
Abstract
In this work, we investigate the ground- and excited-state structures as well as the optical properties of a series of five formazanate dyes using state-of-the-art density-based and wavefunction-based methods. The present work is the first to evaluate the properties of formazanate-BF2 dyes with wavefunction-correlated schemes. Firstly, we show that CC2 provides more twisted ground-state geometries than DFT while both approaches lead to planar excited-state structures. Secondly, we demonstrate that the differences between the transition energies computed at TD-DFT, CIS(D), SOS-CIS(D), ADC(2), and CC2 levels are large and that the optical spectra also significantly depend on the selected geometries. Indeed, CC2 fluorescence energies computed on TD-DFT structures significantly differ from their full-CC2 counterparts. Thirdly, we discuss the importance of solvent effects evaluated with various continuum models. Fourthly, we provide comparisons with experiment.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
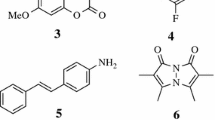
Fluoroborate dyes, which contain a BF2 group tethered between two electronegative carbon atoms in a way to lock the molecular structure, are certainly one of the most successful classes of organic fluorescent derivatives. Indeed, the leading members of this group, namely BODIPYs [1, 2], enjoy an ever-growing popularity due to their ease of synthesis, emission wavelengths tunable by chemical substitutions, large emission quantum yields, and great stabilities in a large panel of environments. Over the years, several other fluoroborate families have been developed [3], the most important being the aza-BODIPYs that present an additional nitrogen atom at the meso position [4]. Aza-BODIPYs present red-shifted optical spectra compared to the corresponding BODIPYs. However, both BODIPYs and aza-BODIPYs have the drawback of generally delivering small Stokes shifts, which limits their applications in several fields. This is why alternative boron(III) dyes [3] have been synthesized and characterized, e.g., iminocoumarins (boricos) [5], boranils [6], hydroxychalcones [7], boronic acid salicylidenehydrazones (bashys) [8], and bis(pyrrolyl-methylene) hydrazones (bophys) [9]. Another class of fluoroborates, using a formazanate core, has recently emerged (see Fig. 1 for representation of selected examples). This family tends to produce quite large Stokes shifts (but sometimes rather small fluorescence quantum yields) and also possesses very interesting electrochemical properties thanks to the presence of four nitrogen atoms in their core. Experimentally, the chemistry of formazanate fluoroborates has been mainly developed by the group of Gilroy [10–15] and by one of us [16, 17], while only a rather limited number of members of this dye family is currently available.

Representation of the compounds investigated in the present work
To our knowledge, almost only vertical time-dependent density functional theory (TD-DFT) calculations have been performed to date to explore the excited-state properties of formazanate fluoroborates [10, 12, 17]. While these calculations are certainly useful to obtain hints into the nature of the low-lying states, they are limited by: (i) the accuracy of TD-DFT transition energies, a particularly important question for fluoroborate derivatives, as correlated wavefunction theoretical methods are often required to reach quantitative estimates of the optical properties of these chromophores [18]; (ii) the vertical approximation in which they have been performed, i.e., there was no exploration of the excited-state potential energy surfaces, which is of course crucial for fluorescent derivatives [19]. Related to the former point, we underline that SOS-CIS(D) simulations of the vertical absorption energies led to significantly different results than TD-DFT for a series of substituted formazanates [17]. In the present contribution, we explore the ground- and excited-state structures of the molecules depicted in Fig. 1 with a focus on methodological aspects. More precisely, we compare the results obtained by correlated wavefunction approaches and by TD-DFT to assess the accuracy of the latter.
Computational details
Following Ref. [20], we have determined all geometrical parameters with the def2-TZVPP atomic basis set, whereas the transition energies have been calculated using the aug-cc-pVTZ atomic basis set. Such choice of very extended bases, containing f orbitals on second-row elements, is a bit overkilling, as much smaller bases are probably sufficient for fluoroborates, at least at the TD-DFT level [18]. However, this selection allows a balanced discussion of methodological effects.
All the DFT and TD-DFT calculations were performed using the Gaussian09 program [21], applying an improved self-consistent field convergence (SCF) threshold (10−8−10−9 a.u.), a tightened optimization criterion (10−5 a.u. on average forces) and the ultrafine pruned (99,590) DFT integration grid. These DFT and TD-DFT calculations relied on the M06-2X hybrid exchange-correlation functional [22] that provides a good correlation with experimental optical spectra though sometimes at the cost of an overestimation of the transitions energies [23–25]. Experimental spectra are available in toluene, an apolar and aprotic medium with a low dielectric constant, so that gas-phase calculations might appear as a suitable approximation. Nevertheless, we have also assessed the impact of solvent effects using the polarizable continuum model (PCM) [26] combined to TD-DFT. For the transition energies, both the linear-response (LR) [27, 28] and the corrected linear-response (cLR) approaches [29] were tested, whereas condensed-phase geometries and vibrational frequencies have been determined using the LR approach. The differences between LR and cLR responses for BODIPYs were discussed elsewhere [30]. We have applied the non-equilibrium (neq) and equilibrium (eq) limits of the PCM-TD-DFT model for transition energies and structures, respectively [26].
The SOS-CIS(D) [31] calculations were performed with Q-Chem [32] using the aug-cc-pVTZ basis set and the corresponding resolution-of-identity (RI) auxiliary basis set. The SCF convergence was set to 10−7 a.u. whereas the two-electron integral cutoff was tightened to 10−11 a.u. The frozen-core approximation was applied.
The CC2 geometry optimizations were achieved with Turbomole [33] using the RI(-JK) approach and correlating all electrons. The thresholds used for the SCF, density matrix, and Cartesian forces convergence limits were tightened to 10−9, 10−7 and 10−5 a.u., respectively, during the geometry optimizations. The aug-cc-pVTZ CIS(D), ADC(2), and CC2 transition energies were obtained with the same program using the RI approximation and applying default thresholds that are sufficient for our purposes. There are examples in the literature in which CC2 has been successfully used to determine the properties of BODIPYs [34–37].
Results and discussion
Ground- and excited-state structures
At the DFT and TD-DFT levels, it was possible to compute the Hessian and to ascertain the nature of the optimized geometries. At the CC2 level, these calculations were computationally out-of-reach and we first systematically started our geometry optimization with both C s and C 2v structures in order to locate the actual minima. It turned out that, at both (TD-)DFT and CC2 levels, the ground-state geometries belong to the former point group with the BF2 group significantly puckered with respect to the plane formed by the four nitrogen atoms, whereas in the excited-state the planar C 2v structures constitute the true minima of the TD-DFT potential energy surface for 1, 2, and 3. In 4, we found a slight deviation of the nitro group at the TD-DFT level, leading to a C 2 symmetry for the excited-state (the imaginary frequency for the C 2v form is i14.7 cm −1 only). In contrast, at the TD-DFT level, the C 2v excited-state structure of 5 is a true minimum. We have therefore examined C s , C 2 and C 2v excited-state geometries with CC2 for both 4 and 5. It turned out that the lowest CC2 energies were reached for the C 2 point group. In these structures, the nitro group is twisted compared to the formazanate plane.
The ground- and excited-state geometries of 1 are given in Fig. 2. On that representation, one clearly notices that the departure from planarity for the former structures is much more significant with CC2 than with DFT, the latter yielding out-of-plane deformations in better agreement with the experimental XRD [10], though this outcome could possibly be related to a compensation between the neglect of solid-state packing effects and the limitations of DFT. At the CC2 level, the energy difference between the C s and C 2v ground-state geometries attains 3.17, 2.65, 2.58, 3.00, and 1.98 kcal.mol −1 for 1, 2, 3, 4, and 5, respectively. The DFT differences were previously reported to be significantly smaller than their CC2 counterparts, of the order of 1 kcal.mol −1 only [10], a trend consistent with the smaller deformations of the ground-state DFT geometries from planarity (see Fig. 2).

Ground- and excited-state geometries of 1 obtained at the (TD-)DFT and CC2 levels of approximation. At the leftmost side, the experimental XRD structure taken from Ref. [10] is given—note that in this structure two non-equivalent molecules are present in each unit cell and that only one is displayed
In Table 1, we report a comparison between the computed and measured geometrical parameters in the core of the formazanate for the five investigated dyes. Overall, the agreement between all approaches is satisfying, but we nevertheless note that DFT provides too short N-N bonds compared to experiment whereas CC2 yields the opposite trend (with a similar absolute deviation compared to the crystal data). For the N-B-N angle, the CC2 values are significantly smaller than the XRD ones, whereas DFT is on the spot, and this is related to the different puckering of the BF2 group mentioned above. For the bond lengths, taking XRD values as references, we determine a mean absolute error (MAE) of 0.012 Å for both DFT and CC2 considering all values in Table 1, whereas for the valence angles the MAE are 1.9 and 3.9 ∘ with DFT and CC2, respectively. Overall, both levels of theory provide accurate ground-state geometries.
As stated above, the excited-state geometries are flat (see Fig. 2) employing both theoretical approaches, but the TD-DFT and CC2 structures nevertheless significantly differ. Notably, the N-N distances are shorter with TD-DFT than with CC2, like in the ground state. In Table 2, we list the variations, between the two electronic states, of the key geometrical parameters for the five molecules. Clearly, the structural modifications in the formazanate core are similar for all dyes, the major variation being the large elongation of the N-N bonds in the excited state. This change is perfectly consistent with the topology of the LUMO (see Fig. 3) that presents a clear anti-bonding character for the N-N bond. We notice that the magnitude of this elongation is slightly smaller with CC2 than with TD-DFT. The variations in the C-N and B-N bond distances are systematically trifling, while the largest changes in the valence angles appear for N-B-N due to the excited-state planarization. The difference between the ground- and excited-state geometries being stronger at the CC2 level of theory, the variation of the N-B-N angle is logically larger at this level of theory than with (TD-)DFT.

DFT HOMO (left) and LUMO (right) of 1
The partial atomic charges obtained with the Merz–Kollman model for 1 are listed in Table 3 for the atomic groups defined in Fig. 4. Interestingly, one notices that the nitrogen atoms tethering the boron (N[BF2]) are positively charged, whereas the other nitrogen atoms (N[C-CN]) bear a negative partial charge. For the ground state, DFT and CC2 yield very similar values (discrepancies < 0.05 e), so that the differences in the N-N distance obtained with these two approaches (see Table 1) are not related to dissimilar charge separations. In the excited state, the N[C-CN] atoms become slightly more negatively charged whereas both the phenyl rings and the C-CN groups becomes slightly more positive, but again, the TD-DFT and CC2 descriptions remain rather similar, but for the N[BF2] (phenyl rings) that are slightly more (less) positively charged with TD-DFT than with CC2.

Representation of the groups used in Table 3
Optical properties
Table 4 lists the vertical absorption and emission energies obtained with six levels of theory for the geometries obtained at the (TD-)DFT and CC2 levels. In this table, the most accurate values are the full CC2 ones on the rightmost column for each state and we use them as references in our analysis.
Let us start by focusing on the transition energies obtained with different methods on a given geometry. As expected, the CIS values are much too large with an overestimation of the order of 0.50 eV compared to the CC2 reference figures, an effect that we attribute to the neglect of electron correlation effects. TD-DFT transition energies are also too large in most cases, which is typical for fluoroborate compounds [18], but the magnitude of the error is much smaller, ca. 0.10 eV. In contrast, ADC(2) undershoots the CC2 values rather significantly, with an average deviation of –0.23 eV across Table 4, which contrasts with most classes of dyes for which ADC(2) and CC2 values match very well [20, 38]. SOS-CIS(D) delivers even worse values, with underestimations often larger than –0.40 eV. In fact, it is CIS(D) that is the closest method from the CC2 references, with only a very small tendency to yield too small values, again contrasting with trends obtained on more diverse sets of compounds [38].
An aspect that has been much less studied in the literature is the impact of using CC2 instead of (TD-)DFT geometries. For the vertical absorption, we notice that choosing CC2 structures instead of their DFT counterparts yields significant increases of the transition energy for the methods that (partly) account for the double excitations, e.g., the ADC(2) absorption energies increase by +0.14 eV on average. This tendency is consistent with the geometries represented in Fig. 2: one expects smaller gaps for more planar structures that are favorable for π-conjugation. In contrast, the CIS absorption energies decrease when going from DFT to CC2 geometries, which is counter-intuitive. Interestingly, the TD-DFT absorption values are almost insensitive to the selected ground-state geometry in the present case. For the vertical emission, although CC2 and TD-DFT excited-state geometries are rather similar, the values obtained for a given method on the CC2 geometries are significantly smaller than the one computed with the TD-DFT structures. This effect is rather large, e.g., −0.22 eV, −0.23 eV, and −0.20 eV on average for the TD-DFT, ADC(2) and CC2 fluorescence energies, respectively. Besides illustrating that formazanate are particularly sensitive to electron correlation effects, these results also come as a warning: computing CC2 transition energies on TD-DFT structures might be insufficient to reach accurate results. This is well illustrated by the Stokes shift of 3 that attains 0.516 eV at the TD-M06-2X level and a similar value, 0.481 eV, when CC2 transition energies are determined on (TD-)DFT structures, but is much larger, 0.740 eV, when CC2 is used for all computational steps.
Solvent effects
To evaluate solvatochromic effects, we have used the TD-DFT level only as refined continuum models can be combined to TD-DFT [29]. Our results are given in Table 5. First, we recall that the reference experiments have been carried out in toluene, an aprotic apolar solvent, so that no drastic solvent effects are expected. For the absorption, we found that using the condensed phase geometries induces a slight increase (ca. + 0.02 eV) of the transition energies, whereas the differences between the results obtained with the eq and neq limits are very small, which is related to the nature of toluene (\(\epsilon _{r} \simeq {n^{2}_{D}}\)). In contrast, using the LR-PCM solvation model leads to a significant decrease of the transition energies (ca. −0.10 eV) compared to the gas-phase reference, an effect that we associate to the limits of this model, as the more refined cLR-PCM approach indeed predicts much smaller variations. Globally, the vertical absorption energies therefore slightly increase (+ 0.02 eV for 1 and + 0.04 eV for 2) when going from gas-phase to toluene. The trends are rather similar for the vertical fluorescence, i.e., the LR-PCM approach overshoots solvation effects, cLR and gas-phase transition energies are similar and the difference between eq and neq limits are trifling. However, using excited-state structures optimized in solution now leads to a decrease of the transition energies (ca. −0.04 eV for 1 and ca. −0.03 eV for 2). This indicates that the estimated Stokes shifts are slightly larger when solvent effects are accounted for.
Comparisons with experiment
Having explored several methodological aspects, we now turn towards a comparison between theoretical and experimental values. To allow physically meaningful comparisons, we determined the theoretical 0–0 energies that can be directly compared to the crossing point between the experimental absorption and emission curves [10, 12]. As the solvent effects are trifling but as the influences of the selected theoretical models are large, we decided to determine 0–0 energies at the gas-phase CC2 level only. The sole TD-DFT term entering in the formulation of the 0–0 energy is the difference between the vibrational energies (ΔE ZPVE) of the two states that is known to be rather independent of the selected level of theory [20]. ΔE ZPVE amounts to −0.069, −0.065, −0.057, −0.074 and −0.063 eV for 1, 2, 3, 4 and 5, respectively, values that are not uncommon for organic dyes [39]. Adding these values to the CC2 adiabatic energies, we obtain theoretical 0-0 energies of 2.319, 2.219, 1.994, 2.312, and 1.923 eV for the five dyes. Their experimental counterparts are 2.29, 2.24, 2.03, 2.28, and 1.99 eV. This yields a mean absolute deviation between theoretical and experimental estimates as small as 0.04 eV and a linear determination coefficient (R 2) as large as 0.99. This undoubtedly demonstrates the quality of the results obtained with the “best” level of theory: both the absolute and relative evolutions of the transition energies are very accurately reproduced.
Conclusions
We have evaluated the ground- and excited-state properties of five fluoroborates dyes presenting a formazanate core, with different approaches. It turned out that the deviation from planarity of the ground-state is much more pronounced with CC2 than with DFT. In the excited state, the two methods foresee a planarization of the core of the formazanate, and hence there is a strong geometrical reorganization between the two electronic states, explaining the rather large Stokes shifts of these dyes. The selected level of theory has not only a strong direct impact on the transition energies—which was expected—but also a large indirect impact through the change of the optimal structures—which is more surprising. Indeed, CC2 emission energies determined on TD-DFT and CC2 excited-state geometries differ by ca. 0.20 eV, which is larger than the differences between TD-DFT and CC2 transition energies computed on a given geometry. Though this outcome might be related to the very specific electronic nature of formazanates, it comes as a warning that mixed wavefunction/TD-DFT methodologies might sometimes be unable to deliver the expected accuracy level. The impact of solvent effects (here toluene) are estimated to be rather weak if not negligible with the corrected linear-response approach, while the linear-response models predict rather large positive solvatochromic effects for both absorption and emission, an inconsistent result given the selected solvent. Using the most accurate level of theory proposed herein (CC2), very small deviation with respect to the experimental 0–0 energies are obtained, with a mean absolute deviation limited to 0.04 eV.
References
Loudet A, Burgess K (2007) Chem Rev 107:4891
Ulrich G, Ziessel R, Harriman A (2008) Angew Chem Int Ed 47:1184
Frath D, Massue J, Ulrich G, Ziessel R (2014) Angew Chem Int Ed 53:2290
Ge Y, O’Shea DF (2016) Chem Soc Rev 45:3846
Frath D, Poirel A, Ulrich G, De Nicola A, Ziessel R (2013) Chem Commun 49:4908
Frath D, Azizi S, Ulrich G, Ziessel R (2012) Org Lett 14:4774
D’Aléo A, Gachet D, Heresanu V, Giorgi M, Fages F (2012) Chem Eur J 18:12764
Santos FMF, Rosa JN, Candeias NR, Carvalho CP, Matos AI, Ventura AE, Florindo HF, Silva LC, Pischel U, Gois PMP (2016) Chem Eur J 22:1631
Tamgho IS, Hasheminasab A, Engle JT, Nemykin VN, Ziegler CJ (2014) J Am Chem Soc 136:5623
Barbon SM, Reinkeluers PA, Price JT, Staroverov VN, Gilroy JB (2014) Chem Eur J 20:11340
Barbon SM, Price JT, Reinkeluers PA, Gilroy JB (2014) Inorg Chem 53:10585
Barbon SM, Staroverov VN, Gilroy JB (2015) J Org Chem 80:5226
Barbon SM, Price JT, Yogarajah U, Gilroy JB (2015) RSC Adv 5:56316
Hesari M, Barbon SM, Staroverov VN, Ding Z, Gilroy JB (2015) Chem Commun 51:3766
Maar RR, Barbon SM, Sharma N, Groom H, Luyt LG, Gilroy JB (2015) Chem Eur J 21:15589
Chang MC, Otten E (2014) Chem Commun 50:7431
Chang MC, Chantzis A, Jacquemin D, Otten E (2016) Dalton Trans 45:9477
Le Guennic B, Jacquemin D (2015) Acc Chem Res 48:530
Santoro F, Jacquemin D (2016) WIREs Comput Mol Sci 6:460
Winter NOC, Graf NK, Leutwyler S, Hattig C (2013) Phys Chem Chem Phys 15:6623
Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Scalmani G, Barone V, Mennucci B, Petersson GA, Nakatsuji H, Caricato M, Li X, Hratchian HP, Izmaylov AF, Bloino J, Zheng G, Sonnenberg JL, Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, Nakajima T, Honda Y, Kitao O, Nakai H, Vreven T, Montgomery Jr JA, Peralta JE, Ogliaro F, Bearpark M, Heyd JJ, Brothers E, Kudin KN, Staroverov VN, Kobayashi R, Normand J, Raghavachari K, Rendell A, Burant JC, Iyengar SS, Tomasi J, Cossi M, Rega N, Millam JM, Klene M, Knox JE, Cross JB, Bakken V, Adamo C, Jaramillo J, Gomperts R, Stratmann RE, Yazyev O, Austin AJ, Cammi R, Pomelli C, Ochterski JW, Martin RL, Morokuma K, Zakrzewski VG, Voth GA, Salvador P, Dannenberg JJ, Dapprich S, Daniels AD, Farkas O, Foresman JB, Ortiz JV, Cioslowski J, Fox DJ (2009) Gaussian 09 revision d.01. Gaussian Inc, Wallingford CT
Zhao Y, Truhlar DG (2008) Theor Chem Acc 120:215
Jacquemin D, Planchat A, Adamo C, Mennucci B (2012) J Chem Theory Comput 8:2359
Isegawa M, Peverati R, Truhlar DG (2012) J Chem Phys 137:244104
Leang SS, Zahariev F, Gordon MS (2012) J Chem Phys 136:104101
Tomasi J, Mennucci B, Cammi R (2005) Chem Rev 105:2999
Cammi R, Mennucci B (1999) J Chem Phys 110:9877
Cossi M, Barone V (2001) J Chem Phys 115:4708
Caricato M, Mennucci B, Tomasi J, Ingrosso F, Cammi R, Corni S, Scalmani G (2006) J Chem Phys 124:124520
Jacquemin D, Chibani S, Le Guennic B, Mennucci B (2014) J Phys Chem A 118:5343
Rhee YM, Head-Gordon M (2007) J Phys Chem A 111:5314
Shao Y, Gan Z, Epifanovsky E, Gilbert AT, Wormit M, Kussmann J, Lange AW, Behn A, Deng J, Feng X, Ghosh D, Goldey M, Horn PR, Jacobson LD, Kaliman I, Khaliullin RZ, Kuś T, Landau A, Liu J, Proynov EI, Rhee YM, Richard RM, Rohrdanz MA, Steele RP, Sundstrom EJ, Woodcock HL, Zimmerman PM, Zuev D, Albrecht B, Alguire E, Austin B, Beran GJO, Bernard YA, Berquist E, Brandhorst K, Bravaya KB, Brown ST, Casanova D, Chang CM, Chen Y, Chien SH, Closser KD, Crittenden DL, Diedenhofen M, DiStasio RA, Do H, Dutoi AD, Edgar RG, Fatehi S, Fusti-Molnar L, Ghysels A, Golubeva-Zadorozhnaya A, Gomes J, Hanson-Heine MW, Harbach PH, Hauser AW, Hohenstein EG, Holden ZC, Jagau TC, Ji H, Kaduk B, Khistyaev K, Kim J, Kim J, King RA, Klunzinger P, Kosenkov D, Kowalczyk T, Krauter CM, Lao KU, Laurent AD, Lawler KV, Levchenko SV, Lin CY, Liu F, Livshits E, Lochan RC, Luenser A, Manohar P, Manzer SF, Mao SP, Mardirossian N, Marenich AV, Maurer SA, Mayhall NJ, Neuscamman E, Oana CM, Olivares-Amaya R, O’Neill DP, Parkhill JA, Perrine TM, Peverati R, Prociuk A, Rehn DR, Rosta E, Russ NJ, Sharada SM, Sharma S, Small DW, Sodt A, Stein T, Stück D, Su YC, Thom AJ, Tsuchimochi T, Vanovschi V, Vogt L, Vydrov O, Wang T, Watson MA, Wenzel J, White A, Williams CF, Yang J, Yeganeh S, Yost SR, You ZQ, Zhang IY, Zhang X, Zhao Y, Brooks BR, Chan GK, Chipman DM, Cramer CJ, Goddard WA, Gordon MS, Hehre WJ, Klamt A, Schaefer HF, Schmidt MW, Sherrill CD, Truhlar DG, Warshel A, Xu X, Aspuru-Guzik A, Baer R, Bell AT, Besley NA, Chai JD, Dreuw A, Dunietz BD, Furlani TR, Gwaltney SR, Hsu CP, Jung Y, Kong J, Lambrecht DS, Liang W, Ochsenfeld C, Rassolov VA, Slipchenko LV, Subotnik JE, Van Voorhis T, Herbert JM, Krylov AI, Gill PM, Head-Gordon M (2015) Mol Phys 113:184
Turbomole v6.6 2014, a development of university of karlsruhe and forschungszentrum karlsruhe gmbh, 1989-2007, turbomole gmbh, since 2007; available from http://www.turbomole.com (accessed 13 june 2016)
Quartarolo AD, Sicilia E, Russo N (2009) J Chem Theory Comput 5(7):1849
Quartarolo AD, Russo N (2011) J Chem Theory Comput 7(4):1073
Momeni MR, Brown A (2015) J Chem Theory Comput 11(6):2619
Momeni MR, Brown A (2016) J Phys Chem A 120(16): 2550
Jacquemin D, Duchemin I, Blase X (2015) J Chem Theory Comput 11:5340
Laurent AD, Jacquemin D (2013) Int J Quantum Chem 113:2019
Acknowledgments
D.J. acknowledges the European Research Council (ERC) and the Région des Pays de la Loire for financial support in the framework of a Starting Grant (Marches - 278845) and the LumoMat project, respectively. This research used resources of (i) the GENCI-CINES/IDRIS; (ii) CCIPL (Centre de Calcul Intensif des Pays de Loire); (iii) a local Troy cluster and (iv) HPC resources from ArronaxPlus (grant ANR-11-EQPX-0004 funded by the French National Agency for Research).
Author information
Authors and Affiliations
Corresponding author
Additional information
It is our pleasure to dedicate this work to Prof. Henry Chermette with whom it has always been a great pleasure to discuss and collaborate. Merci Henry !
This paper belongs to Topical Collection Festschrift in Honor of Henry Chermette
Rights and permissions
About this article

Cite this article
Laurent, A.D., Otten, E., Le Guennic, B. et al. Formazanate boron difluoride dyes: discrepancies between TD-DFT and wavefunction descriptions. J Mol Model 22, 263 (2016). https://doi.org/10.1007/s00894-016-3126-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00894-016-3126-6