
Abstract
Rationale
Prepulse inhibition (PPI) refers to the reduction of the startle response magnitude when a startling stimulus is closely preceded by a weak stimulus. PPI is commonly used to measure sensorimotor gating. In rats, the PPI reduction induced by the dopamine agonist apomorphine can be reversed by systemic administration of nicotine. A high concentration of nicotinic receptors is found in the lateral habenula (LHb), an epithalamic structure with efferent projections to brain regions involved in the modulation of PPI, which has been shown to regulate the activity of midbrain dopamine neurons.
Objectives
The prospective role of nicotinic receptors in the LHb in the regulation of PPI was assessed in this study, using different pharmacological models of sensorimotor gating deficits.
Methods
Interactions between systemic amphetamine and haloperidol and intra-LHb infusions of mecamylamine (10 μg/side) or nicotine (30 μg/side) on PPI were analyzed in Experiments 1 and 2. Intra-LHb infusions of different nicotine doses (25, and 50 μg/side) and their interactions with systemic administration of amphetamine or dizocilpine on PPI were examined in Experiments 3 and 4.
Results
Infusions of nicotine into the LHb dose-dependently attenuated amphetamine-induced PPI deficits but had no effect on PPI disruptions caused by dizocilpine. Intra-LHb mecamylamine infusions did not affect PPI nor interact with dopaminergic manipulations.
Conclusions
These results are congruent with previous reports of systemic nicotine effects on PPI, suggesting a role of the LHb in the attenuation of sensorimotor gating deficits caused by the hyperactivity of dopamine systems.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Sensorimotor gating refers to the modulation of motor responses observed when multiple sensory stimuli are presented in rapid succession (Swerdlow et al. 2000). One form of startle plasticity commonly used to assess sensorimotor gating experimentally is prepulse inhibition (PPI). PPI is a neuropsychological phenomenon in which the motor response to a startling stimulus (pulse) is significantly reduced when that stimulus is closely preceded in time by another usually weaker stimulus (prepulse).
The neural and behavioral mechanisms underlying startle response are well documented. The neural circuit that mediates the acoustic startle response consists of an excitatory pathway in which neurons in the caudal pontine reticular nucleus (PnC) receive acoustic input from cochlear root neurons and project to the motor neurons to produce the startle response (Lingenhöhl and Friauf 1994). The PnC also receives inhibitory projections from neurons in the pedunculopontine tegmental nucleus (PPTg), where acoustic information is relayed from the inferior and superior colliculi (Carlson and Willott 1996; Fendt 1999). This mediatory circuit of the acoustic startle response and PPI (excitatory and inhibitory pathways) receives projections from a number of different brain structures that can modulate PPI, through direct or indirect projections to the PPTg (“modulatory circuit”; Koch 1999). These areas include the nucleus accumbens (NAC; Swerdlow et al. 1990a), ventral tegmental area (VTA; Zhang et al. 1995), hippocampus (Bakshi and Geyer 1998; Japha and Koch 1999), amygdala (Decker et al. 1995), raphe nuclei (RN) (Kusljic et al. 2003), and medial prefrontal cortex (MPFC) (Bubser and Koch 1994). Since patients with various neuropsychiatric disorders exhibit impaired sensorimotor gating as well as dysfunction in brain regions that modulate PPI, studying the contribution of specific neurotransmitters on PPI may aid in the development of treatments with potential therapeutic applications (Swerdlow et al. 2000).
The habenula complex is an epithalamic structure that receives afferent connections from limbic brain regions and projects to brainstem structures (Hikosaka et al. 2008), providing feedback control over the modulation of brainstem dopamine and serotonin systems. The lateral part of the habenula (LHb) has direct projections to dopamine and serotonin brain regions, such as the VTA and RN (Hikosaka et al. 2008), structures that have modulatory influences on PPI (Koch 1999) and that have been linked to anxiety and stress, as well as cognitive function (Lowry et al. 2008). Heldt and Ressler (2006) showed that PPI in mice increased following fear conditioning training, whereas PPI in animals that received electrolytic lesions of the habenula did not change after conditioning. Conditioned fear stress has been shown to increase the concentration of extracellular dopamine in the MPFC (Yoshioka et al. 1996), and dopamine innervation of the MPFC can affect the activity of the NAC (Jaskiw and Weinberger 1987). Infusion of the indirect dopamine agonist amphetamine into the MPFC reduces dopamine in the NAC (Louilot et al. 1989; Jaskiw et al. 1991), and lesions that deplete dopamine from the NAC can reverse the amphetamine-induced disruption of PPI in rats (Swerdlow et al. 1990b). So, stress-induced increases of dopamine in the MPFC could lead to reduced levels of dopamine in the NAC and enhanced PPI levels. Since administration of clozapine to habenula-lesioned mice resulted in PPI levels similar to control animals, Heldt and Ressler’s (2006) results suggest an involvement of the habenula in the regulation and maintenance of appropriate PPI levels through the modulation of monoamine systems.
The LHb contains a high concentration of nicotinic receptors (Rainbow et al. 1984). Sanders et al. (2010) showed that chronic infusion of the nicotinic receptor antagonist mecamylamine into the LHb increases memory errors in a radial arm maze task, an effect that could be reversed by the systemic administration of nicotine. Based on these findings, together with those showing the role of the LHb on the regulation of dopamine systems (e.g., Christoph et al. 1986; Heldt and Ressler 2006) and the efferent connections of this structure to brain areas involved in the modulation of PPI (Hikosaka et al. 2008), the goal of the present study was to analyze the contribution of nicotinic receptors in the LHb on the regulation of PPI using animal models of sensorimotor gating deficits.
The effects on PPI of localized infusions into the LHb of the nicotinic antagonist mecamylamine (Experiment 1) and the nicotinic agonist nicotine (Experiments 2 and 3) were tested against systemic dopaminergic drugs (amphetamine and haloperidol on Experiments 1 and 2; amphetamine on Experiment 3). Previous studies have shown that (a) electrical stimulation of the LHb results in reduced activity of dopamine neurons in the VTA (Christoph et al. 1986), and (b) excitation of LHb neurons can inhibit dopamine neurons, but inhibition of LHb neurons does not trigger excitation of dopamine neurons (Matsumoto and Hikosaka 2007). Therefore, it was hypothesized that nicotine infusions would improve the PPI deficits induced by the amphetamine treatment, and that mecamylamine infusions would have no effect or further decrease PPI. Based on the results of Experiment 2, the nicotine dose range was increased in Experiment 3 and tested against the same systemic amphetamine treatment. In Experiment 4, the effects on PPI of localized infusions into the LHb of nicotine were tested against a systemic NMDA/glutamatergic antagonist drug (dizocilpine), in order to assess the role of the habenula in the modulation of PPI through a dopamine-independent mechanism.
Methods
Subjects
Female Sprague–Dawley rats (one set of animals (n = 12) for Experiments 1 and 2 and a different set (n = 12) for Experiments 3 and 4) were obtained from the Charles River Laboratories (Raleigh, NC, USA). Animals were housed in groups of three per cage (before surgery) or individually (after surgery) at an ambient temperature of 20 °C on a 12:12 h reverse dark–light cycle (lights off at 0700 h) and had ad libitum access to water and food. Rats were acclimated to the housing facilities for 2 weeks after arrival and exposed to the test context and experimental apparatus before testing sessions began. Mean rat weight at the time of surgery was 275 g and varied between 280 and 320 g during behavioral tests.
Experimental design
The interactions between systemic dopaminergic manipulations and nicotinic receptors in the habenula on PPI were analyzed in Experiments 1 and 2. In Experiment 1, animals received subcutaneous (SC) injections of saline, amphetamine, or haloperidol after local LHb infusions of artificial cerebrospinal fluid (ACSF) or nicotinic receptor antagonist mecamylamine (10 μg/side; Decker and Majchrzak 1992). In Experiment 2, rats received local LHb infusions of ACSF or nicotine (30 μg/side; Craft and Millholland 1998) with systemic SC administration of the same drugs used in Experiment 1. In Experiments 3 and 4, the effects on PPI of SC administration of amphetamine (Experiment 3) or dizocilpine (Experiment 4) after local LHb infusions of a larger nicotine dose in the habenula were analyzed. In each experiment, all possible combinations of systemic drug treatment and LHb infusion were given using a repeated-measures counterbalanced (Latin-square) design, in order to (a) test behavioral responses randomly across the different phases of the estrous cycle and (b) reduce the potential impact of drug carryover effects following repeated test sessions. Within each experiment, every animal received all treatment combinations. The animals were maintained according to NIH guidelines, and protocols were approved by the Duke University Animal Care and Use Committee.
Surgery and local lateral habenular infusions
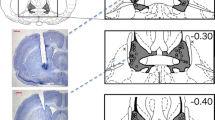
The procedure to implant the chronic guide cannulae took place after two baseline PPI sessions. The coordinates for the LHb nuclei were determined based on Pellegrino et al.’s (1979) atlas: anterioposterior (AP), −1.8 mm from bregma; mediolateral (ML), ± 1 mm from midline; dorsoventral (DV), −4.75 mm from the skull surface. Cannula implantation surgery was done in a manner as described previously (Kutlu et al. 2013). In order to minimize tissue damage, guide cannulae tips were set 1 mm dorsal to the target infusion site. After surgery, animals were allowed to recover for a week before testing started. Before the beginning of each PPI session, drugs were administered using infusion cannulae whose tips extended 1 mm ventrally beyond the end of the guide cannulae. Drugs were dissolved in ACSF (which was administered by itself as control), and the total infusion volume of 1 μl/side/session was delivered over 180 s. For the different experiments, infusion treatments consisted of the following: Experiment 1, ACSF and mecamylamine (mecamylamine HCl; (Sigma, St. Louis, MO, USA); 10 μg/side as of salt weight); Experiment 2, ACSF and nicotine ditartrate (Sigma, St. Louis, MO, USA); 30 μg/side as of salt weight); Experiment 3, ACSF and nicotine ditartrate (25 and 50 μg/side); Experiment 4, ACSF and nicotine ditartrate (50 μg/side). After all PPI sessions were completed, placement of the infusion cannulae was verified by performing histological sections of the animals’ brains.
Systemic drug administration
Following LHb infusions, systemic drugs were administered subcutaneously in a 1 ml/kg volume of saline vehicle, alone or in combination with the drug dose to be analyzed (saline-alone injections were used as control). Injections on consecutive test days were administered on alternating sides of the animal’s body to prevent potential tissue lesions. For the different experiments, systemic drug treatments consisted of the following: Experiments 1 and 2, saline, amphetamine (amphetamine sulfate salt; (Sigma, St. Louis, MO, USA); 1 mg/kg as of salt weight) and haloperidol (haloperidol HCl; (Abbott Labs, Abbott Park, IL, USA); 0.1 mg/kg as of salt weight); Experiment 3, saline and amphetamine (1 mg/kg); Experiment 4, saline and dizocilpine (dizocilpine hydrogen maleate; (Sigma, St. Louis, MO, USA); 0.05 mg/kg as of salt weight). Drugs were administered 10 min before the beginning of the acclimation period of each test session, and test sessions were conducted 2–3 days apart to allow for complete drug washout between successive sessions.
Apparatus
Acoustic startle responses were measured and PPI levels calculated using the Med Associates Startle Reflex System (St. Albans, VT, USA). The equipment included cylindrical acrylic restraining devices to confine the animals (7.6 cm inside diameter), harnessed on top of piezoelectric platforms inside sound-attenuating chambers. Each platform was calibrated using a spinner-type calibrator (Med Associates Startle Calibrator). Data from the platforms measuring animals’ responses were collected in 1-s time windows beginning 200 ms prior to the onset of the startling stimuli and sampled at a 1-KHz frequency. Auditory stimuli were presented through speakers located within the chambers midway along the long axis of the acrylic restraints. The sound intensity of the speaker in each chamber was calibrated before test sessions (Digital Sound Level Meter, Extech Instruments, Wilmington, NC, USA). The white background noise was set at 65 dB.
Startle and prepulse inhibition procedure
Behavioral tests started with a 5-min acclimation period in which animals were exposed only to the 65-dB background white noise. Three blocks of trials were presented following the acclimation period, with trials consisting of either (a) a single startling acoustic stimulus (20-ms, 120-dB white noise burst; pulse-alone trial), or (b) an acoustic prepulse stimulus (20-ms white noise burst of one of three possible intensities: 68, 71, or 77 dB) followed by the startling acoustic noise (prepulse-pulse trial). In prepulse-pulse trials, prepulses were presented 100 ms before the pulse stimuli (lead interval). In Block 1, 6 pulse-alone trials were presented in order to habituate and stabilize the animals’ responses. In Block 2, 48 trials were randomly presented: 12 pulse-alone and 36 prepulse-pulse trials (12 prepulse-pulse trials for each of the 3 prepulse intensities). Block 3 consisted of 5 additional pulse-alone trials. The intertrial interval varied randomly between 10 and 20 s in all blocks. Data from trials in Block 2 were used to determine response amplitudes and PPI. Startle response amplitude was calculated as the difference between the maximum wave amplitude in a 200-ms window after pulse onset and the maximum wave value during the 100-ms interval pre-stimulus presentation (null period). Prepulse inhibition was calculated as the ratio of the difference in response amplitude in pulse-alone and prepulse-pulse trials to the startle response in pulse-alone trials, multiplied by 100. That is, PPI (%) = 100* (pulse-alone response amplitude – prepulse-pulse response amplitude)/pulse-alone response amplitude. Including LHb infusion and drug administration, test sessions lasted approximately 35 min.
Data analysis
Repeated-measures analysis of variance (ANOVA) tests were used to assess the effects of systemic drug administration, local LHb infusion, and their interactions on the startle response amplitude. Analyses of PPI for all experiments included the same factors as those employed for the startle amplitude, with the addition of prepulse intensity. Significant interactions were followed up by post-hoc tests comparing the impact of the individual treatments and their combinations using Tukey’s honest significant difference (HSD) tests. A p value of 0.05 was used as the threshold for statistical significance, and interactions with p < 0.10 were followed up by tests of simple main effects (Snedecor and Cochran 1967). When the number of levels of a repeated-measure factor was larger than two, Mauchly’s test was used to verify the validity of the sphericity assumption and to determine if Greenhouse–Geisser corrections needed.
Results
Experiment 1
Histological verification of cannulae placement after behavioral testing for the first set of 12 rats revealed one unsuccessful infusion case. In addition, one rat did not recover from the surgical procedure. Therefore, only data from 10 animals were included in the subsequent analyses. The combined effects on PPI of mecamylamine infusion into the habenula and systemic dopaminergic treatments are shown in Fig. 1. A 3 × 2 × 3 ANOVA test on PPI (systemic treatment: saline, 1 mg/kg amphetamine, and 0.1 mg/kg haloperidol × local lateral habenular infusion into the habenula: ACSF and 10 μg/side mecamylamine × prepulse intensity: 68, 71, and 77 dB) revealed significant main effects of systemic drug treatment (F(2,18) = 60.46, p < 0.0001; χ 2(2) = 1.82, p > 0.4) and prepulse intensity (F(2,18) = 46.92, p < 0.0001; χ 2(2) = 2.36, p > 0.3), and a trend toward a significant interaction between those factors (F(4,36) = 2.46, p = 0.06; χ 2(9) = 6.4, p > 0.7). Subsequent analyses showed that, when compared to saline, amphetamine decreased PPI (57.15 % and 23.53 % respectively, p < 0.0002). Increasing prepulse intensities resulted in larger inhibition levels (40.23 %, 46.46 %, and 60.29 % for 68, 71, and 77 dB prepulses). Mecamylamine infusion into the habenula did not affect PPI, nor did it interact with any other factors.

Effects on PPI of mecamylamine (Mec) infusion into the habenula and systemic dopaminergic manipulations. Data represent mean values (± SEM). *Denotes significant differences in PPI of systemic treatment from saline condition, p < 0.0002
As shown in Table 1, startle responses were not affected by drug treatment or LHb infusion. A 3 × 2 ANOVA test on response amplitude (systemic treatment: saline, 1 mg/kg amphetamine, and 0.1 mg/kg haloperidol × LHb infusion into the habenula: ACSF and 10 μg/side mecamylamine) revealed no significant main effect of systemic drug administration, LHb infusion, or an interaction between factors.
Experiment 2
Figure 2 shows the combined effects on PPI of nicotine infusion into the habenula and systemic dopaminergic treatments. A 3 × 2 × 3 ANOVA test on PPI (systemic treatment: saline, 1 mg/kg amphetamine, and 0.1 mg/kg haloperidol × local LHb infusion into the habenula: ACSF and 30 μg/side nicotine × prepulse intensity: 68, 71, and 77 dB) revealed significant main effects of systemic drug treatment (F(2,18) = 68.4, p < 0.0001; χ 2(2) = 0.09, p > 0.95) and prepulse intensity (F(2,18) = 93.04, p < 0.0001; χ 2(2) = 2.16, p > 0.34), and a trend toward a significant interaction between systemic treatment and local LHb infusion (F(2,18) = 2.8, p = 0.08; χ 2(2) = 0.67, p > 0.71), mainly due to a non-significant increase in PPI after amphetamine administration when nicotine was infused into the LHb compared to the ASCF condition (p = 0.09). Subsequent analyses showed that, when compared to saline, amphetamine decreased PPI (71.0 % and 31.65 % respectively, p < 0.0002), and that increasing prepulse intensities resulted in larger inhibition levels (44.76 %, 55.94 %, and 70.62 % for 68, 71, and 77 dB prepulses). Nicotine infusion into the habenula did not significantly affect overall PPI, nor did it interact with prepulse intensity. No other significant interactions were found.

Effects on PPI of nicotine (Nic) infusion into the habenula and systemic dopaminergic manipulations. Data represent mean values (± SEM). *Denotes significant differences in PPI of systemic treatment from saline condition, p < 0.0002
Nicotine infusion caused a reduction in startle response magnitude (Table 1). A 3 × 2 ANOVA test on response amplitude (systemic treatment: saline, 1 mg/kg amphetamine, and 0.1 mg/kg haloperidol × local lateral habenular infusion into the habenula: ACSF and 30 μg/side nicotine) revealed a significant main effect of nicotine infusion (F(1,9) = 6.49, p < 0.04) but not of systemic drug administration or an interaction between factors.
Experiment 3
Histological verification of cannulae placement after behavioral testing for the second set of 12 rats revealed two unsuccessful infusion cases. Therefore, only data from 10 animals were included in the subsequent analyses. The effects on PPI of nicotine infusions into the habenula and systemic amphetamine administration are shown in Fig. 3. A 2 × 3 × 3 ANOVA test on PPI (systemic treatment: saline and 1 mg/kg amphetamine × local lateral habenular infusion into the habenula: ACSF, 25 μg/side, and 50 μg/side nicotine × prepulse intensity: 68, 71, and 77 dB) revealed significant main effects of systemic amphetamine administration (F(1,9) = 107.07, p < 0.0001), prepulse intensity (F(2,18) = 29.76, p < 0.0001; χ 2(2) = 1.50, p > 0.47), and a significant interaction between amphetamine and nicotine infusion into the habenula (F(2,18) = 5.95, p < 0.011; χ 2(2) = 0.41, p > 0.81). Overall PPI was lower after amphetamine (17.1 %) than saline administration (57.4 %), and subsequent analyses showed that increasing prepulse intensities resulted in larger inhibition levels (27.67 %, 35.87 %, and 48.19 % for 68, 71, and 77 dB prepulses). With ACSF infusions, amphetamine administration decreased PPI levels when compared to the saline condition (4.66 % and 61.24 %, p < 0.0002). This amphetamine-induced decrease in PPI was significantly counteracted when nicotine was infused into the habenula (26.13 %, p < 0.05), though this improvement in PPI was not sufficient to reach control levels (p < 0.001). Nicotine infusion did not significantly affect overall PPI, nor did it interact with prepulse intensity. No other significant interactions were found.

Effects on PPI of nicotine (Nic) infusions into the habenula and systemic amphetamine administration. Data represent mean values (± SEM). *Denotes significant differences in PPI of systemic treatment from saline condition, p < 0.0001. + p < 0.0002 compared with the saline/ACSF condition (three-way ANOVA followed by Tukey’s HSD test). x p < 0.05 compared with the amphetamine/ACSF condition (three-way ANOVA followed by Tukey’s HSD test)
As shown in Table 1, startle responses were not affected by systemic drug treatment or local lateral habenular infusion. A 2 × 3 ANOVA test on response amplitude (systemic treatment: saline and 1 mg/kg amphetamine × local lateral habenular infusion into the habenula: ACSF, 25 μg/side, and 50 μg/side nicotine) revealed no significant main effect of systemic drug administration, local lateral habenular infusion, or an interaction between factors.
Experiment 4
Figure 4 shows the combined effects on PPI of nicotine infusions into the habenula and systemic dizocilpine administration. A 2 × 2 × 3 ANOVA test on PPI (systemic treatment: saline and 0.05 mg/kg dizocilpine × local lateral habenular infusion into the habenula: ACSF and 50 μg/side nicotine × prepulse intensity: 68, 71, and 77 dB) revealed significant main effects of systemic dizocilpine administration (F(1,9) = 34.09, p < 0.0005), prepulse intensity (F(2,18) = 24.24, p < 0.0001; χ 2(2) = 1.80, p > 0.4), and a significant interaction between dizocilpine and nicotine infusion into the habenula (F(1,9) = 11.29 p < 0.01). Overall PPI was lower after dizocilpine (42.92 %) than saline administration (63.31 %), and subsequent analyses showed that increasing prepulse intensities resulted in larger inhibition levels (43.55 %, 52.66 %, and 63.14 % for 68, 71, and 77 dB prepulses). With ACSF infusions, dizocilpine administration decreased PPI levels when compared to the saline condition (38.8 % and 67.9 %, p < 0.0005). This dizocilpine-induced decrease in PPI was not counteracted when nicotine was infused into the habenula. Nicotine infusion did not significantly affect overall PPI, nor did it interact with prepulse intensity. No other significant interactions were found.

Effects on PPI of nicotine (Nic) infusions into the habenula and systemic dizocilpine administration. Data represent mean values (± SEM). *Denotes significant differences in PPI of systemic treatment from saline condition, p < 0.0005
Startle responses were not affected by systemic drug treatment or LHb infusion (Table 1). A 2 × 2 ANOVA test on response amplitude (systemic treatment: saline and 0.05 mg/kg dizocilpine × local LHb infusion into the habenula: ACSF and 50 μg/side nicotine) revealed no significant main effect of systemic drug administration, nicotine infusion, or an interaction between factors.
Discussion
When tested against dopaminergic models of sensorimotor gating deficits, nicotine has been shown to dose-dependently reverse the deficits in PPI induced by dopamine agonist apomorphine (Suemaru et al. 2004). In agreement with these findings, nicotine infusion into the LHb in the present experiments did not affect overall PPI levels but significantly attenuated the PPI deficits induced by systemic administration of amphetamine. In Experiment 3, when animals were administered amphetamine, 50 μg/side nicotine infusions significantly increased PPI when compared to ACSF infusions (p < 0.05), whereas the effects of amphetamine on PPI was not significantly altered by 25 μg/side nicotine infusions (Experiment 2). These results suggest that the LHb may be a brain structure involved in the observed beneficial effects of systemic nicotine administration on sensorimotor gating deficits caused by dysregulation of midbrain dopamine systems.
The NAC is a brain region where several neurotransmitter systems converge (Koch 1999), and therefore a main structure in the regulation of PPI and the most likely substrate for the PPI deficits observed in systemic treatments with dopamine agonists (Swerdlow et al. 1992). One important source of dopaminergic input to the NAC is the VTA (Swanson 1982), a target structure of the LHb (Hikosaka et al. 2008). Using a “reward-based visual saccade task”, Matsumoto and Hikosaka (2007) reported that in unrewarded trials neural activity of LHb neurons increased and was followed by a decrease of activity of midbrain dopamine neurons. Similarly, Christoph et al. (1986) found that electrical stimulation of LHb neurons in anesthetized rats inhibited dopamine neurons in the VTA and substantia nigra compacta. Therefore, the activation of nicotinic receptors in the LHb after nicotine infusions in Experiments 2 and 3 could have resulted in the attenuation of the amphetamine-induced PPI deficits by inhibiting dopamine neurons in the VTA, decreasing dopamine levels in the NAC and increasing PPI.
Nicotine infusion into the LHb, despite being able to significantly attenuate the PPI deficit induced by amphetamine, could not restore PPI to control levels. It is possible that infusions of higher doses of nicotine than those used in Experiments 2 and 3 could result in a more complete restoration of PPI, as suggested by the increased enhancement of PPI observed with increasing doses of nicotine. Another possibility is that the complete restoration of apomorphine-induced PPI deficits after systemic administration of nicotine (Suemaru et al. 2004) is achieved by the activation of nicotinic receptors in brain regions other than the LHb, or by the interaction of nicotine with other neurotransmitter systems that affect PPI (such as glutamate; Toth et al. 1993). Future experiments will be needed in order to determine the neural mechanisms by which nicotine can fully counteract the deleterious effects of dopamine agonists on PPI.
In Experiment 1, mecamylamine infusions into the LHb did not affect overall PPI levels, in line with previous studies showing that mecamylamine administration does not produce significant changes in PPI (Curzon et al. 1994; Jones and Shannon 2000), nor did it interact with systemic dopaminergic manipulations. In Matsumoto and Hikosaka’s (2007) study, inhibition of habenula neurons did not trigger the excitation of midbrain dopamine neurons. Therefore, inhibition of LHb neurons by mecamylamine in Experiment 1 may have not altered dopamine levels in the NAC, accounting for the absence of an interaction between mecamylamine infusions and dopaminergic manipulations on PPI. The absence of significant effects of mecamylamine in Experiment 1 could also be due to a lack of tonic cholinergic activation, rendering blockade of nicotinic receptors in the LHb ineffective in the modulation of PPI.
Dizocilpine has been shown to strongly disrupt PPI (e.g., Mansbach and Geyer 1989), with different brain regions mediating this effect, including the amygdala, dorsal hippocampus and to a lesser degree, the MPFC (Bakshi and Geyer 1998). Systemic administration of nicotine has been shown to reverse apomorphine-induced PPI deficits but not PPI deficits caused by NMDA receptor antagonist phencyclidine (Suemaru et al. 2004). In line with these findings, the results of Experiment 4 show that dizocilpine effectively disrupted PPI, and that 50 μg/side nicotine infusions into the LHb could not reverse this disruption. Furthermore, since PPI deficits caused by NMDA receptor antagonists are not mediated by dopamine systems (Keith et al. 1991), the results of the present experiments provide additional support to the view that the LHb can attenuate sensorimotor gating deficits through the regulation of dopaminergic systems. It should be noted that the dizocilpine-induced PPI deficit in Experiment 4 was weaker than those induced by amphetamine and may have reduced the ability to detect potential effects of habenular nicotine infusions. However, there was a 29 % decrease in PPI after dizocilpine administration when compared to saline with ACSF infusions, a disruption greater to that observed in previous studies in which dizocilpine-induced PPI deficits were effectively counteracted by idazoxan (e.g., Larrauri and Levin 2012a, 20 % PPI decrease).
Previous studies have shown that PPI deficits induced by increased dopamine activity in the NAC are mediated by GABAergic projections from the NAC to the ventral pallidum (VP), which, in turn, modulates PPI though descending GABAergic projections to the PPT (Swerdlow et al. 1990c; Kodsi and Swerdlow 1997). Since the LHb exerts inhibitory control over serotonin neurons in the RN (Hikosaka et al. 2008), it is possible that the observed attenuation of amphetamine-induced PPI deficits in Experiments 2 and 3 may reflect reduced serotonin levels in the VP from RN neurons after nicotine infusions into the LHb. The results of Experiment 4 are consistent with this view, since PPI deficits induced by NMDA receptor antagonists are not mediated by the NAC-VP projections (Kretschmer and Koch 1998) and thus, nicotine receptors on the LHb would not be able to modulate dizocilpine-induced PPI deficits through the RN-VP pathway. Future studies will be needed in order to determine the potential involvement of serotonin transmission in the habenula-mediated attenuation of PPI deficits.
Female rats were used in the experiments in order to maintain consistency with previous studies analyzing the contribution of different neurotransmitter systems in the regulation of PPI (e.g., Larrauri and Levin 2012a). Female Sprague–Dawley rats exhibit similar startle responses and PPI levels to those of male animals (Swerdlow et al. 1993) but maintain a relatively more constant weight throughout adulthood (Bell and Zucker 1971), leading to more stable response amplitudes. PPI in Sprague–Dawley rats has been shown to be lower during proestrous when compared to estrous or diestrous (Koch 1998), but the phase of the estrous cycle becomes a random variable when a Latin-square counterbalanced design is used, minimizing potential confounding effects (Larrauri and Levin 2012b).
The results of these experiments show, in sum, that activation of nicotinic receptors in the LHb can attenuate PPI deficits induced by dopamine agonists. Nicotine has been reported to normalize sensory gating in schizophrenic patients and their relatives by improving the diminished gating of P50 evoked potentials to auditory stimuli (Adler et al. 1992, 1993), and the potential beneficial effects of nicotine in schizophrenic patients have been supported by studies using PPI models of sensorimotor gating deficits in schizophrenia (Suemaru et al. 2004). However, the exact role of nicotinic receptor systems in the modulation of sensorimotor gating remains unclear. The present results suggest that the habenula may play an important role in the regulation of dopamine neurons in the midbrain and contribute to the attenuation of deficits in sensorimotor gating caused by the hyperactivity of dopamine systems.
References
Adler LE, Hoffer LJ, Griffith J, Waldo MC, Freedman R (1992) Normalization by nicotine of deficient auditory sensory gating in the relatives of schizophrenics. Biol Psychiatry 32:607–616
Adler LE, Hoffer LD, Wiser A, Freedman R (1993) Normalization of auditory physiology by cigarette smoking in schizophrenic patients. Am J Psychiatry 150:1856–1861
Bakshi VP, Geyer MA (1998) Multiple limbic regions mediate the disruption of prepulse inhibition produced in rats by the noncompetitive NMDA antagonist dizocilpine. J Neurosci 18:8394–8401
Bell DD, Zucker I (1971) Sex differences in body weight and eating: organization and activation by gonadal hormones in the rat. Physiol Behav 7:27–34
Bubser M, Koch M (1994) Prepulse inhibition of the acoustic startle response of rats is reduced by 6-hydroxydopamine lesions of the medial prefrontal cortex. Psychopharmacology (Berl) 113:487–492
Carlson S, Willott JF (1996) The behavioral salience of tones as indicated by prepulse inhibition of the startle response: relationship to hearing loss and central neural plasticity in C57BL/6J mice. Hear Res 99:168–175
Christoph GR, Leonzio RJ, Wilcox KS (1986) Stimulation of the lateral habenula inhibits dopamine-containing neurons in the substantia nigra and ventral tegmental area of the rat. J Neurosci 6:613–619
Craft RM, Millholland RB (1998) Sex differences in cocaine- and nicotine-induced antinociception in the rat. Brain Res 809:137–140
Curzon P, Kim DJB, Decker MW (1994) Effect of nicotine, lobeline, and mecamylamine on sensory gating in the rat. Pharmacol Biochem Behav 49:877–882
Decker MW, Majchrzak MJ (1992) Effects of systemic and intracerebroventricular administration of mecamylamine, a nicotinic cholinergic antagonist, on spatial memory in rats. Psychopharmacology (Berl) 107:530–534
Decker MW, Curzon P, Brioni JD (1995) Influence of separate and combined septal and amygdala lesions on memory, acoustic startle, anxiety, and locomotor activity in rats. Neurobiol Learn Mem 64:156–168
Fendt M (1999) Enhancement of prepulse inhibition after blockade of GABA activity within the superior colliculus. Brain Res 833:81–85
Heldt SA, Ressler KJ (2006) Lesions of the habenula produce stress- and dopamine-dependent alterations in prepulse inhibition and locomotion. Brain Res 1073–4:229–239
Hikosaka O, Sesack SR, Lecourtier L, Shepard PD (2008) Habenula: crossroad between the basal ganglia and the limbic system. J Neurosci 28:11825–11829
Japha K, Koch M (1999) Picrotoxin in the medial prefrontal cortex impairs sensorimotor gating in rats: reversal by haloperidol. Psychopharmacology (Berl) 144:347–354
Jaskiw GE, Weinberger DR (1987) The prefrontal cortex-accumbens circuit: who’s in charge? Behav Brain Sci 10:217–218
Jaskiw GE, Weinberger DR, Crawley JN (1991) Microinjection of apomorphine into the prefrontal cortex of the rat reduces dopamine metabolite concentrations in microdialysate from the caudate nucleus. Biol Psychiatry 29:703–706
Jones CK, Shannon HE (2000) Muscarinic cholinergic modulation of prepulse inhibition of the acoustic startle reflex. J Pharmacol Exp Ther 294:1017–1023
Keith VA, Mansbach RS, Geyer MA (1991) Failure of haloperidol to block the effects of phencyclidine and dizocilpine on prepulse inhibition of startle. Biol Psychiatry 30:557–566
Koch M (1998) Sensorimotor gating changes across the estrous cycle in female rats. Physiol Behav 64:625–628
Koch M (1999) The neurobiology of startle. Prog Neurobiol 59:107–128
Kodsi MH, Swerdlow NR (1997) Regulation of prepulse inhibition by ventral pallidal projections. Brain Res Bull 43:219–228
Kretschmer BD, Koch M (1998) The ventral pallidum mediates disruption of prepulse inhibition of the acoustic startle response induced by dopamine agonists, but not by NMDA antagonists. Brain Res 798:204–210
Kusljic S, Copolov DL, van den Buuse M (2003) Differential role of serotonergic projections arising from the dorsal and median raphe nuclei in locomotor hyperactivity and prepulse inhibition. Neuropsychopharmacology 28:2138–2147
Kutlu MG, Burke D, Slade S, Hall BJ, Rose JE, Levin ED (2013) Role of insular cortex D1 and D2 dopamine receptors in nicotine self-administration in rats. Behav Brain Res 256:273–278
Larrauri JA, Levin ED (2012a) The α2-adrenergic antagonist idazoxan counteracts prepulse inhibition deficits caused by amphetamine or dizocilpine in rats. Psychopharmacology (Berl) 219:99–108
Larrauri JA, Levin ED (2012b) Differential effects of the antidepressant mirtazapine on amphetamine- and dizocilpine-induced PPI deficits. Pharmacol Biochem Behav 102:82–87
Lingenhöhl K, Friauf E (1994) Giant neurons in the rat reticular formation: a sensorimotor interface in the elementary acoustic startle circuit? J Neurosci 14:1176–1194
Louilot A, Le Moal M, Simon H (1989) Opposite influences of dopaminergic pathways to the prefrontal cortex or the septum on the dopaminergic transmission in the nucleus accumbens. An in vivo voltammetric study. Neuroscience 29:45–56
Lowry CA, Hale MW, Evans AK, Heerkens J, Staub DR, Gasser PJ, Shekhar A (2008) Serotonergic systems, anxiety, and affective disorder: focus on the dorsomedial part of the dorsal raphe nucleus. Ann NY Acad Sci 1148:86–94
Mansbach RS, Geyer MA (1989) Effects of phencyclidine and phencyclidine biologs on sensorimotor gating in the rat. Neuropsychopharmacology 2:299–308
Matsumoto M, Hikosaka O (2007) Lateral habenula as a source of negative reward signals in dopamine neurons. Nature 447:1111–1115
Rainbow TC, Schwartz RD, Parsons B, Kellar KJ (1984) Quantitative autoradiography of nicotinic [3H]acetylcholine binding sites in rat brain. Neurosci Lett 50:193–196
Sanders D, Simkiss D, Braddy D, Baccus S, Morton T, Cannady R, Weaver N, Rose JE, Levin ED (2010) Nicotinic receptors in the habenula: importance for memory. Neuroscience 166:386–390
Snedecor GW, Cochran WG (1967) Statistical methods. Ames, Iowa
Suemaru K, Yasuda K, Umeda K, Araki H, Choshi T, Hibino S, Gomita Y (2004) Nicotine blocks apomorphine-induced disruption of prepulse inhibition of the acoustic startle in rats: possible involvement of central nicotinic α7 receptors. Br J Pharmacol 142:843–850
Swanson LW (1982) The projections of the ventral tegmental area and adjacent regions: a combined fluorescent retrograde tracer and immunofluorescence study in the rat. Brain Res Bull 9:321–353
Swerdlow N, Braff D, Masten V, Geyer M (1990a) Schizophrenic-like sensorimotor gating abnormalities in rats following dopamine infusion into the nucleus accumbens. Psychopharmacology (Berl) 101:414–420
Swerdlow NR, Mansbach RS, Geyer MA, Pulvirenti L, Koob GF, Braff DL (1990b) Amphetamine disruption of prepulse inhibition of acoustic startle is reversed by depletion of mesolimbic dopamine. Psychopharmacology (Berl) 100:413–416
Swerdlow NR, Braff DL, Geyer MA (1990c) GABAergic projection from nucleus accumbens to ventral pallidum mediates dopamine-induced sensorimotor gating deficits of acoustic startle in rats. Brain Res 532:146–150
Swerdlow NR, Caine SB, Braff DL, Geyer MA (1992) The neural substrates of sensorimotor gating of the startle reflex: a review of recent findings and their implications. J Psychopharmacol 6:176–190
Swerdlow NR, Auerbach P, Monroe SM, Hartston H, Geyer MA, Braff DL (1993) Men are more inhibited than women by weak prepulses. Biol Psychiatry 34:253–260
Swerdlow NR, Braff DL, Geyer MA (2000) Animal models of deficient sensorimotor gating: what we know, what we think we know, and what we hope to know soon. Behav Pharmacol 11:185–204
Toth E, Vizi ES, Lajtha A (1993) Effect of nicotine on levels of extracellular amino acids in regions of the rat brain in vivo. Neuropharmacology 32:827–832
Yoshioka M, Matsumoto M, Togashi H, Saito H (1996) Effect of conditioned fear stress on dopamine release in the rat prefrontal cortex. Neurosci Lett 209:201–203
Zhang J, Engel JA, Hjorth S, Svensson L (1995) Changes in the acoustic startle response and prepulse inhibition of acoustic startle in rats after local injection of pertussis toxin into the ventral tegmental area. Psychopharmacology (Berl) 119:71–78
Acknowledgments
This research was sponsored by the Wallace Research Foundation. Supported by NIDA P50 grant DA027840.
Conflicts of interest
None.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Larrauri, J.A., Burke, D.A., Hall, B.J. et al. Role of nicotinic receptors in the lateral habenula in the attenuation of amphetamine-induced prepulse inhibition deficits of the acoustic startle response in rats. Psychopharmacology 232, 3009–3017 (2015). https://doi.org/10.1007/s00213-015-3940-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00213-015-3940-z