexes
The optimized structures of the NHC ligand coordinated to the M[Cl(CO)2] (M = Mn and Re) of A, B1 and B2 ones showed discrepancies with regard to the stability order between isomers in each case (Table 2, Fig. 3). The Mn and Re metals of group VII have not the same ground state as illustrated in table and figure, where numerous works have been reported with Mn and Re in low oxidation state +I [11]. For the manganese element, the ground state corresponds to a triplet state whatever the coordinated site obtained by both BP86 and B3LYP methods. Indeed, the Mn–B2(T) triplet structure obtained without symmetry constraints is found as the global minimum calculated more stable than its homolog of singlet state by 17.5 (BP86) and 17.9 kcal/mol (B3LYP). Both Mn–B2(S) and Mn–B2(T) exhibit comparable square planar geometry around the Mn(I) metal cation corresponding to a deficient 14-MVE configuration. Furthermore, the symmetrical isomers of conformation B1, merely Mn–B1(S) and Mn–B1(T) are found less stable than the global minimum by 20.7 and 5.9 (BP86) or 28.7 and 6.5 kcal/mol (B3LYP), respectively. Unlike, for the rhenium structures, the ground state is the symmetrical Re–B1(S) of singlet spin state exhibiting large HOMO–LUMO gap of 1.00 (BP86) or 2.27 eV (B3LYP), in accordance with the ∆ES/T energy splitting of 34.2 (BP86) or 38.0 kcal/mol (B3LYP). The structures B2 are obtained from those of B1 ones by rotation giving rise to structures less stable regardless the spin state. Indeed, the Re–B1(S) and Re–B2(S) species of the same conformation adopt a pseudo-tetrahedral geometry around the Re(I) ion.

BP86-optimized structures for (NHC)M[Cl(CO)2] (M = Mn, Re) complexes. S and T designate the singlet and triplet spin states, respectively. Relative energies between isomers ΔE1 (BP86) and ΔE2 (B3LYP) are given in kcal/mol
The energy barriers of the rotation of the Re[Cl(CO)2] fragment between the isomers of singlet state are 25.0 kcal/mol; the rotational movement changes the stability order between the isomers favouring the symmetrical structure. Thereby, the stability order is shifted in favour of the symmetrical Re–B1(S) singlet state, which adopts a pseudo-tetrahedral geometry around the Re(I) metal cation, with an open C(1)–Re–Cl angle of 119 and 118° and C(1)–Re–CO angle of 90 and 126°, respectively.
The elongation of N–C bond lengths from 1.378 Å in free ligand to 1.392 Å in Mn–B1(S) is consistent with the π-backdonation from the filled orbitals of the Mn[Cl(CO)2] metallic fragment into the vacant π * C–N ones of the carbene fragment. Similarly, the lengthening of the N–C bond distances from 1.376 Å in Re–B2(S) to 1.389 Å in Re–B1(S) highlights clearly the π-backdonation from the occupied orbitals of the Mn[Cl(CO)2] metallic fragment into the vacant π * C–N ones of the carbene fragment.
The full η6-coordination of the Mn[Cl(CO)2] fragment to the external C6 ring in Mn–A(S) is found high in energy than the global minimum structure by 20.4 (BP86) or 37.8 kcal/mol (B3LYP) displaying Mn–C bond distances in the range 2.163–2.278 Å giving rise to a 18-MVE configuration of the Mn(I) cation.
6 Cobalt and rhodium complexes
For both BP86 and B3LYP methods, the structures B2 are obtained more stable than those of structures A and B1 regardless the spin state (Table 3, Fig. 4). The BP86 method gave the Co–B2(S) singlet state as global minimum despite its small HOMO–LUMO gap of 0.36 eV. However, the Co–B2(T) triplet state is obtained as global minimum by B3LYP. For both BP86 and B3LYP methods, the ΔELS/HS energy splitting of about 6 kcal/mol suggests their inter-conversion in solution. It is interesting to note the differences between the structures arising from the rotation of the Co[Cl(CO)2] fragment in each case. As can be seen from the structures displayed in Fig. 4, in the Co–B2(S) complex, the Co[Cl(CO)2] moiety adopts a square planar geometry evidenced by C(1)–Co–C′(1) and C–Co–Cl angles of 90° and 176° (linear angle), respectively, while in the Co–B2(T) structure, the Co[Cl(CO)2] moiety is in a distorted tetrahedral geometry. Likewise for Co–B1(S) and Co–B1(T), the Co[Cl(CO)2] moiety is in different environment, which is a distorted square planar in the former placing two of donors ligands above and two others below the plane containing the cobalt anion witnessed by the C(1)–Co–C′(1) of 94° and the C–Co–Cl angles of about 176° and distorted tetrahedral in the latter affording to the ligands to minimize the inter-ligands repulsions. The lengthening of the N–C bond distances from 1.378 Å in free ligand to 1.381 Å in Co–B1(S) points out the π-backbonding from the occupied orbitals of the CoCl(CO)2 metallic fragment into the vacant π * C–N ones of the NHC ligand.

BP86-optimized structures for (NHC)M[Cl(CO)2] (M = Co, Rh) complexes. S and T designate the singlet and triplet spin states, respectively. Relative energies between isomers ΔE1 (BP86) and ΔE2 (B3LYP) are given in kcal/mol
For the Rh species, the most stable structure corresponds to the unsymmetrical Rh–B2(S) one with planar structure due to the square planar geometry around the Rh ion (Cl–Rh–CO angle of 175°). This Rh–B2(S) global minimum displays significant HOMO–LUMO gap of 0.96 (BP86) and 2.48 eV (B3LYP) reproducing the experimental geometrical parameters [55], while all other isomers of conformations A, B1 and B2 are found high in energy as gathered in Table 3 and illustrated in Fig. 4. Both Co(I) and Rh(I) cations attain the 16-MVE configuration.
7 Copper complexes
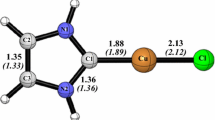
Both BP86 and B3LYP methods gave the singlet spin state Cu–B2(S) structure as the global minimum as clearly shown in Table 4 and Fig. 5. The Cu–B2(S) structure obtained without symmetry constraints is found as the global minimum exhibiting a considerable HOMO–LUMO gap of 1.17 (BP86) or 2.67 eV (B3LYP) giving rise to Cu(I) centre of 18-EVM configuration in a pseudo-tetrahedral environment. The natural population was evaluated to be + 0.79 (BP86) or + 0.78 (B3LYP), in agreement with the depopulation of 4s and 3d orbitals and very weak population of 4p one as illustrated in Table 4. The relatively long Cu–Cl bond distance of 2.389 Å (BP86) is in accordance with the atomic radii of Cu and Cl elements.

BP86-optimized structures for (NHC)Cu[Cl(CO)2] complexes. S and T designate the singlet and triplet spin states, respectively. Relative energies between isomers ΔE1 (BP86) and ΔE2 (B3LYP) are given in kcal/mol
The passage from the global minimum Cu–B2(S) of singlet state to the Cu–B2(T) triplet structure leads to a structural modifications that consist of shortening of V–Cl bond distances from 2.389 to 2.307 Å and corresponding to the opening of C(1)–Cu–C′(1) and Cl–Cu–C′(1) angles giving rise to a square planar geometry around the Cu(I) metal cation. Indeed, this isomer is modelled less stable than the global minimum by 49.7 (BP86) or 55.3 kcal/mol (B3LYP). The rotation of the Cu[Cl(CO)2] moiety leading to the symmetrical Cu–B1(S) isomer is accompanied by a loss of the total bonding energy (Table 4). The symmetrical Cu–B1(S) structure is found less stable by 6.9 (BP86) or 6.6 kcal/mol (B3LYP), showing the influence of the rotation of the Cu[Cl(CO)2], and both isomers are not on a flat potential energy. A comparison of the N–C bond distances within the NHC ligand gives rise to a weak π-backbonding in the Cu–B1(S) (1.370 Å) than in Cu–B2(S) (1.363 Å).
Surprisingly, there is no coordination between the CoCl(CO)2 moiety and the external C6 ring, while a η2-coordination would provide the copper cation the 18-MVE configuration.
8 Energy decomposition analysis and bonding
In order to elucidate the bonding between the NHC ligand and the M[Cl(CO)2] (M = V, Mn, Re, Co, Rh and Cu) metallic fragment, the Morokuma–Ziegler [56,57,58] energy decomposition analysis (EDA) was applied for all complexes of singlet spin state. (The EDA method is well detailed in the literature.) The total bonding energy between fragments is formulated as the sum of three components: the Pauli repulsion (ΔEPauli), the electrostatic interaction energy (ΔEelstat) and the orbital interaction energy (ΔEorb) for BP86 and B3LYP methods (the results are given in Table 5): ΔEint = ΔEpauli + ΔEelstat + ΔEorb.
The ΔEint bonding energies between the interacting fragments are negative synonymous of stabilizing effects for the studied complexes. For the complexes of the first-row transition metals (V, Mn, Co and Cu), the bonding interaction reaches its maximum for the symmetrical Mn–B1(S) complex against its minimum for the Cu–B1(S) one. The evolution of the ΔEint bonding energy in function of the metal valence electrons is represented in Fig. 6a, b, illustrating its diminishing from vanadium to manganese; thereafter, it slightly increases for cobalt, attaining its highest value for copper. As can be seen from Table 5, the positive Pauli repulsive interaction is overbalanced by the attractive interaction regrouping the electrostatic and orbital components. The attractive contribution is governed by the electrostatic one, which ranges from 66% (two-third) to 75% (three quarter) of the stabilizing total attractive energy (ΔEelstat + ΔEorb) comparable to the results obtained for organometallic complexes [59, 60]. However, the findings of Table 5 show strong interactions between fragments of rhenium element in Re–B1(S) complex attaining − 3.96 eV, compared to that encountered in Mn–B1(S) of − 2.88 eV for the Mn element belonging to the same group VI (Fig. 6a). The ΔEint for Re–B1(S) is composed of 9.23, − 8.87 and − 4.79 eV (BP86) as Pauli repulsion, electrostatic and orbital contributions, respectively, which are comparable to those obtained by B3LYP method. Indeed, the ΔEelstat amount of − 8.87 eV accounts 65% of the total attractive energy (ΔEelstat + ΔEorb) against a value of − 4.79 eV as ΔEorb corresponding to 35% of (ΔEelstat + ΔEorb) contribution. Furthermore, the ΔEint increases to − 2.6 eV for the unsymmetrical Re–B2(S) complex displaying its weakness with regard to that of Re–B1(S). For all complexes, the bonding energy strength responds to the following sequence: Cu < V < Rh ≈ Co < Mn < Re (PB86). Table 5 shows that the values of ΔEelstat are more important than those of ΔEorb giving rise to a pronounced ionic character than covalent one for all the complexes. It appears that the Cu[Cl(CO)2] of ΔEorb = − 1.52 eV (25%) is the less covalently bonded to the NHC ligand due probably to the large electronegativity’s difference between copper and the carbon compared to those between the other metals and the carbon atom of the carbene, while for the vanadium, manganese and rhenium complexes, the ΔEorb corresponds to the more covalently ones.

Variation of interaction energies ΔEint (eV) in function of the metal valence electrons for M-B1(S) (a) and M-B2(S) (b) complexes
9 Molecular orbital analysis
The analysis of the molecular orbital populations could provide a deeper understanding into the bonding between the interacting fragments. For both M–B1(S) and M–B2(S) complexes (M = V, Mn, Re, Co, Rh and Cu), the natural populations of the NHC frontier orbitals (Table 5) indicate that only one σ-type orbital (HOMO of the NHC fragment) is involved in the electron donation and thus significant participation of this orbital in the bonding regardless the nature of the metal, while the π-backdonation is relatively more marked for the symmetrical M–B1(S) against weak values for the unsymmetrical M–B2(S) except for the V–B2(S) complex displaying an occupation of 0.12 electrons of the orbital having a π * C–N character. The understanding of what is happening necessitates a comparison of the molecular orbitals diagrams. In fact, we have considered the symmetrical and the unsymmetrical rhenium complexes as examples, where the molecular orbital diagrams are displayed in Figs. 7 and 8, each constructed from NHC fragment Re[Cl(CO)2] of the (NHC)Re[Cl(CO)2] complexes of Cs and C1 symmetries. The lowest occupation of this orbital is 1.39 (21a′), which is donating 0.61 (BP86) electrons to the metallic fragment obtained for the symmetrical Re–B1(S) complex followed by that encountered in Co–B1(S) and Mn–B1(S) of 0.60 and 0.59, respectively, However, these interactions are relatively less strong in the symmetrical analogous species, where their corresponding V, Rh and Cu are weakly interacting with electron donation of 0.47, 0.35 and 0.41. In opposition, one of the lowest vacant orbitals [23a′ of π * (C–N) nature] receives 0.18 (BP86) or 0.16 (B3LYP) electrons from the Re[Cl(CO)2] metallic fragment, in agreement with the elongation of the N–C bond length as discussed above due to the π-backbonding occurred between the occupied d metallic orbital and the orbital of π * character of the NHC fragment. The NHC ligand in Co–B1(S) behaves similarly with regard to the electron donation, but with different capabilities of π-backdonation as given in Table 5. However, the weak π-backdonation values into the vacant orbital of NHC ligand for unsymmetrical complexes M–B2(S) in the range 0.02–0.12 are due to the orientation of the orbital 21a of the Re[Cl(CO)2] fragment, which is almost perpendicular to the orbital 40a of the NHC fragment that does not allow interactions.

Molecular orbital interaction diagram for Re[Cl(CO2)] of Cs symmetry. The populations of the interacting orbitals are in parentheses, and their contribution percentages are given

Molecular orbital interaction diagram for Re[Cl(CO2)] of C1 symmetry. The populations of the interacting orbitals are in parentheses, and their contribution percentages are given
The electron π-backdonation follows the sequence V < Cu < Mn ≈ Co ≈ Rh < Re. Consequently, the orbitals’ populations show that the electron π-backdonation is less important than the electron donation for all studied complexes as gathered in Table 5.
It is worth noting that for the symmetrical M–B1(S) species, the interactions are dominated by those of σ-type (≈ 85%) comparable to those calculated in previous works [14], while those of π ones are less important (10–15%) whose are negligible in the unsymmetrical ones (less than 7%) except for the vanadium complex.
10 Conclusion
The metal cation coordinated to the carbene centre of the five-membered ring of the NHC ligand is in pseudo-tetrahedral, trigonal pyramidal or square planar geometry depending on the nature of the metal, the spin state and the orientation of the M[Cl(CO)2] fragment. The obtained results by BP86 and B3LYP functionals show comparable tendencies relative to the geometrical parameters, the stability of the isomers, energy interactions between fragments and the populations of molecular orbitals. The molecular orbital analysis emphasizes strong σ-type interactions both in symmetrical and unsymmetrical species, but variable π interactions between the metal ions and the carbene π * orbitals. Energy decomposition analysis of the model complexes allowed us to conclude that the interactions are more electrostatic than covalent, where the electrostatic contribution features 65–75% (almost two-third) to the total attractive contribution.
The investigation of π-backbonding from the metallic fragment to the π * orbitals of NHC ligands is helpful to compare structural parameters in the carbenic C3N2 ring in a series of isostructural NHC complexes containing electron-poor metal (V), electron-moderately rich metals (Mn, Re, Co and Rh) and electron-rich metal (Cu).
The average C–N bond lengths increase from 1.378 Å in free ligand to 1.385–1396 Å in symmetrical M–B1(S) complexes. These structural changes corroborate the existence of metal–NHC π-backbonding, while structural modifications are negligible for Cu complexes.
It was found that the π-backbonding interactions in these species contribute to approximate 15–20% amongst orbital interaction energies for the symmetrical species, in agreement with the π-backdonation.
Thus, this π-backbonding from metal to the NHC ligand exists, and it would result in an increase in the C–N bond lengths for the electron-moderately rich metal cations, where the strongest bonding energies are calculated.
It is important to note that there is no π-donation from occupied π N–C orbitals to vacant d ones of the metallic fragment.
References
Bourissou D, Guerret O, Gabbai FP, Bertrand G (2000) Chem Rev 100:39–91
Arduengo AJ III (1999) Looking for stable carbenes: the difficulty in starting anew. Acc Chem Res 32:913–922
Herrmann WA (2002) N-heterocyclic carbenes: a new concept in organometallic catalysis. Angew Chem Int Ed 41:1290–1309
Cavell KJ, McGuinness DS (2004) Review Redox processes involving hydrocarbylmetal (N-heterocyclic carbene) complexes and associated imidazolium salts: ramifications for catalysis. Coord Chem Rev 248:671–681
Peris E, Crabtree RH (2004) Recent homogeneous catalytic applications of chelate and pincer N-heterocyclic carbenes. Coord Chem Rev 248:2239–2246
Weskamp T, Böhm VPW, Herrmann WA (2000) N-heterocyclic carbenes: state of the art in transition-metal-complex synthesis. J Organomet Chem 600:12–22
Wanzlick HW, Schonherr H-J (1968) Electronic structure and reactivity of metal carbenes. Angew Chem Int Engl Ed 7:141–142
Öfele K (1968) Coordination chemistry of organozinc compounds RZnX. J Organomet Chem 12:42–43
Cardin DJ, Ҫetinkaya B, Ҫetinkaya E, Lappert MF (1973) Carbene complexes. Part 1111. Carbon4 3 nuclear magnetic resonance studies of carbene complexes of I,3-diorganoimidazolidin-2-ylidenes. J Chem Soc, Dalton Trans 19:1982–1985
Bellemin-Laponnaz S, Dagorne S (2014) Group 1 and 2 and early transition metal complexes bearing N-heterocyclic carbene ligands: coordination chemistry, reactivity, and applications. Chem Rev 114:8747–8774
Hock SJ, Schaper LA, Herrmann WA, Kühn FE (2013) Group 7 transition metal complexes with N-heterocyclic carbenes. Chem Soc Rev 42:5073–5090
Hahn FE, Jahnke MC, Pape T (2007) Synthesis of pincer-type bis(benzimidazolin-2-ylidene) palladium complexes and their application in C-C coupling reactions. Organometallics 26:150–154
Clarke MJ, Taube H (1975) J Am Chem Soc 97:1397–1403
Boehme C, Frenking G (1998) Organometallics 17:5801–5809
Cavallo L, Correa A, Costabile C, Jacobsen H (2005) Steric and electronic effects in the bonding of N-heterocyclic ligands to transition metals. J Organomet Chem 690:5407–5413
Hu X, Tang Y, Gantzel P, Meyer K (2003) Silver complexes of a novel tripodal N-heterocyclic carbene ligand: evidence for significant metal-carbene π-interaction. Organometallics 22:612–614
Hu X, Castro-Rodriguez I, Olsen K, Meyer K (2004) Group 11 metal complexes of N-heterocyclic carbene ligands: nature of the metal-carbene bond. Organometallics 23:755–764
Hillier AC, Sommer WJ, Yong BS, Petersen JL, Cavallo L, Nolan SP (2003) A combined experimental and theoretical study examining the binding of N-heterocyclic carbenes (NHC) to the Cp * RuCl (Cp * = η5-C5Me5) moiety: insight into stereoelectronic differences between unsaturated and saturated NHC ligands. Organometallics 22:4322–4326
Viciu MS, Navarro O, Germaneau RF, Kelly RA III, Sommer W, Marion N, Stevens ED, Cavallo L, Nolan SP (2004) Synthetic and structural Studies of (NHC)Pd(allyl)Cl complexes (NHC = N-heterocyclic carbene). Organometallics 23:1629–1635
Dorta R, Stevens D, Scott NM, Costabile C, Cavallo L, Hoff CD, Nolan SP (2005) J Am Chem Soc 127:2485
Scott NM, Dorta R, Stevens ED, Correa A, Cavallo L, Nolan SP (2005) Interaction of a bulky N-heterocyclic carbene ligand with Rh(I) and Ir(I). Double C-H activation and isolation of bare 14-electron Rh(III) and Ir(III) complexes. J Am Chem Soc 127:3516–3526
Chekkal F, Zendaoui SM, Zouchoune B, Saillard JY (2013) Structural and spin diversity of M (indenyl) 2 transition-metal complexes: a DFT investigation. New J Chem 37:2293–2302
Farah S, Korichi H, Zendaoui SM, Saillard JY, Zouchoune B (2009) The coordination of azepine to transition-metal complexes: a DFT analysis. Inorg Chim Acta 362:3541–3546
Saiad A, Zouchoune B (2015) Electronic structure and bonding analysis of transition metal sandwich and half-sandwich complexes of the triphenylene ligand. Can J Chem 93:1096–1108
Farah S, Ababsa S, Benhamada N, Zouchoune B (2010) Theoretical investigation of the coordination of dibenzazepine to transition-metal complexes: a DFT study. Polyhedron 29:2722–2730
Benmachiche A, Zendaoui SM, Bouaoud SE, Zouchoune B (2012) Electronic structure and coordination chemistry of phenanthridine ligand in first-row transition metal complexes: a DFT study. Int J Quant Chem 113:985–996
Farah S, Bouchakri N, Zendaoui SM, Saillard JY, Zouchoune B (2010) Electronic structure of bis-azepine transition-metal complexes: a DFT investigation. J Mol Struct Theochem 953:143–150
Zendaoui SM, Zouchoune B (2016) Coordination chemistry of mixed M(benzene)(cyclopendadienyl) sandwich complexes: electronic properties and bonding analysis. New J Chem 40:2554–2564
Zouchoune B, Mansouri L (2017) Electronic structure and UV–Vis spectra simulation of square planar bis(1-(4-methylphenylazo)-2-naphtol)-transition metal complexes [M(L)2]x (M = Ni, Pd, Pt, Cu, Ag). Struct Chem 30(3):691–701
Tabrizi L, Zouchoune B, Zaiter A (2018) Experimental and theoretical investigation of cyclometallated platinum(II) complex containing adamantanemethylcyanamide and 1,4-naphthoquinone derivative as ligands). RSC Adv 9:287–300
Tabrizi L, Zouchoune B, Zaiter A (2019) Theoretical and experimental study of gold (III), palladium (II), and platinum (II) complexes with 3-((4-nitrophenyl) thio) phenylcyanamide and 2,2′-bipyridine ligands. Inorg Chim Acta 499:119211
Merzoug M, Zouchoune B (2018) Molecular models of monometallic-phenazine sandwich complexes M (phz)(2)(M = Ti, Cr, Fe and Ni; phz = C12H8N2): a DFF investigation. J New Tech Mat 8:44–54
ADF2016, SCM. Theoretical Chemistry, VrijeUniversiteit, Amsterdam, The Netherlands
Baerends EJ, Ellis DE, Ros PJCP (1973) Self-consistent molecular Hartree–Fock Slater calculations I. The computational procedure. Chem Phys 2:41–51
TeVelde G, Baerends EJ (1992) Numerical integration for polyatomic systems. J Comput Phys 99:84–98
Guerra CF, Snijders JG, teVelde GT, Baerends EJ (1998) Towards an order-N DFT method. Theor Chem Acc 99(6):391–403
Bickelhaupt FM, Baerends EJ (2000) Kohn–Sham density functional theory: predicting and understanding chemistry. Rev Comput Chem 15:1–86
TeVelde GT, Bickelhaupt FM, Baerends EJ, Fonseca Guerra C, van Gisbergen SJ, Snijders JG, Ziegler T (2001) Chemistry with ADF. J Comput Chem 22:931–967
Vosko SH, Wilk L, Nusair M (1980) Accurate spin-dependent electron liquid correlation energies for local spin density calculations: a critical analysis. Can J Phys 58:1200–1211
Becke AD (1986) Density functional calculations of molecular bond energies. J Chem Phys 84:4524–4529
Becke AD (1988) Density-functional exchange-energy approximation with correct asymptotic behavior. Phys Rev A 38:3098–3100
Perdew JP (1986) Density-functional approximation for the correlation energy of the inhomogeneous electron gas. Phys Rev B 33:8822–8824
Perdew JP (1986) Erratum: density-functional approximation for the correlation energy of the inhomogeneous electron gas. Phys Rev B 34:7406
Becke AD (1993) Becke’s three parameter hybrid method using the LYP correlation functional. J Chem Phys 98:5648–5652
Lee C, Yang W, Parr RG (1988) Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys Rev B 37:785–789
vanLenthe E, Baerends EJ, Snijders JG (1993) Relativistic regular two-component Hamiltonians. J Chem Phys 99:4597–4610
vanLenthe E, Baerends EJ, Snijders JG (1994) Relativistic total energy using regular approximations. J Chem Phys 101:9783–9792
vanLenthe E, Baerends EJ, Snijders JG (1999) Geometry optimizations in the zero order regular approximation for relativistic effects. J Chem Phys 110:8943–8953
Versluis L, Ziegler T (1988) The determination of molecular structures by density functional theory. The evaluation of analytical energy gradients by numerical integration. J Chem Phys 88:322–328
Fan L, Ziegler T (1992) Application of density functional theory to infrared absorption intensity calculations on main group molecules. J Chem Phys 96:9005–9012
Fan L, Ziegler T (1992) Application of density functional theory to infrared absorption intensity calculations on transition-metal carbonyls. J Phys Chem 96:6937–6941
Wiberg KB (1968) Application of the pople-santry-segal CNDO method to the cyclopropylcarbinyl and cyclobutyl cation and to bicyclobutane. Tetrahedron 24:1083–1096
Weinhold F, Landis CR (2005) Valency and bonding: a natural bond orbital donor-acceptor perspective. Cambridge University Press, Cambridge
Weinhold F, Glendening ED (2001) NBO 5.0 program manual: natural bond orbital analysis programs. Theoretical Chemistry Institute and Department of Chemistry, University of Wisconsin, Madison, WI 53706
Sanderson MD, Kamplain JW, Bielawski CW (2006) Quinone-annulated N-heterocyclic carbene transition-metal complexes: observation of p-backbonding using FT-IR spectroscopy and cyclic votammetry. J Am Chem Soc 128:16514–16515
Morokuma K (1971) Molecular orbital studies of hydrogen bonds. III. C = O H–O hydrogen bond in H2CO H2O and H2CO 2H2O. J Chem Phys 55:1236–1244
Ziegler T, Rauk A (1979) Carbon monoxide, carbon monosulfide, molecular nitrogen, phosphorus trifluoride, and methyl isocyanide as. sigma. donors and. pi. acceptors. A theoretical study by the Hartree–Fock–Slater transition-state method. Inorg Chem 18:1755–1759
Ziegler T, Rauk A (1979) A theoretical study of the ethylene-metal bond in complexes between copper (1+), silver (1+), gold (1+), platinum (0) or platinum (2+) and ethylene, based on the Hartree–Fock–Slater transition-state method. Inorg Chem 18:1558–1565
Zaiter A, Zouchoune B (2018) Electronic structure and energy decomposition of binuclear transition metal complexes containing β-diketiminate and imido ligands: spin state and metal’s nature effects. Struct Chem. https://doi.org/10.1007/s11224-018-1112-6
Mecheri S, Zouchoune B, Zendaoui SM (2020) Bonding and electronic structures in dinuclear (X)[(Ind)M2L2] complexes (M = Ni, Pd, L = CO, PEt3, X = Cl, Allyl, Ind = indenyl, Cp = cyclopentadienyl): analogy between four-electron donor ligands. Theor Chem Acc. https://doi.org/10.1007/s00214-019-2526-y
Acknowledgements
The authors acknowledge the Algerian MESRS (Ministère de l’Enseignement Supérieur et de la Recherche Scientifique) and DGRSDT (Direction Générale de la Recherche Scientifique et du Développement Technologique) for Financial support.
Author information
Authors and Affiliations
Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no competing interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Mokrane, Z., Zouchoune, B. & Zaiter, A. Coordination’s preference and electronic structure of N-heterocyclic carbene–monometallic complexes: DFT evaluation of σ-bonding and π-backbonding interactions. Theor Chem Acc 139, 114 (2020). https://doi.org/10.1007/s00214-020-02628-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00214-020-02628-6