
Abstract
Rationale
Prepulse inhibition (PPI) is the reduction in startle response magnitude when intense stimuli are closely preceded by other weak stimuli. Animal models used to investigate sensorimotor gating deficits include both the stimulation of dopamine receptors (e.g., amphetamine or apomorphine) and the blockade of NMDA-glutamate receptors (e.g., dizocilpine or phencyclidine).
Objectives
We assessed the effects of idazoxan (an α2-adrenergic antagonist) on amphetamine- and dizocilpine-induced PPI disruptions in adult female Sprague–Dawley rats.
Methods
In experiment 1, rats were tested for PPI in a bimodal paradigm with an acoustic prepulse and a tactile startle stimulus. Interactions of amphetamine (1 mg/kg) and idazoxan (0.5, 1, and 2 mg/kg) were assessed, with all rats receiving all drug doses in a counterbalanced order. In experiment 2, dizocilpine (0.05 mg/kg) and idazoxan (0.5, 1, and 2 mg/kg) interactions were analyzed.
Results
Amphetamine (1 mg/kg) caused a significant reduction in PPI. Both the 1- and 2-mg/kg doses of idazoxan significantly counteracted this effect. Dizocilpine (.05 mg/kg) effectively inhibited PPI, and the 2-mg/kg idazoxan dose significantly counteracted this impairment.
Conclusions
These results suggest that the effectiveness of atypical antipsychotics such as clozapine in counteracting sensorimotor gating deficits reported in previous studies (e.g., Swerdlow and Geyer, Pharmacol Biochem Behav 44:741–744, 1993; Bakshi et al., J Pharmacol Exp Ther 271:787–794, 1994) may be related to their α2-antagonist effects, which may be a critical mechanism of the therapeutic effects of atypical antipsychotics in schizophrenia.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The startle response is the reflex-like motor reaction elicited by salient sensory stimuli. This response can be produced by acoustic, tactile, or visual stimuli (Landis et al. 1939) and is present in different species ranging from simple invertebrate organisms (Eaton 1984) to mammals including humans (Graham 1975; Koch 1999). The magnitude of the startle response can be modulated by internal factors (e.g., potentiated by fear or attenuated by pleasure; Brown et al. 1951; Schmid et al. 1995) as well as by external variables. For instance, preceding a startling stimulus (or pulse) closely in time by another stimulus (prepulse) can cause a reduction in the magnitude of the startle response when compared to that produced by the startling stimulus alone (Hoffman and Searle 1968), an effect known as prepulse inhibition (PPI). PPI is a neurobehavioral process in which the experience of an initial sensory stimulus attenuates the motoric startle response to a closely following sensory stimulus, a phenomenon that can be used as an operational measure of sensorimotor gating (Swerdlow et al. 2000). Deficits in PPI have been reported in individuals with psychiatric and neurobiological disorders such as schizophrenia (Braff et al. 1978), Tourette's syndrome (Castellanos et al. 1996), obsessive–compulsive disorder (Swerdlow et al. 1993b), and Huntington's disease (Swerdlow et al. 1995). Therefore, studying the neural mechanisms that regulate PPI may provide further information about the physiological bases of these disorders and help in the development of therapeutic treatments for these syndromes.
Hoffman and Ison (1980) put forward a model PPI circuit in which sensory stimuli trigger both fast excitatory and slow inhibitory signals on different pathways that converge to produce the overall startle response. Later studies showed that excitatory information in this “mediatory” PPI circuit reaches the motoneurons through the caudal pontine reticular nucleus (PNC) (Davis et al. 1982), whereas inhibitory signals triggered by stimuli of different sensory modalities converge in the superior colliculus (Meredith et al. 1992), activating the pedunculopontine tegmental nucleus (PPT) (Fendt et al. 1994), and in turn inhibiting the PNC through a cholinergic muscarinic PPT–PNC projection (Koch et al. 1993). In addition, other brain regions not included in the mediatory circuit can modulate PPI by regulating activity in the PPT, such as the ventral pallidum, nucleus accumbens (NAC), entorhinal cortex, medial prefrontal cortex (MPFC), hippocampus, and amygdala (Koch 1999). Animal models of schizophrenia in humans include pharmacological manipulations that target these “modulatory” brain areas in rats and produce deficits in PPI similar to those observed in patients with schizophrenia (Geyer et al. 2001).
Systemic administration of dopamine receptor agonists such as amphetamine or apomorphine reduces PPI, a disruption that can be reversed by the co-administration of the typical antipsychotic haloperidol (Mansbach et al. 1988). Furthermore, direct infusion of dopamine in the NAC in rats produces a dose-dependent decrease in PPI (Swerdlow et al. 1990) suggesting that sensorimotor gating deficits associated with dopamine receptor agonists are associated to overactive dopamine activity. Similarly, it has been shown that systemic administration of NMDA receptor antagonists such as dizocilpine or phencyclidine (PCP) reduces PPI in rats (Mansbach and Geyer 1989). However, these disruptions are not reversed by haloperidol (Keith et al. 1991) indicating that NMDA-receptor antagonist induced PPI disruptions are not mediated by the mesolimbic dopamine system. Bakshi and Geyer (1998) studied the effects of localized bilateral infusions of dizocilpine on PPI in different limbic regions, finding significant decreases in PPI after infusions in the amygdala or dorsal hippocampus, and a trend toward reduction in PPI after administration in the MPFC, but no changes after infusions in the NAC, ventral hippocampus, or dorsomedial thalamus. Therefore, reduction of PPI by pharmacological stimulation of dopamine receptors or blockade of NMDA glutamate receptors can be used to determine the contribution of other receptors in the reinstatement of normal sensorimotor gating properties.
Whereas typical antipsychotics (such as haloperidol) exert strong dopamine receptor blockage, atypical antipsychotics target a wider range of receptors. For instance, the atypical antipsychotic clozapine has complex antagonistic effects on dopamine D1–D4, histamine H1, serotonin 5-HT2A and 5-HT2C, norepinephrine α1 and α2, and acetylcholine muscarinic receptors (Coward 1992). By having a wider range of receptor blockage action, clozapine has been shown to effectively reverse both apomorphine- (Swerdlow and Geyer 1993) and dizocilpine-induced (Bakshi et al. 1994; Bortolato et al. 2005; Bubenikova et al. 2005; Caceda et al. 2005) PPI disruptions in rats. In order to develop improved antipsychotic treatments, it is important to study the effects of drugs that specifically target the same receptors affected by clozapine to determine their potential therapeutic or detrimental effects. In previous studies, we have shown that the H1-antagonist pyrilamine significantly attenuates the PPI impairments caused by either amphetamine (Larrauri and Levin 2010) or dizocilpine (Roegge et al. 2007). We also found that the addition of the 5-HT2A/2C antagonist ketanserin reduced the effectiveness of pyrilamine in counteracting amphetamine-induced PPI deficits (Larrauri and Levin 2010).
As mentioned, clozapine has, among others, antagonistic effects on α2-adrenergic receptors. Litman et al. (1996) showed that the addition of idazoxan (an α2-adrenergic antagonist) to a fluphenazine (a typical antipsychotic) treatment resulted in significant reductions in psychotic symptoms in patients with schizophrenia, when compared to the fluphenazine treatment alone. Furthermore, the reduction in psychosis obtained with the idazoxan and fluphenazine treatment was comparable to the one obtained with a clozapine treatment. Hertel et al. (1999) showed that the combination of idazoxan and raclopride (D2-dopamine antagonist) resulted in an increase of dopamine in the MPFC in rats, an effect similar to the one obtained with clozapine (Moghaddam and Bunney 1990). Overall, these results suggest that the superior antipsychotic effects of clozapine may be related to the α2 receptor blockade by this drug. Therefore, the goal of the present study was to examine the effects of the α2-adrenergic antagonist idazoxan on amphetamine-induced (“Experiment 1”) and dizocilpine-induced (“Experiment 2”) PPI disruptions. Based on the reported beneficial effects of idazoxan in the treatment of schizophrenia with typical antipsychotics (Litman et al. 1996), an improvement in sensorimotor gating (as indexed by PPI) after its disruption with amphetamine or dizocilpine is hypothesized.
Methods
Subjects
Adult female Sprague–Dawley rats (n = 12 for “Experiment 1” and n = 11 for “Experiment 2”) obtained from Taconic Farms (Germantown, NY, USA) were tested for auditory PPI of the tactile startle response. Animals were housed in groups of three per cage at an ambient temperature of 20°C on a 12:12-h reverse dark:light cycle (lights off at 0700 h) and had ad-libitum access to water and food. In “Experiment 1”, animals' body weights ranged between 242 and 325 g and were 93 days old at the start of test sessions. In “Experiment 2”, animals were 98 days old at the beginning of test sessions, and their body weights varied between 242 and 298 g. Rats were allowed to acclimate to the housing facilities for 2 weeks after arrival and were acclimated to the test room and experimental apparatus before testing sessions began. Before the beginning of test sessions, animals received eight handling sessions (daily), followed by five sessions of exposure to the testing situation (apparatus and stimuli) conducted 2 days apart. Testing started 2 days after the last exposure session. Animals were maintained according to NIH guidelines, and protocols were approved by the Duke University Animal Care and Use Committee.
Apparatus
Tactile startle responses and prepulse inhibition were measured using the SR-Lab Startle Response System manufactured by San Diego Instruments (SDI). Plexiglas cylinders (9 cm inside diameter), each with a piezoelectric accelerometer attached to its base, were used to confine the animals inside the sound-attenuating test chambers. All piezoelectric accelerometer responses were standardized using the SDI calibrator. Data from the accelerometers gauging the animals' responses were collected in 1-s time windows beginning at the onset of the startling stimuli and sampled at a 1-MHz frequency. The startle response amplitude was calculated as the difference between the peak response in the 200-ms time window after the stimulus onset and the pre-stimulus activity. Auditory stimuli were presented through speakers located in the test chambers above the restraining tubes, and tactile stimuli (air puffs) were delivered through copper tubes (4 mm inner diameter, 75 cm long) connected to a compressed air tank that entered each Plexiglas tube through a small opening on the top of the cylinder. Auditory stimuli in all chambers were calibrated using a digital sound level meter (Extech Instruments), and the air-puff pressure was controlled at 207 kPa (30 PSI, which produced an 82-dB noise signal) through a standard gas regulator connected between the main air pipe and the copper tubes. The background noise throughout test sessions was a 65-dB broad-band noise.
Startle and prepulse inhibition procedure
At the beginning of each test session, there was a 5-min acclimation period where animals were only exposed to the background 65-dB broad-band noise. Following this period, three blocks of trials were presented, in which trials consisted of either a startling tactile stimulus (50-ms air puff) alone (i.e., pulse-alone trial) or an acoustic prepulse stimulus (a broad-band noise of any of three possible intensities, 68, 71, or 77 dB) followed by the startling tactile stimulus (i.e., prepulse–pulse trial). In Block 1, 6 pulse-alone trials (tactile air-puff stimulus) were presented in order to habituate and stabilize the animals' startle responses. During Block 2, 48 trials were randomly presented, 12 pulse-alone and 36 prepulse–pulse trials (i.e., 12 prepulse–pulse trials for each prepulse intensity). Block 3 comprised an additional 5 pulse-alone trials. For all blocks, the intertrial interval varied randomly between 10 and 20 s. In prepulse–pulse trials, auditory prepulse stimuli had a 2-ms rise/fall time and a 20-ms duration, and the lead interval (time difference between the onsets of the prepulse and pulse stimuli) was 100 ms. Data from trials in Block 2 were used to determine percent PPI, which was calculated as the ratio of the difference in response amplitude in pulse-alone and prepulse–pulse trials to the startle response amplitude in pulse-alone trials, multiplied by 100. That is, PPI [%] = 100 × (pulse-alone startle response − prepulse–pulse startle response) / pulse-alone startle response. The entire test period (including the 10-min interval between drug administration and the beginning of the acclimation period) lasted approximately 34 min.
Drug administration
Drugs were administered in the form of cocktail solutions. Before the beginning of each study, all drug combinations (eight cocktails per experiment) were prepared by mixing the appropriate drug amounts in saline solutions. Ten minutes before the beginning of the acclimation period of each test session, drugs were injected subcutaneously in a 1-ml/kg volume of the corresponding drug combination to be analyzed. In “Experiment 1”, drug treatments consisted of combinations of amphetamine (amphetamine sulfate salt; 0 and 1 mg/kg as of salt weight) and idazoxan (idazoxan hydrochloride; 0, 0.5, 1, and 2 mg/kg as of salt weight). In “Experiment 2”, combinations of dizocilpine (dizocilpine hydrogen maleate; 0 and 0.05 mg/kg as of maleate weight) and idazoxan (idazoxan hydrochloride; 0, 0.5, 1, and 2 mg/kg as of salt weight) were administered. All drugs were procured from Sigma, St. Louis, Mo., USA, and drug combinations were prepared in 0.9% saline solution. Saline-alone injections (cocktails with 0 mg/kg doses of both drugs) were used as the control condition. In both experiments, the drug treatments were given using a repeated-measures counterbalanced (Latin-square) design, in order to (a) test startle responses randomly across the different phases of the estrous cycle, and (b) reduce the potential impact of drug carryover effects following repeated test sessions. In addition, test sessions were conducted two to three days apart to allow for complete drug washout between successive sessions.
Data analysis
Percent prepulse inhibition of the tactile startle response for the three acoustic prepulse intensities examined was determined for each drug combination. Repeated-measures analysis of variance (ANOVA) tests were used to assess the effects of the drugs and their interactions on PPI and on the startle response in pulse-alone trials. In “Experiment 1”, interactions between the dopamine agonist amphetamine and the α2-receptor antagonist idazoxan with regard to PPI and startle response amplitude were investigated. In “Experiment 2”, interactions between the NMDA-receptor antagonist dizocilpine and idazoxan were analyzed. The PPI analyses included the same factors than those employed for those of startle responses, with the addition of prepulse intensity. Significant interactions were followed up by tests of simple main effects comparing the impact of the individual drugs and combinations thereof using Fisher's LSD tests. Reduced degrees of freedom (Greenhouse–Geisser) were used when appropriate to offset violations of the sphericity assumption underlying repeated-measures ANOVA tests. Statistical analyses were performed using SuperANOVA (Abacus Concepts, Inc, Berkeley, CA, USA). A p value of 0.05 was used as the threshold for statistical significance.
Results
Experiment 1
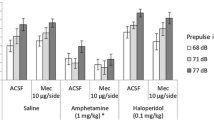
The effects of amphetamine and idazoxan on PPI for the three different prepulse intensities analyzed are shown in Fig. 1. A 2 × 4 × 3 ANOVA (amphetamine, 0 and 1 mg/kg × idazoxan, 0, 0.5, 1, and 2 mg/kg × prepulse intensity, 68, 71, and 77 dB) test on PPI levels revealed a significant main effect of prepulse intensity (F(2,22) = 30.24, p < 0.000005, ε = 0.9), a significant amphetamine × idazoxan interaction (F(3,33) = 3.16, p < 0.05, ε = 0.82), and a trend towards a significant amphetamine effect (F(1,11) = 3.71, p = 0.08), but no significant main effect of idazoxan (F(3,33) = 1.076, p > 0.37). Post hoc analyses indicated that 68-dB prepulses produced less overall inhibition (4.1 ± 3.1%, mean ± SEM) than 71-dB prepulses (18.5 ± 2.6%; p < 0.00005), which in turn caused less overall inhibition than 77-dB prepulses (25.1 ± 3.0%; p < 0.05). Amphetamine-only administration significantly reduced mean PPI (−4.1 ± 6.2%; p < 0.005) when compared to the control saline condition (24.7 ± 5.1%). This disruption in inhibition was effectively counteracted by the 1 mg/kg (14.7 ± 7.5%; p < 0.05) and 2 mg/kg (23.2 ± 4.6%; p < 0.005) idazoxan doses, which increased mean PPI values to levels that were not statistically significantly different from those in the control saline condition (p > 0.26 and p > 0.87, respectively). In addition, compared to the control condition, PPI was not significantly reduced after administration of idazoxan (0.5, 1, and 2 mg/kg) without amphetamine (p > 0.86, p > 0.21, and p > 0.51, respectively). No significant interactions were found between prepulse intensity and amphetamine (p > 0.32) or idazoxan (p > 0.15).

Idazoxan interactions with amphetamine-induced PPI disruption by prepulse intensity. The control saline condition corresponds to the 0-mg/kg amphetamine–0-mg/kg idazoxan treatment. Amphetamine-only administration reduced PPI levels when compared to the control saline condition (p < 0.005), and both 1 and 2 mg/kg doses of idazoxan effectively counteracted that disruption (*p < 0.05 and **p < 0.005, respectively). These effects resulted in a significant amphetamine × idazoxan interaction (p < 0.05). Increasing prepulse intensity levels enhanced PPI (p < 0.000005), and no significant interactions were found between prepulse intensity and amphetamine and/or idazoxan. Data represent mean values (±SEM)
Figure 2 presents the effects of amphetamine and idazoxan on the magnitude of the tactile startle response in pulse-alone trials. A 2 × 4 ANOVA (amphetamine, 0 and 1 mg/kg × idazoxan, 0, 0.5, 1, and 2 mg/kg) on the startle amplitude yielded significant main effects of amphetamine (F(1,11) = 31.52, p < 0.0002) and idazoxan (F(3,33) = 9.32, p < 0.002, ε = 0.65), indicating that both drugs enhanced overall responses. In addition, a significant interaction between amphetamine and idazoxan (F(3,33) = 4.98, p < 0.015, ε = 0.75) on the startle amplitude was found. Administration of 0.5 mg/kg idazoxan without amphetamine caused a significant increase in startle (377.9 ± 69.9) when compared to the control saline condition (258.3 ± 40.8; p < 0.05), whereas co-administration of amphetamine and 0.5, 1, or 2 mg/kg idazoxan resulted in larger startle amplitudes (524.0 ± 51.7; 499.4 ± 64.4; 461.9 ± 63.4; respectively) when compared to the amphetamine-only condition (214.2 ± 29.9; ps < 0.00005). For the conditions in which co-treatment of amphetamine and idazoxan resulted in increased levels of PPI (when compared to the amphetamine-only condition; i.e., amphetamine with 1 and 2 mg/kg idazoxan), correlation tests revealed no significant relationship between those changes in PPI and the observed increases in the amplitude of the startle response (t(10) = 0.14, p > 0.44 and t(10) = 1.09, p > 0.15, respectively). Furthermore, median split analyses including the effects on startle as a factor in the ANOVA tests on PPI yielded significant amphetamine × idazoxan interactions for the 1 and 2 mg/kg doses (F(1,10) = 4.9, p < 0.05 and F(1,10) = 5.1, p < 0.05, respectively) but no significant startle effect × amphetamine × idazoxan interactions (F(1,10) = 0.03, p > 0.85 and F(1,10) = 0.078, p > 0.78), suggesting that the drugs' effects on the startle magnitude were distinct from those observed on PPI.

Effects of idazoxan and amphetamine interactions on the tactile startle response (mean ± SEM). The control saline condition corresponds to the 0-mg/kg amphetamine–0-mg/kg idazoxan treatment. Both amphetamine and idazoxan significantly affected the magnitude of the startle response (p < 0.0002 and p < 0.002, respectively). Compared to the control saline condition, 0.5 mg/kg of idazoxan caused an increase in response amplitude (*p < 0.05). Amphetamine co-administration with 0.5, 1, and 2 mg/kg doses of idazoxan enhanced the startle response when compared to the amphetamine-only condition (+ p < 0.00005). These effects resulted in a significant amphetamine × idazoxan interaction (p < 0.015)
Experiment 2
The effects of dizocilpine and idazoxan on PPI for the three different prepulse intensities examined are shown in Fig. 3. A 2 × 4 × 3 ANOVA (dizocilpine, 0 and 0.05 mg/kg × idazoxan, 0, 0.5, 1, and 2 mg/kg × prepulse intensity, 68, 71, and 77 dB) test on PPI levels revealed significant main effects of dizocilpine (F(1,10) = 8.81, p < 0.015) and prepulse intensity (F(2,20) = 36.76, p < 0.000001, ε = 0.87), and significant dizocilpine × idazoxan (F(3,30) = 3.26, p < 0.05, ε = 0.82) and dizocilpine × prepulse intensity (F(2,20) = 8.21, p < 0.005, ε = 0.94) interactions. No significant main effect of idazoxan (F < 1) or dizocilpine × idazoxan × prepulse intensity interaction (F < 1) on PPI was found. Post hoc analyses indicated that 68-dB prepulses caused less overall inhibition (13.2 ± 2.8%) than 71-dB prepulses (23.3 ± 2.6%; p < 0.0015), which in turn produced less overall inhibition than 77-dB prepulses (41.2 ± 2.3%; p < 0.0001); and that dizocilpine significantly reduced overall PPI for 68-dB (p < 0.000001) and 71-dB (p < 0.00025) prepulses, but not for 77-dB prepulses (p > 0.2). Dizocilpine-only administration significantly reduced mean PPI (13.9 ± 4.8%) when compared to the control saline condition (33.9 ± 5.9%; p < 0.01). This disruption was effectively counteracted by the 2 mg/kg idazoxan dose (32.5 ± 3.7%; p < 0.015), which increased mean PPI to levels that were not statistically significantly different from those in the control saline condition (p > 0.83). When compared to the control condition, administration of idazoxan (0.5, 1 and 2 mg/kg) without dizocilpine did not cause a significant decrease in PPI (p > 0.71, p > 0.60, p > 0.55). No significant interaction between prepulse intensity and idazoxan (F < 1) was found.

Idazoxan interactions with dizocilpine-induced PPI disruption by prepulse intensity (mean ± SEM). The control saline condition corresponds to the 0-mg/kg dizocilpine–0-mg/kg idazoxan treatment. Dizocilpine significantly reduced PPI levels (p < 0.015). Dizocilpine-only administration reduced PPI levels when compared to the control saline condition (p < 0.01), and the 2-mg/kg dose of idazoxan effectively counteracted that disruption (# p < 0.015). These effects resulted in a significant dizocilpine × idazoxan interaction (p < 0.05). Increasing prepulse intensity levels enhanced PPI (p < 0.000001), and a significant dizocilpine × prepulse interaction (p < 0.005) revealed that dizocilpine disrupted inhibition more effectively for prepulses of lower intensity
Figure 4 presents the effects of dizocilpine and idazoxan on the magnitude of the tactile startle response in pulse-alone trials. A 2 × 4 ANOVA (dizocilpine, 0 and .05 mg/kg × idazoxan, 0, 0.5, 1, and 2 mg/kg) on the startle amplitude revealed a significant main effect of dizocilpine (F(1,10) = 15.89, p < 0.005). No significant main effect of idazoxan (F(3,30) = 1.7, p > 0.2, ε = 0.72) or an interaction between the drugs (F(3,30) = 2.13, p > 0.14, ε = 0.68) was found. Whereas co-administration of dizocilpine with 2 mg/kg idazoxan resulted in increased PPI levels (when compared to the dizocilpine-only condition), correlation tests revealed no significant relationship between the increase in PPI and the changes in the amplitude of the startle response in this condition (t(9) = 0.26, p > 0.39). In addition, median split analyses including the effects on startle as a factor in the ANOVA test on PPI yielded a significant dizocilpine × idazoxan interaction for the 2 mg/kg dose (F(1,9) = 8.5, p < 0.05), but no significant startle effect × dizocilpine × idazoxan interaction (F(1,9) = 0.076, p > 0.78), suggesting that the drugs' effect on the startle magnitude was distinct from that observed on PPI.

Effects of idazoxan and dizocilpine interactions on the tactile startle response (mean ± SEM). Dizocilpine significantly increased the magnitude of the startle response (p < 0.005), but no significant main effect of idazoxan (p > 0.2) or an interaction between these drugs (p > 0.14) on the tactile startle response was found
Discussion
In agreement with previous findings using unimodal acoustic prepulse and acoustic startle stimuli (e.g., Mansbach et al. 1988; Sills 1999; Swerdlow et al. 2005), amphetamine-only administration in “Experiment 1” caused a significant decrease in acoustic PPI of the tactile startle response when compared to the control saline condition but had no effect on the magnitude of startle on pulse-alone trials (Sills 1999). Idazoxan administration did not have a significant effect on acoustic PPI by itself, but effectively counteracted the amphetamine-induced PPI disruption in a dose-dependent manner. Both the 1 and 2 mg/kg idazoxan doses significantly counteracted the amphetamine-induced PPI disruption, whereas the 0.5 mg/kg idazoxan dose did not significantly alter the amphetamine-induced PPI reduction. Only the lowest dose (0.5 mg/kg) of idazoxan tested in “Experiment 1” caused a significant increase in startle response in pulse-alone trials when compared to the control saline condition, whereas response amplitude did not significantly change relative to the control saline condition with the 1 and 2 mg/kg idazoxan doses. In addition, co-administration of amphetamine with all doses of idazoxan resulted in an increase in startle response compared to the amphetamine-only condition. Even when significant amphetamine × idazoxan interactions on PPI and startle magnitude were found on “Experiment 1”, the opposing effects of idazoxan on the amphetamine-induced PPI disruption by idazoxan do not seem to result as a consequence of the observed increase in startle response. In fact, co-administration of amphetamine with the lowest idazoxan dose (0.5 mg/kg) caused the largest increase in the startle response magnitude (see Fig. 2), but the amphetamine-induced PPI impairment was not counteracted in this condition (p > 0.15). Furthermore, correlation tests between increases in PPI and changes in the amplitude of the startle response for the cases in which co-treatment of amphetamine and idazoxan (1 and 2 mg/kg) resulted in increased PPI levels (with respect to the amphetamine-only condition) yielded no significant relationships between these factors (p > 0.44 and p > 0.15, respectively), indicating that idazoxan counteracts the amphetamine-induced sensorimotor gating deficit independently of its effects on the startle response magnitude.
A possible mechanism of action for the observed reversal of the PPI disruption by idazoxan may be through a change in dopaminergic transmission due to an interaction between α2 receptors and dopaminergic systems (Litman et al. 1996). Grenhoff et al. (1993) showed that idazoxan increased neural firing in the locus coeruleus (LC), modulating mesolimbic and mesocortical dopaminergic activity. Since reduced dopamine levels in the MPFC result in lower PPI values (Bubser and Koch 1994) it is possible that idazoxan counteracts the amphetamine-induced PPI disruption by increasing the levels of cortical dopamine (Hertel et al. 1999). In addition, marked increases in dopamine output in the MPFC are also caused by the antipsychotic clozapine (Moghaddam and Bunney 1990), which also effectively reverses PPI deficits (Bakshi et al. 1994; Swerdlow and Geyer 1993).
Results from “Experiment 2” showed that, in accord with previous reports, dizocilpine administration caused a significant decrease in PPI and increased startle responses (Mansbach and Geyer 1989; Roegge et al. 2007). A significant prepulse intensity × dizocilpine interaction revealed that the disruptive effects of dizocilpine on PPI were more marked for lower prepulse intensities. Idazoxan administration by itself did not significantly affect PPI. However the highest idazoxan dose tested (2 mg/kg) significantly attenuated the PPI impairment caused by dizocilpine. Neither of the lower idazoxan doses (05 or 1 mg/kg) significantly altered the dizocilpine-induced PPI impairment. In addition, a correlation test between increases in PPI and changes in the amplitude of the startle response for the dizocilpine with 2 mg/kg idazoxan condition (for which PPI levels increased with respect to the dizocilpine-only condition) yielded no significant relationships between these factors (p > 0.39), indicating that the counteracting effects of idazoxan on the dizocilpine-induced PPI deficit were independent of those observed in the startle response amplitude.
Previous studies have provided evidence showing the involvement of the MPFC in the dizocilpine-induced PPI disruption. Bakshi and Geyer (1998) found a trend toward a decrease in PPI after local infusion of dizocilpine in the MPFC, and Schwabe and Koch (2004) reported that dizocilpine failed to disrupt PPI in rats with ibotenic acid lesions in the MPFC. Idazoxan can increase the levels of cortical dopamine (Hertel et al. 1999), which may reduce dopamine release in the NAC by enhancing inhibition of the MPFC glutamatergic neurons that project to the VTA (Karreman and Moghaddam 1996), thus decreasing dopamine levels in the NAC (Koch 1999) and counteracting the dizocilpine-induced PPI disruption.
Studies analyzing the effects of α2-receptor antagonists on PPI disruptions induced by NMDA-receptor antagonists have yielded mixed results. For instance, Bakshi and Geyer (1997) showed that the α1-receptor antagonist prazosin, but not the α2-receptor antagonist RX821002, was able to reverse PCP-induced PPI deficits. In contrast, Palsson et al. (2008) showed that agmatine (a potent α2-adrenergic and imidazoline receptor antagonist) effectively reverses PCP disruptions of PPI. In addition, Ballmaier et al. (2001) analyzed the effects of a combination of idazoxan and the D2/D3 receptor antagonist raclopride on PCP-induced PPI deficits and found no reversal of the PPI disruption with those drugs. However, only one dose of idazoxan was used in their experiment (1.5 mg/kg), and the results of “Experiment 2” suggest that there may be a threshold dose for which the opposing effects of idazoxan on the dizocilpine-induced PPI deficit can be detected (i.e., 2 mg/kg). Therefore, that the lack of effect of idazoxan in reversing PPI deficits caused by NMDA-receptor antagonists reported by Ballmaier et al. (2001) might be attributed to the lower idazoxan dose used in their study. Finally, and in agreement with the results of “Experiment 2”, Sallinen et al. (2007) showed that JP-1302 (a selective α2C-adrenoceptor antagonist) effectively antagonized PCP-induced PPI deficits in rats.
In addition to its ability to restore amphetamine or dizocilpine-induced PPI deficits, idazoxan (as well as other α2-adrenergic antagonists) can reverse haloperidol-induced catalepsy effects in rats (Invernizzi et al. 2003; Kleven et al. 2005) and modulate the effects of amphetamine on locomotion and stereotypies in mice (Luttinger and Durivage 1986). These results seem to further validate the prospective use of α2-adrenergic antagonists for the treatment of some psychiatric conditions. Clozapine is a complex drug with antagonistic effects on several different neurotransmitter receptors, including histamine, serotonin, dopamine, norepinepherine, and muscarinic (Coward 1992), and therefore the study of the effects of these different neurotransmitters on animals models of schizophrenia (such as amphetamine- and dizocilpine-induced PPI deficits; Geyer et al. 2001) might help to elucidate their individual contributions for therapeutic treatments. The results of “Experiments 1” and “2” showing that idazoxan is able to restore both amphetamine- and dizocilpine-induced PPI deficits suggest that the previously reported effectiveness of clozapine in counteracting both apomorphine- (Swerdlow and Geyer 1993) and dizocilpine-induced PPI deficits (Bubenikova et al. 2005; Levin et al. 2007) may be related to clozapine's α2-receptor blockage properties. However, previous studies have also shown that histamine H1-receptor blockage can counteract dizocilpine- (Roegge et al. 2007) and amphetamine-induced PPI deficits (Larrauri and Levin 2010). Thus, it is possible that complex interactions among the different receptors bound by clozapine are responsible for its superior antipsychotic effects, and further studies will be needed to clarify them.
Besides a strong α2-adrenoceptor antagonist affinity (70–80% specific binding; Mallard et al. 1992), idazoxan has also weaker imidazoline-receptor antagonist properties. Thus, it is possible that the restoring effects of idazoxan observed in “Experiments 1” and “2” may be related to its antagonist action on imidazoline receptors. Though no studies have analyzed the specific effects of these receptors on PPI, the stronger specific α2-receptor antagonist affinity of idazoxan, as well as experimental results showing that specific α2C-antagonists can restore PCP-induced PPI deficits (Sallinen et al. 2007), suggest that the observed restoring effects of idazoxan on amphetamine- and dizocilpine-induced PPI deficits may related to the effects of idazoxan on α2-receptors.
A few methodological issues in this study deserve additional consideration. A cross-modal PPI paradigm was used because it allows the determination of the generality of neurotransmitter system involvement in sensorimotor plasticity. Changes in startle responses to auditory stimuli presented after auditory prepulses may arise as a consequence of repeated stimulation of the same sensory neural pathways. The cross-modal approach avoids this particular problem (Levin et al. 2007). The effects of the different pharmacological manipulations on PPI were studied on female rats because (a) women comprise a large percentage of the population with schizophrenia (42%; McGrath 2006), and (b) female rats have an advantage over males for long-term chronic studies inasmuch as their adult body weight stays relatively constant throughout adulthood whereas males continually increases (Bell and Zucker 1971). Also, sexual dimorphism in pharmacokinetics has been reported in rats with respect to amphetamine (female rats show increased locomotor activity and focal stereotypies; Milesi-Halle et al. 2007) and dizocilpine (longer-lasting behavioral changes in female animals; Honack and Loscher 1993). Studies comparing responses of male and female Sprague–Dawley rats have not found gender differences in PPI or startle amplitude (Swerdlow et al. 1993a), and similar effects of clozapine in reversing dizocilpine-induced deficits in PPI have been found in both male and female Sprague–Dawley rats (Bakshi et al. 1994; Levin et al. 2007). Even when female Sprague–Dawley rats show a decrease in PPI during proestrous when compared to estrous or diestrous (Koch 1998), the phase of the cycle becomes a random variable when using a counterbalanced design. The results of Koch (1998) also show that Sprague–Dawley males and females (diestrous or estrous) do not differ in terms of PPI. Concerning the carryover effects inherent of a repeated-measures design, the use of a counterbalanced design protects against potential carryover effects of the drugs. In addition, test sessions were conducted 2 to 3 days apart to allow for the drug effects to disappear between sessions.
In sum, our results show that the α2-adrenergic antagonist idazoxan can effectively attenuate the amphetamine- or dizocilpine-induced PPI disruption in rats. Since the antipsychotic drug clozapine also has antagonistic effects at this receptor type and has previously been shown to counteract both apomorphine- (Swerdlow and Geyer 1993) and dizocilpine-induced PPI deficits (Bubenikova et al. 2005; Levin et al. 2007), the results of our experiments suggest that the α2 antagonistic effects of clozapine may contribute to its efficacy in reversing PPI deficits. By individually analyzing the different systems that affect PPI, a better comprehension of the effects of atypical antipsychotics might be attained, ultimately leading to an improved understanding of sensorimotor gating deficits in neuropsychiatric disorders, such as schizophrenia. Sensory over-responsiveness is observed in individuals with disorders that also entail PPI deficits (such as autism; Perry et al. 2007, or Asperger's syndrome; McAlonan et al. 2002), and can as well be seen without concomitant psychotic symptoms, such as the case with sensory processing disorder (Miller et al. 2009). In this group for which the full activity of a complex antipsychotic drug such as clozapine is not appropriate for treatment, identification of a more selective compound which effectively improves deficient sensory processing without undue side effects is essential.
References
Bakshi VP, Geyer MA (1997) Phencyclidine-induced deficits in prepulse inhibition of startle are blocked by prazosin, an alpha-1 noradrenergic antagonist. J Pharmacol Exp Ther 283:666–674
Bakshi VP, Geyer MA (1998) Multiple limbic regions mediate the disruption of prepulse inhibition produced in rats by the noncompetitive NMDA antagonist dizocilpine. J Neurosci 18:8394–8401
Bakshi VP, Swerdlow NR, Geyer MA (1994) Clozapine antagonizes phencyclidine-induced deficits in sensorimotor gating of the startle response. J Pharmacol Exp Ther 271:787–794
Ballmaier M, Zoli M, Mazzoncini R, Gennarelli M, Spano F (2001) Combined alpha 2-adrenergic/D2 dopamine receptor blockade fails to reproduce the ability of clozapine to reverse phencyclidine-induced deficits in prepulse inhibition of startle. Psychopharmacol (Berl) 159:105–110
Bell DD, Zucker I (1971) Sex differences in body weight and eating: organization and activation by gonadal hormones in the rat. Physiol Behav 7:27–34
Bortolato M, Aru GN, Fa M, Frau R, Orru M, Salis P, Casti A, Luckey GC, Mereu G, Gessa GL (2005) Activation of D1, but not D2 receptors potentiates dizocilpine-mediated disruption of prepulse inhibition of the startle. Neuropsychopharmacology 30:561–574
Braff D, Stone C, Callaway E, Geyer M, Glick I, Bali L (1978) Prestimulus effects on human startle reflex in normals and schizophrenics. Psychophysiology 15:339–343
Brown JS, Kalish HI, Farber IE (1951) Conditioned fear as revealed by magnitude of startle response to an auditory stimulus. J Exp Psychol 41:317–328
Bubenikova V, Votava M, Horacek J, Palenicek T, Dockery C (2005) The effect of zotepine, risperidone, clozapine and olanzapine on MK-801-disrupted sensorimotor gating. Pharmacol Biochem Behav 80:591–596
Bubser M, Koch M (1994) Prepulse inhibition of the acoustic startle response of rats is reduced by 6-hydroxydopamine lesions of the medial prefrontal cortex. Psychopharmacol (Berl) 113:487–492
Caceda R, Kinkead B, Owens MJ, Nemeroff CB (2005) Virally mediated increased neurotensin 1 receptor in the nucleus accumbens decreases behavioral effects of mesolimbic system activation. J Neurosci 25:11748–11756
Castellanos FX, Fine EJ, Kaysen D, Marsh WL, Rapoport JL, Hallett M (1996) Sensorimotor gating in boys with Tourette's syndrome and ADHD: preliminary results. Biol Psychiatry 39:33–41
Coward DM (1992) General pharmacology of clozapine. Br J Psychiatry Suppl (17):5–11
Davis M, Gendelman DS, Tischler MD, Gendelman PM (1982) A primary acoustic startle circuit: lesion and stimulation studies. J Neurosci 2:791–805
Eaton RC (1984) Neural mechanisms of startle behavior. Plenum, New York
Fendt M, Koch M, Schnitzler HU (1994) Sensorimotor gating deficit after lesions of the superior colliculus. Neuroreport 5:1725–1728
Geyer MA, Krebs-Thomson K, Braff DL, Swerdlow NR (2001) Pharmacological studies of prepulse inhibition models of sensorimotor gating deficits in schizophrenia: a decade in review. Psychopharmacol (Berl) 156:117–154
Graham FK (1975) Presidential Address, 1974. The more or less startling effects of weak prestimulation. Psychophysiology 12:238–248
Grenhoff J, Nisell M, Ferre S, Aston-Jones G, Svensson TH (1993) Noradrenergic modulation of midbrain dopamine cell firing elicited by stimulation of the locus coeruleus in the rat. J Neural Transm Gen Sect 93:11–25
Hertel P, Fagerquist MV, Svensson TH (1999) Enhanced cortical dopamine output and antipsychotic-like effects of raclopride by alpha2 adrenoceptor blockade. Science 286:105–107
Hoffman HS, Ison JR (1980) Reflex modification in the domain of startle: I. Some empirical findings and their implications for how the nervous system processes sensory input. Psychol Rev 87:175–189
Hoffman HS, Searle JL (1968) Acoustic and temporal factors in the evocation of startle. J Acoust Soc Am 43:269–282
Honack D, Loscher W (1993) Sex differences in NMDA receptor mediated responses in rats. Brain Res 620:167–170
Invernizzi RW, Garavaglia C, Samanin R (2003) The alpha 2-adrenoceptor antagonist idazoxan reverses catalepsy induced by haloperidol in rats independent of striatal dopamine release: role of serotonergic mechanisms. Neuropsychopharmacology 28:872–879
Karreman M, Moghaddam B (1996) The prefrontal cortex regulates the basal release of dopamine in the limbic striatum: an effect mediated by ventral tegmental area. J Neurochem 66:589–598
Keith VA, Mansbach RS, Geyer MA (1991) Failure of haloperidol to block the effects of phencyclidine and dizocilpine on prepulse inhibition of startle. Biol Psychiatry 30:557–566
Kleven MS, Barret-Grevoz C, Slot LB, Newman-Tancredi A (2005) Novel antipsychotic agents with 5-HT(1A) agonist properties: role of 5-HT(1A) receptor activation in attenuation of catalepsy induction in rats. Neuropharmacology 49:135–143
Koch M (1998) Sensorimotor gating changes across the estrous cycle in female rats. Physiol Behav 64:625–628
Koch M (1999) The neurobiology of startle. Prog Neurobiol 59:107–128
Koch M, Kungel M, Herbert H (1993) Cholinergic neurons in the pedunculopontine tegmental nucleus are involved in the mediation of prepulse inhibition of the acoustic startle response in the rat. Exp Brain Res 97:71–82
Landis C, Hunt WA, Strauss H (1939) The startle pattern. Farrar & Rinehart, New York
Larrauri JA, Levin ED (2010) PPI deficit induced by amphetamine is attenuated by the histamine H1 antagonist pyrilamine, but is exacerbated by the serotonin 5HT2 antagonist ketanserin. Psychopharmacology 212:551–558
Levin ED, Caldwell DP, Perraut C (2007) Clozapine treatment reverses dizocilpine-induced deficits of pre-pulse inhibition of tactile startle response. Pharmacol Biochem Behav 86:597–605
Litman RE, Su TP, Potter WZ, Hong WW, Pickar D (1996) Idazoxan and response to typical neuroleptics in treatment-resistant schizophrenia. Comparison with the atypical neuroleptic, clozapine. Br J Psychiatry 168:571–579
Luttinger D, Durivage ME (1986) Alpha 2-adrenergic antagonists effect on amphetamine-induced behaviors. Pharmacol Biochem Behav 25:155–160
Mallard NJ, Hudson AL, Nutt DJ (1992) Characterization and autoradiographical localization of non-adrenoceptor idazoxan binding sites in the rat brain. Br J Pharmacol 106:1019–1027
Mansbach RS, Geyer MA (1989) Effects of phencyclidine and phencyclidine biologs on sensorimotor gating in the rat. Neuropsychopharmacology 2:299–308
Mansbach RS, Geyer MA, Braff DL (1988) Dopaminergic stimulation disrupts sensorimotor gating in the rat. Psychopharmacol (Berl) 94:507–514
McAlonan GM, Daly E, Kumari V, Critchley HD, van Amelsvoort T, Suckling J, Simmons A, Sigmundsson T, Greenwood K, Russell A, Schmitz N, Happe F, Howlin P, Murphy DG (2002) Brain anatomy and sensorimotor gating in Asperger's syndrome. Brain 125:1594–1606
McGrath JJ (2006) Variations in the incidence of schizophrenia: data versus dogma. Schizophr Bull 32:195–197
Meredith MA, Wallace MT, Stein BE (1992) Visual, auditory and somatosensory convergence in output neurons of the cat superior colliculus: multisensory properties of the tecto-reticulo-spinal projection. Exp Brain Res 88:181–186
Milesi-Halle A, McMillan DE, Laurenzana EM, Byrnes-Blake KA, Owens SM (2007) Sex differences in (+)-amphetamine- and (+)-methamphetamine-induced behavioral response in male and female Sprague–Dawley rats. Pharmacol Biochem Behav 86:140–149
Miller LJ, Nielsen DM, Schoen SA, Brett-Green BA (2009) Perspectives on sensory processing disorder: a call for translational research. Front Integr Neurosci 3:1–12
Moghaddam B, Bunney BS (1990) Acute effects of typical and atypical antipsychotic drugs on the release of dopamine from prefrontal cortex, nucleus accumbens, and striatum of the rat: an in vivo microdialysis study. J Neurochem 54:1755–1760
Palsson E, Fejgin K, Wass C, Klamer D (2008) Agmatine attenuates the disruptive effects of phencyclidine on prepulse inhibition. Eur J Pharmacol 590:212–216
Perry W, Minassian A, Lopez B, Maron L, Lincoln A (2007) Sensorimotor gating deficits in adults with autism. Biol Psychiatry 61:482–486
Roegge CS, Perraut C, Hao X, Levin ED (2007) Histamine H1 receptor involvement in prepulse inhibition and memory function: relevance for the antipsychotic actions of clozapine. Pharmacol Biochem Behav 86:686–692
Sallinen J, Hoglund I, Engstrom M, Lehtimaki J, Virtanen R, Sirvio J, Wurster S, Savola JM, Haapalinna A (2007) Pharmacological characterization and CNS effects of a novel highly selective alpha2C-adrenoceptor antagonist JP-1302. Br J Pharmacol 150:391–402
Schmid A, Koch M, Schnitzler HU (1995) Conditioned pleasure attenuates the startle response in rats. Neurobiol Learn Mem 64:1–3
Schwabe K, Koch M (2004) Role of the medial prefrontal cortex in N-methyl-d-aspartate receptor antagonist induced sensorimotor gating deficit in rats. Neurosci Lett 355:5–8
Sills TL (1999) Amphetamine dose dependently disrupts prepulse inhibition of the acoustic startle response in rats within a narrow time window. Brain Res Bull 48:445–448
Swerdlow NR, Geyer MA (1993) Clozapine and haloperidol in an animal model of sensorimotor gating deficits in schizophrenia. Pharmacol Biochem Behav 44:741–744
Swerdlow NR, Braff DL, Masten VL, Geyer MA (1990) Schizophrenic-like sensorimotor gating abnormalities in rats following dopamine infusion into the nucleus accumbens. Psychopharmacol (Berl) 101:414–420
Swerdlow NR, Auerbach P, Monroe SM, Hartston H, Geyer MA, Braff DL (1993a) Men are more inhibited than women by weak prepulses. Biol Psychiatry 34:253–260
Swerdlow NR, Benbow CH, Zisook S, Geyer MA, Braff DL (1993b) A preliminary assessment of sensorimotor gating in patients with obsessive compulsive disorder. Biol Psychiatry 33:298–301
Swerdlow NR, Paulsen J, Braff DL, Butters N, Geyer MA, Swenson MR (1995) Impaired prepulse inhibition of acoustic and tactile startle response in patients with Huntington's disease. J Neurol Neurosurg Psychiatry 58:192–200
Swerdlow NR, Braff DL, Geyer MA (2000) Animal models of deficient sensorimotor gating: what we know, what we think we know, and what we hope to know soon. Behav Pharmacol 11:185–204
Swerdlow NR, Kuczenski R, Goins JC, Crain SK, Ma LT, Bongiovanni MJ, Shoemaker JM (2005) Neurochemical analysis of rat strain differences in the startle gating-disruptive effects of dopamine agonists. Pharmacol Biochem Behav 80:203–211
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Larrauri, J.A., Levin, E.D. The α2-adrenergic antagonist idazoxan counteracts prepulse inhibition deficits caused by amphetamine or dizocilpine in rats. Psychopharmacology 219, 99–108 (2012). https://doi.org/10.1007/s00213-011-2377-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00213-011-2377-2