
Abstract
Aiming to pinpoint an atomic basis set providing accurate transition energies at a minimal computational cost, we investigate the evolution with basis set size of the energy of low-lying excited states in nine representative conjugated dyes with a wide panel of theoretical approaches, namely TD-DFT, SOPPA, ADC(2), CIS(D), CC2, EOM-CCSD, as well as several scaled opposite spin variants, namely SOS-CIS(D), SOS-CIS(D0) and SOS-ADC(2). At the exception of TD-DFT that displays the lowest basis set dependence, it turns out that the changes obtained when increasing the size of the basis set are rather independent of the selected wavefunction model, but strongly change according to the nature of the excited state considered. Reasonable compromises between accuracy and computational burden can be attained with 6-311+G(2d,p) that allows much faster calculations than the typical reference basis set, namely aug-cc-pVTZ, for an average loss of accuracy limited to ca. 0.02 eV.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Introduction
The simulations of excited-state (ES) properties with quantum-mechanical methods remain a topic of extremely intense research, not only because of the academic interest of designing appropriate approaches for challenging cases, but also due to the need of complementing spectroscopic measurements that are often difficult to interpret with standard chemical concepts [1]. ES calculations can be performed with a wide range of approach. This includes, on the one hand, the highly accurate but computationally demanding wavefunction approaches, e.g., CAS-PT2 (complete active space second-order perturbation theory) [2], MR-CI (multi-reference configuration interaction) [3], EOM-CC (equation-of-motion coupled cluster) [4–6], ADC (algebraic diagrammatic construction) [7–9] and SAC-CI (symmetry adapted cluster CI) [10, 11], and, on the other hand, more qualitative but less demanding methods, e.g., the semi-empirical ZINDO (Zerner’s intermediate neglect of differential overlap) [12] as well as the ab initio CIS (CI singles) [13] and TD-DFT (time-dependent density functional theory) [14, 15]. The latter approach undoubtedly remains the most popular approach for ES, as it allows a rapid estimation of transition energies and ES properties for a limited computational effort, even for relatively large molecules (ca. 100–400 atoms) [16]. Despite considerable successes, especially in designing new dyes, TD-DFT suffers from a series of limitations, e.g., its inadequacy for ES presenting a significant double-excitation character and its strong functional dependence—the obtained ES energies and properties are strongly dependent on the selected exchange-correlation functional [17]. The continuous increase in computational power accompanied by the developments of efficient implementation, e.g., the RI (resolution of identity) [18] and the Cholesky decomposition techniques [19–21], has now made possible the treatment of ES of medium-sized molecules with approaches such as ADC(2) (second-order ADC) [7], CC2 (the simplest EOM-CC scheme) [18, 22] and CIS(D) (CIS with a perturbative correction for double excitations) [23, 24]. These methods can provide accurate transition energies in cases in which TD-DFT is less adequate, e.g., for ES presenting a significant multi-excitation nature. There are indeed, an increasing number of applications of these approaches for non-trivial molecules [25–39].
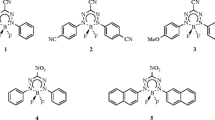
Both the computational time and the quality of the results obtained with these second-order approaches are sensitive to the size of the selected atomic basis set (BS). It is indeed accepted that these schemes, as their wavefunction ground-state counterparts, tend to require larger BS to attain convergence than TD-DFT. This may seriously limit the applicability to large molecules. Therefore, as these wavefunction methods become more widely applied for low-lying excited states of medium-size molecules, it is certainly worth to select the most compact BS leading sufficiently accurate results so to avoid two pitfalls: consuming computational time with oversized BS and analyzing results so far from BS convergence that they become meaningless. As a rule of thumb, one could state that the selected BS should have an effect limited to ca. 30 % of the method accuracy. As ADC(2), CIS(D) and CC2 are generally recognized to provide ES energies with a typical deviations in the 0.05–0.15 eV range (for non-exotic compounds) [9, 29, 37], a BS effect limited to ca. 0.03 eV is enough for most purposes. Surprisingly, despite the large number of valuable studies published to date (see next section), we could not find in the literature an investigation aiming to define the smallest possible BS providing accurate results for the low-lying ES of “real-life” organic dyes and the present paper aims to fill this gap. We have selected the nine molecules shown in Fig. 1 to perform our assessment. This set includes representative members of four of the most important classes of organic fluorophores, namely boron-dipyrromethene (2), coumarin (3), bimane (6), 1,8-napththalimide (7) as well as biphenyl (a typical hydrocarbon, 1), and a highly conjugated stilbene (5) and three compact chromogens relevant for dye chemistry representative of the thiocrabonyl (4), nitroso (8) and diazo (9) classes. This set has been also designed to include representative members of \(\pi \rightarrow \pi ^\star\) valence (1, 6 and 7), \(n \rightarrow \pi ^\star\) valence (4, 8 and 9), charge-transfer (3 and 5) and cyanine (2) excitation categories.

Representation of the systems investigated herein
2 Literature survey
In this section, we summarize previous studies focussing on the determination of low-lying excited states, employing several wavefunction methods and using at least three atomic basis sets.
Grimme and Ugorodina carried out an extensive study of the 0–0 transition energies in small organic molecules with the TD-DFT, CIS(D) and SCS-CIS(D) methods [40]. Using CIS(D) with three BS [TZV(d,p), TZV(2df,2pd) and aug-cc-pVTZ], they benchmarked the \(B_u\) state of the acene series and deviations of −0.24 eV (benzene), −0.22 eV (naphthalene) and −0.20 eV (anthracene) have been obtained when going from TZV(d,p) to aug-cc-pVTZ. In contrast, for other covalent states, TZV(d,p) was found sufficient with deviations of ca. 0.05 eV or less. In addition, these authors observed that for \(n \rightarrow \pi ^{\star }\) (\(\pi \rightarrow \pi ^{\star }\)) states, the excitation energies increase (decrease) with extension of the BS. They also found that the BS effects are slightly smaller with SCS-CIS(D) than with CIS(D).
Thiel’s group addressed the BS dependency in his well-known benchmark studies (CAS-PT2, CC2, CC3, CCSD) of 223 electronic states determined in 28 medium-sized organic molecules [41–43]. In their first study, this team tested 12 different BSs, from cc-pVDZ to d-aug-cc-pV5Z (see Fig 2. in Ref. [41]) for ethene and concluded that the TZVP BS provides transition energies to the lowest ES within 0.1 eV of BS convergence [41]. In contrast, they found that for both hybrid valence-Rydberg and higher-lying Rydberg states, the inclusion of diffuse functions was necessary to reach BS convergence. In two further studies, they concluded, using a much more extended set of molecules, that the TZVP and aug-cc-pVTZ energies are typically within 0.1 eV from each other [42, 43].
Lischka et al. investigated excited-state intramolecular proton transfer within five small molecules and observed variations of ca. 0.1 eV of the RI-CC2 vertical excitation energies when going from SVP to TZVP, the deviations being smaller with TD-DFT [44]. In another work, they studied charge-transfer state in two DNA base dimers using RI-CC2, RI-ADC(2) and TD-DFT (B3LYP, PBE0, M06-2X and M06-HF). Comparing SVP and TZVP BS, they concluded that the flexibility of TZVP was needed for both RI-ADC(2) and RI-CC2 calculations [45]. An investigation of BS effects at the ADC(2) level was also performed on poly-(\(p\)-phenylenevinylene) oligomers, using SV, SV(P), TZVP and TZVPP for chains up to seven unit cells (the largest BS was applied up to the tetramer) [46]. It was concluded that going from SVP to TZVP induces a decrease in the excitation energies by ca. 0.3 eV, irrespective of the chain length, whereas TZVPP yields a further decrease of 0.1 eV, clearly hinting a quite slow convergence with BS size.
In the same vein, Russo and his collaborators published several articles comparing TD-DFT and RI-CC2 performances and assessing BS effects in quite large molecules [26, 31]. Indeed, they carried out benchmarks on both squaraines (ca. 16–35 non-hydrogen atoms) [26] and pyranoanthocyanins (25 or more non-hydrogen atoms) [31]. The impact of BS size on the excited-state energies has been studied for representative compounds with SV(P), SVP, DZ, DZP, TZVP, TZVPP, cc-pVDZ, cc-pTVZ, aug-cc-pVDZ and aug-cc-pTVZ. They concluded that TD-DFT and RI-CC2 are not sensitive to the BS for these cyanine-like derivatives. Indeed, for a small squaraine, a limited decrease in 0.04 (0.06) eV is found when going from SV(P) or SVP to aug-cc-pVTZ with TD-DFT (RI-CC2), whereas for the peonidin-3-glucoside pyruvic acid, the variation between SV(P) or SVP and the triple-\(\zeta\) TZVP or cc-pVTZ is 0.02 eV (0.04 eV) with TD-DFT (RI-CC2).
In 2009, Starcke, Wormit and Dreuw considered open shell molecules [47, 48] and compared the results obtained with the unrestricted formalisms of ADC(2) and EOM-CCSD for the pentadienyl neutral radical and the hexatriene radical cation using 6-31G, 6-31G(d), 6-31+G(d), 6-311G(d) and cc-pVTZ. Probably because their focus was low-lying excited states, they found that 6-31G(d) was a valid compromise to model medium-sized cases [neutral radical of phenoxyl, phenyl, anilino and (methyl)benzyl; radical cation naphthalene, dihydroacenaphthylene and pyracene; radical anion benzoquinone]. Krauter et al. [9] employed Thiel’s set and discussed BS effects using ADC combined to aug-cc-pVDZ, cc-pVTZ and aug-cc-pVTZ. Neglecting diffuse functions induced significant variations (increase in the mean error from 0.03 to 0.14 eV) while going from triple- to double-\(\zeta\) introduced only minor changes (decrease in the mean error from 0.03 to 0.01 eV) for singlet excited states. Very recently, the same group investigated several variations of the ADC approach in the framework of a solvatochromic benchmark [49]. The impact of the BS [(aug-)cc-pVDZ and (aug-)cc-pVTZ] has been investigated for nitrobenzene, a charge-transfer molecule. At the ADC(3) level, they noticed quite large BS effects, with a variation larger than 0.3 eV when shifting from cc-pVDZ to aug-cc-pVTZ.
Kerkines et al. [50] computed the low-lying excited-state energies of pyrene, 1,6-dithiapyrene and tetrathiafulvalene with 6-31G(d), (aug-)TZVPP, aug-cc-pVDZ, aug-cc-pV(D+d)Z and aug-cc-pV(T+d)Z . They underlined that TZVPP or aug-TZVPP have to be employed to reach reasonably accurate estimates of the RI-CC2 absorption and emission spectra, whereas 6-31G(d) is sufficient for TD-B3LYP calculations [50].
Though, not focussed on BS effects, it is worth to briefly review recent studies using extended BS to model the excited states of non-trivial molecules, in order to illustrate the growing interest for such simulations.
In 2007, Rhee and Head-Gordon applied their SOS-CIS(D) approach with the 6-311(2+,2+)G(d,p) BS on a set of molecules proposed by Grimme [24]. The following year, Corral et al. [51] simulated the absorption of anthracene-9,10-endoperoxide with MS-CAS-PT2//CAS-SCF(14,12)/ANO-S as well as with TD-DFT and RI-CC2 approaches using both 6-311G(d,p) and cc-pVTZ. In 2010, Pino et al. [52] explored the reactivity of phenol and its complexes with ammonia in a joint experimental–theoretical study. They computed ES energies at the RI-CC2/aug-cc-pVDZ, this large BS being justified by the Rydberg nature of the investigated excited states. In 2011, Guthmuller used TD-DFT/6-311++G(2df,p), CC2/def2-QZVPP as well as SCS-CC2/def2-QZVPP approaches to evaluate excitation energies of \(o\)-nitrophenol [30]. Studying the charge-transfer induced excited-state intramolecular proton transfer in the chromophore of green fluorescent protein, Cui, Lan and Thiel selected aug-cc-pVTZ for TD-DFT (B3LYP, CAM-B3LYP, \(\omega\)B97XD and M06-2X) computations and TZVP for RI-CC2 calculations [33]. In a recent extensive benchmark paper, Hättig’s group compared 0–0 electronic transition energies in 66 organic molecules from catechol to Zn-tetraphenylporphyrine with a large panel of methods [B3LYP, ADC(2), CC2, SOS-CC2 and SCS-CC2] [37]. They optimized the structures at the TZVP (DFT) and TZVPP (wavefunction) schemes, while they computed all transition energies with aug-cc-pVTZ, an exceptional feat. Already, in the early 2000, the same group performed RI-CC2 calculations with quite larger basis set, namely TZVPP [18] and aug-cc-pVTZ [53] for specific molecules. Krylov et. al. discussed the singlet and triplet \(\pi \rightarrow \pi ^{*}\) states of a model of the GFP chromophore, namely the 4-hydroxybenzylidene-2,3-dimethylimidazolinone (HBDI) anion using CIS, SOS-CIS(D), EOM-EE-CCSD [54]. As the excited states of HBDI anion are lying below the electron detachment energy, the use of compact basis sets created discrete states and artificially excluded the detached states. Increasing the number of diffuse function was crucial to bring the continuum down, the lowest excited state corresponding to the detached state when a large BS is used (see Fig. 4 and Table 2 in Ref. [54]). Adding sets of diffuse functions results lowered the CIS and TD-DFT excitation energy of the bright state by 0.1 eV, whereas going from aug-cc-pVDZ to aug-cc-pVTZ decreased the excitation energy by 0.13 eV.
As can be concluded from this survey, a systematic BS benchmark considering several types of valence states and methods in \(\pi\)-conjugated compounds, and aiming not only to estimate deviations but also to determine the best “accuracy/cost” BS is missing, and the present work is an effort in that direction with a focus on molecules relevant for organic chemistry.
3 Methodology
All calculations presented below have been made using optimal ground-state geometries determined at the M06-2X/6-31+G(d) level of approximation. These optimizations as well as the subsequent TD-DFT [14, 15] and CIS(D) [23] calculations were carried out with the latest version of the Gaussian 09 program package [55]. For the TD-DFT part, we have selected the M06-2X functional [56] and used the so-called ultrafine pruned (99,590) grid. The scaled opposite spin CIS(D) [SOS-CIS(D)] [24], SOS-CIS(D0) [57], ADC(2) [7] and SOS-ADC(2) [8, 9] transition energies were determined with the Q-Chem package using the Resolution of the Identity (RI) scheme [58, 59]. For the most demanding cases, TURBOMOLE was also used to perform some ADC(2) calculations [60]. CC2 [18, 22] and second-order polarization propagator approximation (SOPPA) [61] calculations were performed with the Dalton code [62]. Note that EOM-CCSD [4, 6] transition energies can be computed with three out of the four programs, and test calculations indicated that the variations from one implementation to the other were negligible (max deviation noted 0.001 eV). Consequently, EOM-CCSD calculations have therefore been performed with the three codes, depending on which one was the most efficient for a given molecule and BS. Similar comparisons have been performed for ADC(2) calculations computed with Q-Chem and TURBOMOLE and trifling differences were noted. We have checked that for both ADC(2) and EOM-CCSD, the RI approximation has no significant influence by performing non-RI calculations for a dozen of cases. No deviations larger than 0.001 eV could be detected, and we have therefore processed all further calculations with the RI approximation when available. In the present work, we tested several popular atomic BS with or without diffuse orbitals, namely Pople’s 6-31G(d), 6-311G(d,p), 6-31+G(d), 6-31+G(d,p), 6-31+G(2d), 6-311+G(2d,p), 6-311++G(d,p), 6-311++G(2df,2p) and 6-311++,G(3df,3pd); Dunning’s cc-pVDZ, cc-pVTZ, aug-cc-pVDZ and aug-cc-pVTZ; as well as def2-TZVP.
4 Results and discussion
Let us first underline that our goal is not to perform comparisons with experiments as we considered vertical transition energies in the present contribution. Our results are listed in Tables 1, 2, 3, 4 and 5. It appears that the two largest BSs, namely 6-311++G(3df,3pd) and aug-cc-pVTZ, provide very similar values, independently of the considered theoretical approach. Indeed, considering all data, the mean absolute deviation between the results of these two BS is as small as 0.008 eV, whereas the three largest discrepancies are obtained for compound 4 using SOS-CIS(D0) (0.033 eV), CC2 (0.026 eV) and EOM-CCSD (0.025 eV). For this reason, we performed EOM-CCSD calculations for 4 using larger Dunning BSs. The results are shown in the Supporting Information (SI). The difference between the aug-cc-pVTZ and the d-aug-cc-pVQZ transition energies is as small as 0.002 eV. One can therefore consider that the aug-cc-pVTZ data are close to BS convergence and that they can be used as reference values. The deviations listed in the following have therefore to be understood as “with respect to aug-cc-pVTZ.”
It is obvious from the data of Tables 1, 2, 3, 4 and 5 that increasing the size of the BS almost systematically induces a decrease in the excitation energies, a result typical of low-lying transitions. This can be qualitatively understood: the excited state is more delocalized than the ground state and therefore tends to be more stabilized by the addition of supplementary atomic functions. The very few exceptions that can be found (for which an increase in the BS size provokes an increase in the transition energy) are quantitatively negligible. Indeed, the upshifts are tiny, typically ca. 0.005 eV, the largest effect being +0.036 eV between the TD/aug-cc-pVDZ and TD/aug-cc-pVTZ energies of 8. It is also clear from the data listed in all tables that 6-31G(d), which is often a reasonable choice for geometry optimization at the DFT level, is significantly too small, as it yields overestimations of the transition energies by ca. 0.1–0.3 eV depending on the molecule and method considered.

Errors made when using the cc-pVDZ BS (using aug-cc-pVTZ values obtained with the same method as reference) for the three first molecules [from top to bottom: 1 (\(\pi \rightarrow \pi ^\star\) valence), 2 (cyanine), 3 (charge transfer) and 4 (\(n \rightarrow \pi ^\star\) valence)] considering all nine theoretical approaches
Another expected finding is that the BS effects depend not only on the applied method but also on the selected molecule. Let us first discuss the former dependency. We note that the BS effects are (almost always) smaller with TD-DFT than with the wavefunction approaches, as expected from previous works. This fact is illustrated in Fig. 2 for cc-pVDZ, a representative example of the BS behaviors. Complete representations for all methods and BS are available in the SI. We highlight that for the cyanine-like 2, for which BS effects are rather small (see below), the EOM-CCSD results constitute an exception to that general trend; i.e., TD-DFT BS effects are larger than their EOM-CCSD counterpart for 2. Nevertheless, the BS variations obtained with TD-DFT can generally be regarded as lower bounds of the variations reached with other approaches. While the BS effects tend to be larger for wavefunction methods than for TD-DFT, they are globally of the same order of magnitude for all wavefunction schemes, as illustrated in Fig. 2. However, we note that the variations obtained with SOS-CIS(D) tend to be systematically larger than those measured with CIS(D); e.g., the differences between 6-31+G(d) and aug-cc-pVTZ are 0.171 [0.202] eV, 0.035 [0.078] eV, 0.139 [0.170] eV, 0.012 [0.055] eV, 0.169 [0.193] eV, 0.060 [0.090] eV, 0.141 [0.181] eV, 0.052 [0.132] eV and 0.111 [0.166] eV with CIS(D) [SOS-CIS(D)] for 1, 2, 3, 4, 5, 6, 7, 8 and 9, respectively. On the contrary, these 6-31+G(d)—aug-cc-pVTZ differences become more similar when comparing ADC(2) and SOS-ADC(2), that is 0.161 [0.168] eV, 0.040 [0.039] eV, 0.103 [0.088] eV and 0.035 [0.032] eV for the first four compounds, respectively. One can additionally conclude that cyanine states are particularly unsensitive to BS effects which contrasts with \(\pi \rightarrow \pi ^\star\) and \(n \rightarrow \pi ^\star\) valence and charge-transfer states (see Sect.2 and below). It is interesting that SOS-CIS(D) that allows faster implementations of the double corrections than its canonical counterpart suffers from being more sensitive to BS effects. It is also striking from Fig. 2 that the BS effects are not systematically larger with EOM-CCSD than with CC2, though the latter can be viewed as a less correlated version of the former.
Let us now discuss the variations obtained for the nine molecules. As stated above, the BS effects are weak for 2 (<0.12 eV with the considered panel of BS), and this can be ascribed to the cyanine nature of the excited state in BODIPY [17, 26, 31, 63]. By contrast, 3 and 5 that are characterized by charge-transfer like electronic transitions are much more sensitive to the size of the BS, which is consistent with previous studies (see Sect. 2), and the addition of diffuse functions becomes important to attain reasonably accurate results, e.g., the difference between ADC(2)/cc-pVTZ and ADC(2)/aug-cc-pVTZ attains 0.099 eV for 5. For 1, 6 and 7 that all present a valence-like \(\pi \rightarrow \pi ^\star\) excited state, the BS effects are large with the wavefunction models in two (1 and 7) out of three cases; 6-31G(d) yields errors larger than 0.3 eV. For \(n \rightarrow \pi ^\star\) states, the convergence of the transition energies with BS size seems slower than for \(\pi \rightarrow \pi ^\star\) states; e.g., for 9 with CIS(D), the 6-311++G(d,p) result is still too large by 0.069 eV, whereas for both 4 and 8 going from double-\(\zeta\) to triple-\(\zeta\) induces a strong decrease in the energies with the SOS-CIS(D) approach. We could not find evident correlations between the size of the treated system and the amplitude of the BS effects, the nature of the state is obviously more crucial.

Evolution with BS size of the ADC(2) transition energies for all compounds. From left to right: 1, 2 and 3 (top); 4, 5 and 6 (center) and 7, 8 and 9 (bottom). The horizontal black line indicates the reference aug-cc-pVTZ results, whereas the red line gives the border for a 0.03 eV error. The smallest BS providing an error smaller than this limit is indicated
Let us now try to determine an “optimal” BS keeping in mind that a 0.03 eV error can be viewed as acceptable for most purposes (see Sect. 1). As all approaches (but TD-DFT for all states and SOS-CIS(D)/SOS-CIS(D0) for \(n \rightarrow \pi ^\star\) transitions) undergo rather similar BS dependencies, we have only represented the BS effects with the ADC(2) approach in Fig. 3—ADC(2) was selected because it delivers a rather average behavior over all the wavefunction methods (see Fig. 2). Complete data can be found in the SI. First, diffuse functions are obviously mandatory in all cases (see Tables 1, 2, 3, 4, 5), not only because they induce a strong decrease in the transition energies, but also because they allow a faster convergence with BS size. For instance, cc-pVTZ yields significant deviations, e.g., exceeding 0.1 eV for the \(\pi \rightarrow \pi ^\star\) transition in 1, despite its size. Likewise, def2-TZVP often delivers errors significantly exceeding the 0.03 eV limit. Consequently, in terms of computational efficiency, it is adequate to limit our search of the optimal BS in the diffuse-containing subgroup. In that panel, two major components appear to be necessary: a second set of \(d\) polarization functions and the selection of a triple-\(\zeta\) BS. In other words, the smallest reasonable BS are, on the one hand, 6-311+G(2d,p) in Pople’s series and, on the other hand, aug-cc-pVDZ in Dunning’s series. One can indeed see from Fig. 3 that these two BSs are often the smallest to be within 0.03 eV of the aug-cc-pVTZ reference. The maximal deviations are 0.082 eV [SOS-CIS(D) on 9] and 0.123 eV [SOPPA on 4] for 6-311+G(2d,p) and aug-cc-pVDZ, respectively. In addition 30 and 57 % of the estimates undergo an error exceeding the 0.03 eV mark for 6-311+G(2d,p) and aug-cc-pVDZ, respectively. Interestingly by excluding the three \(n \rightarrow \pi ^\star\) dyes from the set, these values fall to 10 and 46 %, respectively. Despite their similar sizes, it turns out that 6-311+G(2d,p) is overall the most effective of this pair of BS. Indeed, the average absolute errors considering all approaches and molecules (56 cases) are 0.024 and 0.040 eV for 6-311+G(2d,p) and aug-cc-pVDZ, respectively. To significantly improve over 6-311+G(2d,p), that is to cut the remaining error by a factor of ca. 2, one needs to apply a much larger BS, namely 6-311++G(2df,2p), but the computational cost becomes significantly higher. Table 6 provides a statistical analysis for all BSs allowing one to estimate the error made with a specific BS selection. We underline that these data have been obtained considering all methods and states. As stated above, these two parameters significantly influence the final accuracy of a given BS.
Though it is not our main goal here to define the merits of the different quantum chemical approaches applied, we can briefly comment on the relative energies obtained with all approaches. One notices that the ordering of the transition energies tend to follow CIS(D) \(>\) SOS-CIS(D0) \(>\) SOS-CIS(D) and ADC(2) \(>\) SOS-ADC(2), the SOS effect being particularly marked for 2 and relatively weak for all other molecules. EOM-CCSD results are larger than their CC2 counterparts for both 1 and 3, but are similar for 2 and smaller for 4. It is also striking that the SOPPA transition energies are always significantly below the one computed with the other theories.
5 Conclusions
We have performed a complete investigation of BS effects involved in the calculation of low-lying excited states of conjugated dyes, selecting 14 bases, 9 significant molecules and 9 theoretical methods. This study was stimulated by a literature search indicating that methods like CC2 or ADC(2) are often used either with compact diffuse-less single- or double-\(\zeta\) BS (for large compounds) or with aug-cc-pVTZ (for medium-sized molecules), so that defining a valuable compromise between these two rather extreme choices would be of interest. Fixing the acceptable margin of BS error to 0.03 eV, we found that 6-311+G(2d,p) is the lightest BS able to match this limit in ca. 70 % of the tested cases. aug-cc-pVDZ that presents a similar size delivers larger average errors. Choosing 6-31+G(d,p), a quite popular BS for excited-state calculations, induces significant deviations (0.084 eV on average), with only 18 % of the tested cases within the 0.03 eV limit. Among all tested theoretical methods, TD-DFT [SOS-CIS(D)] appeared to be the least [most] sensitive to BS size, though all wavefunction theories provide rather similar evolutions with BS size for a given compound. Obviously, the nature of the excited state also plays a crucial role on the BS effects, and cyanine (charge transfer) transitions tend to require small (large) BS, whereas triple-\(\zeta\) BS is to be recommended for \(n\,\rightarrow\,\pi ^\star\) transitions. It is our hope that this work will allow future excited-state calculations to be performed on larger molecules with a well-controlled accuracy.
References
González L, Escudero D, Serrano-Andrès L (2012) Chem Phys Chem 13:28
Andersson K, Malmqvist P, Roos BO (1992) J Chem Phys 96:1218
Buenker RJ, Peyerimhoff SD (1968) Theor Chim Acta 12:183
Stanton JF, Bartlett RJ (1993) J Chem Phys 98:7029
Kállay M, Gauss J (2004) J Chem Phys 121:9257
Caricato M (2013) J Chem Phys 139:114103
Schirmer J, Trofimov AB (2004) J Chem Phys 120:11449
Hellweg A, Grün SA, Hättig C (2008) Phys Chem Chem Phys 10:4119
Krauter CM, Pernpointner M, Dreuw A (2013) J Chem Phys 138:044107
Nakatsuji H (1991) J Chem Phys 94:6716
Nakatsuji H, Ehara M (1993) J Chem Phys 98:7179
Ridley JE, Zerner M (1973) Theor Chim Acta 32:111
Förner W (1992) Int J Quantum Chem 43:221
Runge E, Gross EKU (1984) Phys Rev Lett 52:997
Casida ME (1995) Time-dependent density-functional response theory for molecules (World Scientific, Singapore). Recent Adv Density Funct Methods 1:155–192
Laurent AD, Adamo C, Jacquemin D (2014) Phys Chem Chem Phys 16:14334
Laurent AD, Jacquemin D (2013) Int J Quantum Chem 113:2019
Hättig C, Weigend F (2000) J Chem Phys 113:5154
Aquilante F, Malmqvist PÅ, Pedersen TB, Ghosh A, Roos BO (2008) J Chem Theory Comput 4(5):694
Boström J, Delcey MG, Aquilante F, Serrano-Andrés L, Pedersen TB, Lindh R (2010) J Chem Theory Comput 6:747
Epifanovsky E, Zuev D, Feng X, Khistyaev K, Shao Y, Krylov AI (2013) J Chem Phys 139:134105
Christiansen O, Koch H, Jørgensen P (1995) Chem Phys Lett 243:409
Head-Gordon M, Maurice D, Oumi M (1995) Chem Phys Lett 246:114
Rhee YM, Head-Gordon M (2007) J Phys Chem A 111:5314
Plasser F, Barbatti M, Aquino AJA, Lischka H (2009) J Phys Chem A 113:8490
Quartarolo AD, Sicilia E, Russo N (2009) J Chem Theory Comput 5:1849
Aittala PJ, Cramariuc O, Hukka TI (2010) J Chem Theory Comput 6:805
Plötner J, Tozer DJ, Dreuw A (2010) J Chem Theory Comput 6:2315
Goerigk L, Grimme S (2010) J Chem Phys 132:184103
Guthmuller J (2011) J Chem Theory Comput 7:1082
Quartarolo AD, Russo N (2011) J Chem Theory Comput 7:1073
Send R, Kühn M, Furche F (2011) J Chem Theory Comput 7:2376
Cui G, Lan Z, Thiel W (2012) J Am Chem Soc 134:1662
Friese DH, Hattig C, Ruud K (2012) Phys Chem Chem Phys 14:1175
Bousquet D, Fukuda R, Maitarad P, Jacquemin D, Ciofini I, Adamo C, Ehara M (2013) J Chem Theory Comput 9:2368
Ehara M, Fukuda R, Adamo C, Ciofini I (2013) J Comput Chem 34:2498
Winter NOC, Graf NK, Leutwyler S, Hattig C (2013) Phys Chem Chem Phys 15:6623
Daengngern R, Kungwan N (2014) Chem Phys Lett 609:147
Li H, Nieman R, Aquino AJA, Lischka H, Tretiak S (2014) J Chem Theory Comput 10:3280
Grimme S, Izgorodina EI (2004) Chem Phys 305:223
Schreiber M, Silva-Junior MR, Sauer SPA, Thiel W (2008) J Chem Phys 128:134110
Silva-Junior MR, Sauer SPA, Schreiber M, Thiel W (2010) Mol Phys 108:453
Silva-Junior MR, Schreiber M, Sauer SPA, Thiel W (2010) J Chem Phys 133:174318
Aquino AJA, Lischka H, Hattig C (2005) J Phys Chem A 109:3201
Aquino AJA, Nachtigallova D, Hobza P, Truhlar DG, Hattig C, Lischka H (2011) J Comput Chem 32:1217
Panda AN, Plasse F, Aquino AJA, Burghardt I, Lischka H (2013) J Phys Chem A 117:2181
Starcke JH, Wormit M, Dreuw A (2009) J Chem Phys 131:144311
Starcke JH, Wormit M, Dreuw A (2009) J Chem Phys 130:024104
Mewes JM, You ZQ, Wormit M, Kriesche T, Herbert JM, Dreuw A (2015) J Phys Chem A. doi:10.1021/jp511163y
Kerkines ISK, Petsalakis ID, Theodorakopoulos G, Klopper W (2009) J Chem Phys 131:224315
Corral I, González L (2008) J Comput Chem 29:1982
Pino GA, Oldani AN, Marceca E, Fujii M, Ishiuchi SI, Miyazaki M, Broquier M, Dedonder C, Jouvet C (2010) J Chem Phys 133:124313
Fliegl H, Köhn A, Hättig C, Ahlrichs R (2003) J Am Chem Soc 125:9821
Epifanovsky E, Polyakov I, Grigorenko B, Nemukhin A, Krylov AI (2009) J Chem Theory Comput 5:1895
Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Scalmani G, Barone V, Mennucci B, Petersson GA, Nakatsuji H, Caricato M, Li X, Hratchian HP, Izmaylov AF, Bloino J, Zheng G, Sonnenberg JL, Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, Nakajima T, Honda Y, Kitao O, Nakai H, Vreven T, Montgomery JA Jr., Peralta JE, Ogliaro F, Bearpark M, Heyd JJ, Brothers E, Kudin KN, Staroverov VN, Kobayashi R, Normand J, Raghavachari K, Rendell A, Burant JC, Iyengar SS, Tomasi J, Cossi M, Rega N, Millam JM, Klene M, Knox JE, Cross JB, Bakken V, Adamo C, Jaramillo J, Gomperts R, Stratmann RE, Yazyev O, Austin AJ, Cammi R, Pomelli C, Ochterski JW, Martin RL, Morokuma K, Zakrzewski VG, Voth GA, Salvador P, Dannenberg JJ, Dapprich S, Daniels AD, Farkas O, Foresman JB, Ortiz JV, Cioslowski J, Fox DJ (2009) Gaussian 09 revision D.01 (2009). Gaussian Inc, Wallingford
Zhao Y, Truhlar DG (2008) Theor Chem Acc 120:215
Head-Gordon M, Oumi M, Maurice D (1999) Mol Phys 96:593
Krylov AI, Gill PM (2013) Wiley Int Rev Comput Mol Sci 3:317
Shao Y, Gan Z, Epifanovsky E, Gilbert AT, Wormit M, Kussmann J, Lange AW, Behn A, Deng J, Feng X, Ghosh D, Goldey M, Horn PR, Jacobson LD, Kaliman I, Khaliullin RZ, Kuś T, Landau A, Liu J, Proynov EI, Rhee YM, Richard RM, Rohrdanz MA, Steele RP, Sundstrom EJ, Woodcock HL, Zimmerman PM, Zuev D, Albrecht B, Alguire E, Austin B, Beran GJO, Bernard YA, Berquist E, Brandhorst K, Bravaya KB, Brown ST, Casanova D, Chang CM, Chen Y, Chien SH, Closser KD, Crittenden DL, Diedenhofen M, DiStasio RA, Do H, Dutoi AD, Edgar RG, Fatehi S, Fusti-Molnar L, Ghysels A, Golubeva-Zadorozhnaya A, Gomes J, Hanson-Heine MW, Harbach PH, Hauser AW, Hohenstein EG, Holden ZC, Jagau TC, Ji H, Kaduk B, Khistyaev K, Kim J, Kim J, King RA, Klunzinger P, Kosenkov D, Kowalczyk T, Krauter CM, Lao KU, Laurent AD, Lawler KV, Levchenko SV, Lin CY, Liu F, Livshits E, Lochan RC, Luenser A, Manohar P, Manzer SF, Mao SP, Mardirossian N, Marenich AV, Maurer SA, Mayhall NJ, Neuscamman E, Oana CM, Olivares-Amaya R, O’Neill DP, Parkhill JA, Perrine TM, Peverati R, Prociuk A, Rehn DR, Rosta E, Russ NJ, Sharada SM, Sharma S, Small DW, Sodt A, Stein T, Stück D, Su YC, Thom AJ, Tsuchimochi T, Vanovschi V, Vogt L, Vydrov O, Wang T, Watson MA, Wenzel J, White A, Williams CF, Yang J, Yeganeh S, Yost SR, You ZQ, Zhang IY, Zhang X, Zhao Y, Brooks BR, Chan GK, Chipman DM, Cramer CJ, Goddard WA, Gordon MS, Hehre WJ, Klamt A, Schaefer HF, Schmidt MW, Sherrill CD, Truhlar DG, Warshel A, Xu X, Aspuru-Guzik A, Baer R, Bell AT, Besley NA, Chai JD, Dreuw A, Dunietz BD, Furlani TR, Gwaltney SR, Hsu CP, Jung Y, Kong J, Lambrecht DS, Liang W, Ochsenfeld C, Rassolov VA, Slipchenko LV, Subotnik JE, Van Voorhis T, Herbert JM, Krylov AI, Gill PM, Head-Gordon M (2015) Mol Phys 113(2):184
TURBOMOLE v6.6 (2014) A development of university of karlsruhe and forschungszentrum karlsruhe gmbh, 1989–2007, TURBOMOLE GmbH, since 2007. Available from http://www.turbomole.com
Nielsen ES, Jorgensen P, Oddershede J (1980) J Chem Phys 73:6238
Aidas K, Angeli C, Bak KL, Bakken V, Bast R, Boman L, Christiansen O, Cimiraglia R, Coriani S, Dahle P, Dalskov EK, Ekström U, Enevoldsen T, Eriksen JJ, Ettenhuber P, Fernández B, Ferrighi L, Fliegl H, Frediani L, Hald K, Halkier A, Hättig C, Heiberg H, Helgaker T, Hennum AC, Hettema H, Hjertenæs E, Høst S, Høyvik IM, Iozzi MF, Jansík B, Jensen HJA, Jonsson D, Jørgensen P, Kauczor J, Kirpekar S, Kjærgaard T, Klopper W, Knecht S, Kobayashi R, Koch H, Kongsted J, Krapp A, Kristensen K, Ligabue A, Lutnæs OB, Melo JI, Mikkelsen KV, Myhre RH, Neiss C, Nielsen CB, Norman P, Olsen J, Olsen JMH, Osted A, Packer MJ, Pawlowski F, Pedersen TB, Provasi PF, Reine S, Rinkevicius Z, Ruden TA, Ruud K, Rybkin VV, Sałek P, Samson CCM, de Merás AS, Saue T, Sauer SPA, Schimmelpfennig B, Sneskov K, Steindal AH, Sylvester-Hvid KO, Taylor PR, Teale AM, Tellgren EI, Tew DP, Thorvaldsen AJ, Thøgersen L, Vahtras O, Watson MA, Wilson DJD, Ziolkowski M, Ågren H (2014) Wiley Int Rev Comput Mol Sci 4(3):269
Chibani S, Le Guennic B, Charaf-Eddin A, Maury O, Andraud C, Jacquemin D (2012) J Chem Theory Comput 8:3303
Acknowledgments
The authors wish to dedicate this work to Dr. M. Wormit who very kindly helped us for the ADC calculations. They also acknowledge A. Dreuw, D. Escudero and C. Sergentu for fruitful discussions. D. J. acknowledges the European Research Council (ERC) and the Région des Pays de la Loire for financial support in the framework of a Starting Grant (Marches-278845) and a recrutement sur poste stratégique, respectively. This research used resources of (1) the GENCI-CINES/IDRIS, (2) CCIPL (Centre de Calcul Intensif des Pays de Loire) and (3) a local Troy cluster.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
214_2015_1676_MOESM1_ESM.pdf
Supporting Information: Dunning BS tests for 4 and figures for all methods, basis sets and molecules. (420KB) 214_2015_1676_MOESM1_ESM.pdf
Rights and permissions
About this article

Cite this article
Laurent, A.D., Blondel, A. & Jacquemin, D. Choosing an atomic basis set for TD-DFT, SOPPA, ADC(2), CIS(D), CC2 and EOM-CCSD calculations of low-lying excited states of organic dyes. Theor Chem Acc 134, 76 (2015). https://doi.org/10.1007/s00214-015-1676-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00214-015-1676-9