
Abstract
Rationale
Accumulating evidence indicates that brain kappa-opioid receptors (KORs) and dynorphin, the endogenous ligand that binds at these receptors, are involved in regulating states of motivation and emotion. These findings have stimulated interest in the development of KOR-targeted ligands as therapeutic agents. As one example, it has been suggested that KOR antagonists might have a wide range of indications, including the treatment of depressive, anxiety, and addictive disorders, as well as conditions characterized by co-morbidity of these disorders (e.g., post-traumatic stress disorder) A general effect of reducing the impact of stress may explain how KOR antagonists can have efficacy in such a variety of animal models that would appear to represent different disease states.
Objective
Here, we review evidence that disruption of KOR function attenuates prominent effects of stress. We will describe behavioral and molecular endpoints including those from studies that characterize the effects of KOR antagonists and KOR ablation on the effects of stress itself, as well as on the effects of exogenously delivered corticotropin-releasing factor, a brain peptide that mediates key effects of stress.
Conclusion
Collectively, available data suggest that KOR disruption produces anti-stress effects and under some conditions can prevent the development of stress-induced adaptations. As such, KOR antagonists may have unique potential as therapeutic agents for the treatment and even prevention of stress-related psychiatric illness, a therapeutic niche that is currently unfilled.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Background
Neuropsychiatric conditions ranging from depressive disorders to addiction can be caused by environmental factors (e.g., life experiences), genetic factors, or interactions of the two. One environmental factor that can serve as a common trigger for all of these conditions is stress. Severe stress has many damaging effects; as one example, it can have acute cognitive-disrupting effects (Campeau et al. 2011; Putnam 2013) that lead to injury or death. There is compelling evidence that even a single exposure to a severe stressor can cause chronic psychiatric illnesses such as major depressive disorder, generalized anxiety disorder, and post-traumatic stress disorder (PTSD) (Kendler et al. 1999; Kessler 1997, 2000; Pine et al. 2002). Stress can also promote substance abuse and addiction (Koob and Volkow 2010), which are often co-morbid with depressive and anxiety disorders (Brown and Wolfe 1994; Koob and Kreek 2007; Logrip et al. 2012). In addition to causing new cases of psychiatric illness, stress can exacerbate existing illnesses (Pittenger and Duman 2008) and trigger relapse of drug-seeking behaviors in humans and laboratory animals (Beardsley et al. 2005; Marchant et al. 2013; Shaham and Stewart 1995; Wee and Koob 2010). Collectively, the disorders caused or exacerbated by stress are costly and frustrating because they tend to be debilitating, persistent, and resistant to existing treatments.
There is accumulating evidence that brain kappa-opioid receptors (KORs) play an important role in transducing the effects of stress. Activation of KORs produces aversive and depressive-like states in humans (Pfeiffer et al. 1986) and in laboratory animals (Carlezon et al. 2006; Mague et al. 2003; Todtenkopf et al. 2004) that may mimic, at least in part, those caused by stress (Land et al. 2008; McLaughlin et al. 2003a). Although the mechanisms by which KOR activation produces stress-like effects are not understood, recent studies suggest that interactions with brain corticotropin-releasing factor (CRF) systems are critical. CRF is a neuropeptide that is released in the brain in response to stress (Koob 1999), and administration of exogenous CRF produces many of the same physiological and behavioral effects as stress (Hauger et al. 2009). Interestingly, key behavioral and molecular effects of stress and CRF are blocked by selective KOR antagonists (Land et al. 2008; McLaughlin et al. 2003a; Van’t Veer et al. 2012), which is consistent with other evidence that these agents have antidepressant-like (Mague et al. 2003; Pliakas et al. 2001) and anxiolytic-like effects, including attenuation of fear-potentiated startle (Knoll et al. 2007; Knoll et al. 2011) and stress-induced reinstatement of drug-seeking behavior (Beardsley et al. 2005; Graziane et al. 2013). A broad effect of reducing the impact of stress may explain how KOR antagonists can have efficacy in such a wide variety of animal models that would appear to represent different disease states.
Regardless of mechanism, KOR antagonist-induced blockade of stress effects may serve as the basis for improved medications that relieve the signs and symptoms of depressive, anxiety, and addictive disorders. It may also represent an opportunity to develop an entirely new therapeutic area: prevention of certain types of psychiatric illness (i.e., those directly caused by stress). In this review, we describe existing data from studies utilizing behavioral pharmacology and genetic engineering to manipulate KORs or their endogenous agonist, dynorphin (Chavkin et al. 1982). We highlight strengths and limitations of existing studies, and identify gaps in current knowledge that should be filled.
Overview of stress effects
Stress is an organism’s response to internal or external challenges (stressors) and can negatively impact psychological and physical well-being. Acutely, stressors lead to involuntary hormonal (e.g., increased free fatty acid generation, inhibition of the immune system), autonomic (e.g., increased heart and breathing rate, increased blood flow to the brain and muscle), and behavioral (e.g., feelings of anxiety and fear, heightened vigilance) changes—often collectively called “the stress response”—that prepare the body to maintain homeostasis in response to a real or perceived threat (Chrousos and Gold 1992). This adaptive response is generally activating and protective in the short-term (Keay and Bandler 2001), but can become impairing with increasing intensity, duration, and frequency of the stressor (Buydens-Branchey et al. 1990; Miczek et al. 2008; Sapolsky 1996). Further, predictability and controllability of the stressor are key parameters of the stress response as even brief, low intensity stressors can have negative effects if unpredictable and uncontrollable (Adell et al. 1988; Foa et al. 1992). Severe or sustained stressors can disrupt cognitive processes and cause confusion (Campeau et al. 2011; Janis and Mann 1977; Keinan 1987; Putnam 2013; Shaham et al. 1992), and often precede the development of anxiety disorders, clinical depression, and substance abuse (Fox et al. 2007; Kendler et al. 1999; Kessler 1997; Koob and Kreek 2007; Pine et al. 2002; Volkow and Li 2004). In humans, clinical depression in characterized by depressed mood, anhedonia, and reduced energy while anxiety disorders entail excessive worrying that is difficult to control and problems concentrating. Some signs of psychiatric illness can be observed; thus testing in animals to quantify, for example, hedonic state, avoidance, escape, and physiologic state can be used to infer depression- and anxiety-like behaviors. In laboratory settings, stress procedures often trigger depressive- and anxiety-like behaviors and drug-seeking in animal models. Discrete stressors including footshock, maternal deprivation, and restraint induce depressive-like behaviors including increased immobility in the forced swim test [FST] (Aisa et al. 2008; Platt and Stone 1982), an effect that is opposite to that of antidepressants (Detke et al. 1995; Porsolt et al. 1977) and thus interpreted to indicate a prodepressive-like effect (Pliakas et al. 2001), as well as elevations in brain reward thresholds (Zacharko and Anisman 1991) that indicate anhedonia. More naturalistic stressors also produce similar outcomes: as an example, subordinate mice in a chronic social defeat stress (CSDS) paradigm—an ethologically relevant stressor involving daily exposure to an aggressor—show anxiogenic-like responses such as spending less time in the lit area of a light/dark box and the open arms of an elevated plus maze (EPM) (Keeney and Hogg 1999; Slattery et al. 2012), as well as decreases in social interaction with other mice (Avgustinovich et al. 2005; Berton et al. 2006). These studies demonstrate how models of stress in rodents may provide valuable insights into the mechanisms of stress-induced illness in humans.
Several mediators have been implicated in the stress response, including the CRF, catecholamine, serotonin, and vasopressin systems (see Carrasco and Van de Kar 2003; Tsigos and Chrousos 2002). Here, we focus on CRF because it plays a well-characterized role in stress-induced behaviors, its function can be dysregulated in people with psychiatric illness, and its effects are linked to KOR systems. First described by Vale and colleagues (1981), CRF is the principal regulator of the stress response (Majzoub 2006; Spiess et al. 1981). The peptide is produced by cells in the paraventricular nucleus of the hypothalamus (PVN) and triggers hormonal stress responses by activating the hypothalamic–pituitary–adrenal (HPA) axis, which leads to the release of adrenocorticotropic hormone (ACTH) from the pituitary (Antoni 1986). In turn, ACTH stimulates glucocorticoid release from the adrenal glands, which produces subsequent metabolic and cardiovascular changes (Fig. 1). Glucocorticoid actions are mediated by two receptors: glucocorticoid receptors (GRs) and mineralocorticoid receptors (MRs) which are expressed throughout the brain including areas involved in emotion, memory, and behavior such as the septum, hippocampus (HIP), and prefrontal cortex (PFC) (Ahima and Harlan 1990; Cintra et al. 1994; Fuxe et al. 1985; Morimoto et al. 1996; Reul and de Kloet 1985; Viengchareun et al. 2007). GRs and MRs regulate hormonal, autonomic and behavioral responses to stress via their widespread expression (Munck et al. 1984), and trigger negative feedback circuits that terminate HPA axis activation following stress (Autelitano et al. 1990; Herman et al. 1989; Swanson and Simmons 1989).

HPA axis and neuronal inputs. Stress causes the release of CRF and AVP from parvocellular neurons in the PVN that project to the anterior pituitary. ACTH secretion then leads to glucocorticoid synthesis and release from the adrenal cortex. Glucocorticoid actions are mediated by GRs and MRs throughout the brain and periphery. Glucocorticoids activate negative feedback loops within the PVN, pituitary and HIP denoted with minus signs in the illustration. Neuronal inputs from the HIP, BNST, PFC and AMY regulate HPA axis (red arrows) activity. Dashed lines represent indirect connections to the PVN. KORs are expressed in organs of the HPA axis and brain regions that influence HPA axis activation. ACTH adrenocorticotropic hormone, AMY amygdala, AVP arginine vasopressin, BNST bed nucleus of the stria terminalis, CRF corticotropin-releasing factor, GR glucocorticoid receptor, HIP hippocampus, HPA hypothalamic–pituitary–adrenal, MR mineralocorticoid receptor, PFC prefrontal cortex, PVN paraventricular nucleus of the hypothalamus
HPA axis regulation is achieved through actions integrated within the PVN. Afferents from cicumventricular organs, brainstem nuclei, and hypothalamic–basal forebrain systems can directly activate PVN neurons (Ziegler and Herman 2002) and relay information on the state of the body such as cardiovascular tone, blood oxygenation, arousal and osmotic state. In particular, the bed nucleus of the stria terminalis (BNST) sends projections from multiple subregions (Dong et al. 2001; Dong and Swanson 2004; Ziegler and Herman 2002), suggesting that this region plays a crucial role in regulation of PVN activity. Inputs to the BNST include regions demonstrated to regulate the HPA axis such as the amygdala (AMY), HIP and PFC despite having sparse or no direct connections with the PVN (Canteras et al. 1995; Crane et al. 2003; Cullinan et al. 1993; Gray et al. 1989; Herman et al. 2005; Ziegler and Herman 2002), suggesting the BNST may represent a relay where limbic information feeds in and is passed to the PVN (Cullinan et al. 1993; Herman et al. 2005). Other regions with direct input to the PVN (e.g., nucleus of the solitary tract) may also relay information from afferent connections (e.g., AMY) (Beaulieu et al. 1987; Schwaber et al. 1982; Xu et al. 1999; Ziegler and Herman 2002). Additionally, CRF receptors are expressed within circuits implicated in motivation and emotion (Dautzenberg and Hauger 2002; De Souza et al. 1985; Millan et al. 1986; Van Pett et al. 2000), such as the mesocorticolimbic system, where they can alter behavior by modulating reward, anxiety, and depressive responses independent of HPA axis activation (Dautzenberg and Hauger 2002; Koob et al. 1993; Merchenthaler 1984; Sakanaka et al. 1987; Swanson et al. 1983). Indeed, CRF is implicated in the detrimental consequences of prolonged stress, and hypersecretion of CRF has been hypothesized to be the primary contributing factor in the development of depressive and anxiety disorders (De Souza 1995; Nemeroff 1992; Owens et al. 1993). In humans, major depressive disorder has been associated with higher levels of cerebrospinal fluid CRF (Arato et al. 1989; Kasckow et al. 2001; Nemeroff et al. 1984; Widerlov et al. 1988), and CRF levels are also elevated in patients with PTSD (Baker et al. 1999; Bremner et al. 1997; de Kloet et al. 2008).
Administration of exogenous CRF induces anxiety- and depressive-like behavior in laboratory animals, enabling studies of cause–effect relationships between stress and behaviors that reflect the signs of psychiatric illness. For example, socially housed non-human primates exhibit depressive-like behaviors such as huddling and wall-facing after intracerebroventricular CRF infusion (Kalin 1990; Strome et al. 2002). In rodents, CRF administration or genetic overexpression also precipitates depressive- and anxiety-like behaviors (Britton et al. 1982; Dunn and File 1987; Liang et al. 1992; Stenzel-Poore et al. 1994; Swiergiel et al. 2008; van Gaalen et al. 2002). In contrast, antagonism or genetic knockout of CRF receptors may produce antidepressant- and anxiogenic-like effects in laboratory animals (Britton et al. 1986; Deak et al. 1999; Griebel et al. 2002; Smith et al. 1998; Timpl et al. 1998), especially when animals are exposed to acute stress before testing (Zorrilla et al. 2013). For example, administration of a CRF receptor antagonist to non-human primates decreases anxiety and fear behavior and increases exploratory and sexual behavior when animals are exposed to stressful stimuli (Habib et al. 2000). Similarly, CRF antagonists block drug withdrawal-induced anxiety-like behaviors (Basso et al. 1999; Heinrichs et al. 1995; Rassnick et al. 1993; Sarnyai et al. 1995) and reduce self-administration to drugs including cocaine, nicotine and heroin in dependent animals (George et al. 2007; Goeders and Guerin 2000; Greenwell et al. 2009; Specio et al. 2008).
Despite these promising effects in preclinical studies, the development of CRF receptor antagonists as therapeutics has been hindered by the high lipophilicity of initial drugs, although several novel compounds are in clinical trials (Zorrilla and Koob 2010). Nevertheless, further investigation into the systems downstream of CRF receptor activation may provide new targets for treatment. One mediator of stress effects that may provide unique drug targets is the KOR system.
Links between stress and KORs
It is well established that endogenous opioid systems play important roles in stress, reward processing, and mood regulation. These systems consist of the neuropeptides endorphins, enkephalins, and dynorphins and their cognate receptors (mu, delta, and kappa, respectively). Biologically active peptides for all opioid receptors are derived from inactive prohormones that are post-translationally processed. The dynorphin family of peptides arises from processing of prodynorphin (Pdyn) into major products (see Bruijnzeel 2009; Schwarzer 2009) that preferentially bind to and activate KORs (Chavkin et al. 1982), although with differing potency (James et al. 1984).
KORs are G-protein-coupled receptors that mainly interact with inhibitory Gα subunits (Law et al. 2000). KOR activation by endogenous or synthetic agonists can produce inhibition of adenylate cyclase activity (Attali et al. 1989; Konkoy and Childers 1989; 1993; Lawrence and Bidlack 1993) and can decrease cell excitability and neurotransmitter release by altering calcium and potassium currents (Gross et al. 1990; Henry et al. 1995; Hjelmstad and Fields 2003; Rusin et al. 1997; Simmons and Chavkin 1996; Tallent et al. 1994). KOR activation has also been shown to activate mitogen activated protein kinase (MAPK) pathways in neurons and astrocytes (Belcheva et al. 1998, 2005; Bohn et al. 2000; Bruchas et al. 2006, 2007a; Kam et al. 2004). The MAPK family includes several kinases that respond to a variety of cell stimuli and regulate diverse functions such as proliferation, differentiation, apoptosis, and gene expression. Thus KOR-mediated effects on ion channels and signaling cascades allows for rapid effects on cell excitability and neurotransmitter release that may underlie acute stress effects, while delayed effects such as gene expression may play a role in conditions of chronic stress (Knoll and Carlezon 2010).
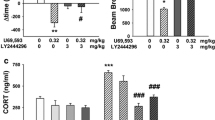
Consistent with a role in mediating stress effects, moderate to high levels of dynorphin and KOR mRNA expression have been detected in stress-responsive brain regions including the PVN, AMY, HIP, and BNST of rodents (Lin et al. 2006; Mansour et al. 1987, 1988; Meng et al. 1993; Merchenthaler et al. 1997; Morris et al. 1986; Peng et al. 2012). A similar expression profile exists in human brain (Hurd 1996; Nikoshkov et al. 2005; Simonin et al. 1995; Zhu et al. 1995), suggesting the KOR system plays an evolutionary conserved role. The expression pattern of KORs and its overlap with the systems traditionally implicated in the stress response raises the possibility that they may participate in HPA axis regulation (Fig. 1). Indeed, stress induces dynorphin release and activation of KORs with synthetic agonist increases corticosterone (CORT) levels in rats (Hayes and Stewart 1985; Iyengar et al. 1986) and cortisol levels in rhesus monkeys and humans (Pascoe et al. 2008; Ur et al. 1997). The mechanism through which KORs activate the HPA axis is unclear, but likely involves stimulation of CRF release in the hypothalamus as well as CRF-independent mechanisms (Buckingham and Cooper 1986; Calogero et al. 1996; Nikolarakis et al. 1987). Data from these studies suggest that disruption of KOR function may reduce HPA axis activation. Consistent with this possibility, CORT levels are reduced in both Pdyn knockouts and wild-type mice treated with the prototypical KOR antagonist nor-binaltorphimine (norBNI). Furthermore, injection stress-induced increases in CORT are attenuated in these knockouts (Wittmann et al. 2009) suggesting KOR activation facilitates HPA axis activation. Reduced CORT levels are also observed in norBNI-treated rats in response to food restriction and cocaine challenge (Allen et al. 2013). However, it does not appear that KOR activation is necessary for all stress-induced HPA axis activation. For example, there are no differences between norBNI-treated or Pdyn knockouts and wild-type mice at either baseline or following forced swim stress (McLaughlin et al. 2006a). In yet a third Pdyn knockout strain, CORT levels following exposure to an elevated zero maze reach a lower peak concentration, but are prolonged compared to controls (Bilkei-Gorzo et al. 2008). These differences may arise from the type of stress used, blood collection time points, differences in targeting construct or strain differences, but overall suggest the KOR system can modulate stress-induced glucocorticoid release in some instances. As many of these studies used systemic KOR agonist treatment and constitutive Pdyn knockouts, additional studies are needed to identify the sites in which KORs regulate HPA axis activation.
Role of kappa-opioid receptors in stress-induced behaviors
Key aspects of KOR-mediated behaviors resemble those observed following stress or CRF administration, suggesting common mechanisms of action. Like stress or CRF, KOR agonists elevate brain reward thresholds (Carlezon et al. 2006; Dinieri et al. 2009; Todtenkopf et al. 2004; Tomasiewicz et al. 2008), and produce depressive-like effects including increased immobility in the FST (Carlezon et al. 2006; Mague et al. 2003). In humans, selective KOR agonists produce negative mood states including dysphoria, anxiety, and abnormal behavior along with psychotomimesis at higher doses (Pfeiffer et al. 1986). There is now considerable evidence that KOR antagonists block KOR agonist effects and have antidepressant- and anxiolytic-like effects on their own. For example, KOR antagonism produces anxiolytic-like effects in the EPM, fear-potentiated startle (FPS), novelty-induced hypophagia and defensive burying tests (Carr and Lucki 2010; Knoll et al. 2007; Wiley et al. 2009), suggesting that KOR activation is necessary for the acquisition and/or expression of anxiety-like behavior. Anxiolytic-like effects have been observed in KOR system-deficient mice (Van’t Veer et al. 2013; Wittmann et al. 2009), although it is important to note that some lines of constitutive KOR knockout mice do not differ from controls in measures of anxiety-like behavior (Filliol et al. 2000; Simonin et al. 1998), and that Pdyn ablation can reportedly increase anxiety (Bilkei-Gorzo et al. 2008). These discrepant results may be explained by differences in compensatory changes that occur during development in these lines, differences in genetic background, as well as differences in the stressfulness of the procedures, environment and factors including husbandry (e.g., Crabbe et al. 1999) among labs. Indeed, restricting KOR ablation to dopamine systems produces a clearer anxiolytic-like phenotype, with increased center exploration in an open field and shorter latencies to enter the lit compartment of the light/dark box (Van’t Veer et al. 2013). Furthermore, effects of KOR blockade may not be apparent until after an initial stressor, at which time putative KOR-dependent neuroadaptations occur (for review, see Knoll and Carlezon 2010). For instance, KOR antagonists and KOR system gene disruption reduce immobility in the FST, but the effects are typically detected during the second exposure to forced swim stress (Mague et al. 2003; McLaughlin et al. 2003a, 2006a; Pliakas et al. 2001, but see Carey et al. 2009). Antidepressant-like effects are also observed when KOR antagonists are administered after the first swim sessions suggesting that blockade of KOR activation following stress may be sufficient to prevent neural adaptations that facilitate immobility during the second swim session (Beardsley et al. 2005; Carr et al. 2010; Wiley et al. 2009). Similarly, central administration of norBNI following footshock stress produces antidepressant-like effects in the learned helplessness paradigm (Newton et al. 2002). The mechanisms of KOR-dependent behaviors are not yet understood, but may involve distinct neural circuits and reflect the differences in immediate actions of KOR activation versus delayed effects such as gene expression changes (see Bruchas and Chavkin 2010; Knoll and Carlezon 2010), which may be especially important in animal models involving initially normal (i.e., non-depressed) subjects.
In addition to forced swimming, stressors including footshock and social defeat also activate the KOR system (Beardsley et al. 2005; Land et al. 2008; McLaughlin et al. 2003a, 2006b; Redila and Chavkin 2008), suggesting it plays an important role in generalized stress effects. In conditioned place preference (CPP) tests, the rewarding effects of a treatment (e.g., a drug of abuse) become associated with the environment in which it was paired, thereby causing a preference for this environment during subsequent drug-free exposure. This effect can be extinguished by repeated access to the testing apparatus, and then rapidly reinstated by stress or drug priming. Disruption of KOR signaling blocks stress-induced but not drug-primed reinstatement (Aldrich et al. 2009; Carey et al. 2007; Eans et al. 2013; Jackson et al. 2013; Redila and Chavkin 2008), suggesting the KOR system plays a highly specific role in mediating the motivational effects of stress. Exposure to stress can also increase the magnitude of drug reward, as measured in the CPP test. Potentiation of drug-induced CPP following social defeat and forced swim stress is blocked by the KOR antagonist norBNI and absent in Pdyn and KOR knockout mice (McLaughlin et al. 2003a, 2006a, b; Schindler et al. 2010; Smith et al. 2012; Sperling et al. 2010). Further, disruption of KOR signaling attenuates the potentiation of cocaine-induced locomotor sensitization by stress (Allen et al. 2013). In contrast, activation of KORs mimics the effects of stress on reward (McLaughlin et al. 2006a; Schindler et al. 2010), demonstrating that KOR activation is a necessary and sufficient element of at least certain stress effects on behavior. These data, together with findings that KOR antagonists block stress-induced drug-seeking behavior in drug self-administration models (Beardsley et al. 2005, 2010; Graziane et al. 2013) and withdrawal-induced anxiogenic- and depressive-like behavior (Chartoff et al. 2012; Jackson et al. 2010; Valdez and Harshberger 2012), without reward-related effects in drug self-administration tests (Beardsley et al. 2005; Todtenkopf et al. 2004), raise the possibility that KOR antagonist treatment may reduce relapse in drug abusers attempting to abstain from use.
Dynorphin release appears critical for encoding the aversive (dysphoric) effects of stress. It is known that mice will develop a conditioned place aversion (CPA) to an odorant that was previously paired with stress. The avoidance behavior is abolished by pretreatment with norBNI before stress (forced swim or footshock) and absent in Pdyn knockout mice (Land et al. 2008), suggesting reduced aversions. Treatment with norBNI blocks both psychological (Takahashi et al. 1990) and physical stress-induced antinociception (McLaughlin et al. 2003a, 2006a) a phenomenon in which stress reduces sensitivity to pain. Both norBNI and Pdyn gene disruption block stress-induced antinociception observed immediately after the first and subsequent days of social defeat (McLaughlin et al. 2006b). During CSDS sessions, rodents display characteristic immobility and social defeat postures, which tend to increase progressively (Miczek et al. 2004). Postures reflecting social defeat are reduced in norBNI-treated and Pdyn knockout mice, suggesting that KOR blockade produces signs of stress resilience. However, these differences are not apparent proceeding the first day of stress, revealing that KOR signaling is not necessary for initial defeat-induced postures in this paradigm, but instead in the progressive, neuroadaptive effects of chronic stress.
Stress produces KOR-dependent effects in learning and memory tasks which may reveal a role for KORs in stress-induced deficits in cognitive function. Mice subjected to repeated forced swim stress or systemic KOR agonist show a deficit in novel object recognition that can be prevented with KOR antagonist treatment and is absent in Pdyn knockout mice (Carey et al. 2009; Paris et al. 2011). Further, chicks treated with a KOR agonist show impairments in a one-trial peck avoidance task while norBNI treatment facilitated performance (Colombo et al. 1992). Pdyn and KOR knockout mice have enhanced performance in the spatial Morris water maze (MWM), suggesting that KOR activation, perhaps induced by swim, may inhibit performance (Jamot et al. 2003; Nguyen et al. 2005). KOR effects on memory may depend, at least in part, on receptors in the HIP as intra-CA3 HIP infusions of KOR agonist induce deficits in the MWM (Daumas et al. 2007). These results are consistent with the hypothesis that stress-induced dynorphin release impairs memory, although there is evidence that KOR activation can improve memory (Hiramatsu and Hoshino 2004; Hiramatsu et al. 1996; Kuzmin et al. 2006). These improvements may be the result of non-KOR effects (Hiramatsu and Hoshino 2004, 2005; Kuzmin et al. 2006) or may represent acute activating effects of KORs as is seen with stressors. Regardless, considering that memory impairments are a symptom of a variety of mood disorders and other psychiatric illnesses, therapeutic agents that mitigate them may have broad indications that cut across conditions previously conceptualized as being unrelated (Morris and Cuthbert 2012).
As described above, CRF is the primary regulator of the stress response; when centrally administered, it can recapitulate many of the behavioral, hormonal and autonomic effects of stress, including dynorphin release. CRF has been shown to stimulate release of dynorphin from spinal cord (Song and Takemori 1992), hypothalamus (Almeida et al. 1986; Nikolarakis et al. 1986), globus pallidus and striatum (Sirinathsinghji et al. 1989). It also produces increases in KOR phosphorylation—a marker of receptor activation (McLaughlin et al. 2003b)—in components of stress and anxiety circuits including the striatum, dorsal raphe nucleus (DRN), AMY, HIP, and NAc that are reduced or absent in norBNI pretreated mice and Pdyn knockouts (Land et al. 2008). The fact that CRF and KOR agonists produce aversive and anxiogenic-like effects raises the possibility that CRF effects may be mediated by the KOR system. To address this question, Land et al. (2008) examined the effect of KOR blockade on CRF-induced CPAs in mice. In these experiments, central CRF administration induced aversion to the context in which mice were placed following infusion. CRF-induced CPA was abolished with norBNI pretreatment and in Pdyn knockout mice (Land et al. 2008), suggesting that CRF receptor activation promotes dynorphin release and subsequent KOR activation to mediate the aversive component of stress. Important interactions also exist between the CRF and KOR systems as measured in the 5-choice serial reaction time task (5CSRTT), a test of cognitive behavior analogous to the continuous performance task used to study attention in humans (Beck et al. 1956; Robbins 2002). CRF dose-dependently disrupts several performance measures in the 5CSRTT, and these deficits are attenuated by systemic administration of the KOR antagonist JDTic at a dose without effects of its own (Van’t Veer et al. 2012). These findings further demonstrate that KOR antagonists can prevent acute CRF-related effects, including those that degrade performance in tasks requiring attention.
Brain regions implicated in KOR-mediated effects
Identifying the brain regions in which drugs have their effect has become a crucial element of neuroscience research, in part because characterizing the substrates and mechanisms of drug action may ultimately lead to dramatic improvements in therapeutics. It is important to emphasize that it is already established that systemic administration of KOR agonists produce depressive-like effects whereas KOR antagonists have antidepressant- and anxiolytic-like effects, as well as general anti-stress effects that can attenuate reinstatement of drug-seeking behaviors. The efficacy of KOR antagonists in these various preclinical models has provided sufficient rationale for moving them into human studies; indeed, there have been early clinical trials of JDTic, as well as novel proprietary agents from Alkermes, Lilly, and Pfizer (see Carroll and Carlezon 2013). Unfortunately, peer-reviewed reports on these trials are yet to appear in the literature. There is a report that buprenorphine—which has weak partial KOR agonist effects (Zhu et al. 1997)—has antidepressant effects (Bodkin et al. 1995), but broad use of this agent to treat depressive illness is limited by mu agonist effects that may engender abuse liability. A better understanding of the brain regions in which KOR ligands produce behavioral effects may facilitate the development of new—and potentially non-pharmacological—methods of targeting specific brain areas. Our research has traditionally focused on the mesocorticolimbic system (Fig. 2), which plays an important role in affective behavior despite not being implicated in classical theories of depression and anxiety (Nestler and Carlezon 2006). The neurons of the mesocorticolimbic DA system originate in the VTA and project to the NAc, HIP, AMY, PFC, and BNST (Swanson 1982). Historically, the VTA and its dopaminergic projections have been studied primarily in the context of motivation and reward (Wise and Bozarth 1987). However, accumulating work has led to greater recognition of the role of this system in aversion as well (Carlezon and Thomas 2009; Pezze and Feldon 2004; Salamone 1994). Aversive stimuli can increase DA neuron population activity (Valenti et al. 2011) and activate the mesocorticolimbic system resulting in postsynaptic DA release (Abercrombie et al. 1989; Imperato et al. 1993; Pascucci et al. 2007; Piazza and Le Moal 1998; Thierry et al. 1976) that may promote or antagonize stress effects on behavior. A key area where additional research is needed is how activation of the VTA might participate in both rewarding and aversive stimuli; indeed, even studies utilizing the most modern and sophisticated techniques can provide somewhat conflicting information (Chaudhury et al. 2013; Lammel et al. 2012; Tye et al. 2013). One possibility is that these stimuli have similar effects on the VTA but different effects upon other brain areas, which thereby regulate the activity of the many VTA target regions and/or affect signal gating in the NAc (Carlezon and Thomas 2009).

The KOR system in anxiety- and depressive-like behaviors. Schematic illustration of mesocorticolimbic brain areas involved in KOR effects on depressive- and anxiety-like behaviors in preclinical models. Relevant references are noted on the representation. Regions implicated in KOR effects on anxiety-like behavior are colored orange and those so far only implicated in depressive-like behavior are colored blue. AMY amygdala, BNST bed nucleus of the stria terminalis, DA dopamine, DRN dorsal raphe nucleus, HIP hippocampus, KOR kappa-opioid receptor, PFC prefrontal cortex, NAc nucleus accumbens, VTA ventral tegmental area
Early investigations into the neural substrates of KOR-induced aversion using CPA identified the VTA as a key area of activation (Bals-Kubik et al. 1993). Aversion was postulated to be the result of KOR-mediated decreases in DA release. Indeed, KOR agonists decrease DA release in VTA cell cultures (Dalman and O’Malley 1999; Ronken et al. 1993) and directly inhibit DA cell firing through G-protein-coupled inwardly rectifying potassium channels (GIRKs) in slice (Margolis et al. 2003). In addition to postsynaptically inhibiting DA release through hyperpolarization, KOR activation in the VTA can induce presynaptic inhibition of somatodendritic DA at its release sites (Ford et al. 2007). KORs can also regulate VTA activity through the control of glutamate input (Margolis et al. 2005) demonstrating the broad range of KOR control over DA function. Dynorphin terminals synapse onto both TH-labeled (presumably DA neurons) and unlabeled dendrites as well as terminals and astrocytes in the VTA (Pickel et al. 1993) where KOR activation can produce differential responses. Because several dynorphin-expressing nuclei project to the VTA including those from the hypothalamus, AMY, CPu, and NAc (Fallon and Leslie 1986), VTA cells expressing KORs may be involved in integrating information from multiple brain circuits or have unique responses based on input and/or projection target. For example, KOR-mediated inhibition of DA neurons varies as a function of projection target (Ford et al. 2006; Margolis et al. 2006, 2008). KORs are also located on the terminals of DA projections from the VTA to the NAc and PFC where they can presynaptically inhibit DA release (Carlezon et al. 2006; Di Chiara and Imperato 1988; Grilli et al. 2009; Spanagel et al. 1992; Werling et al. 1988). Indeed, intra-PFC KOR agonist decreases local DA overflow, while KOR antagonist enhances it (Tejeda et al. 2013). Furthermore, direct infusion of KOR agonist into the PFC can cause place aversions (Bals-Kubik et al. 1993) whereas intra-PFC KOR antagonist attenuates place aversions induced by systemic KOR agonism (Tejeda et al. 2013), suggesting these effects may be due at least in part to changes in DA transmission. Similarly, infusion of KOR agonist into the NAc induces depressive-like behaviors in rodents including place aversions and increases brain reward thresholds (Bals-Kubik et al. 1993; Muschamp et al. 2011) while intra-NAc norBNI decreases escape failures in a learned helplessness paradigm, an antidepressant-like effect (Newton et al. 2002; Shirayama et al. 2004). These effects are also observed following intra-HIP infusions of norBNI (Shirayama et al. 2004), although it is unclear whether the KORs mediating this effect are expressed on terminals from the VTA.
Additional evidence for a role of the mesocorticolimbic system in KOR effects on behavior has come from studies utilizing the transcription factor cAMP-response-element-binding protein (CREB) (for review, see Carlezon et al. 2005 and Muschamp and Carlezon 2013). Cell signaling events can activate CREB which in turn alters expression of CREB-regulated genes, including Pdyn. Stress has been shown to cause behaviors characteristic of depression such as anhedonia, behavioral despair, and dysphoria in rats (Land et al. 2008; Moreau et al. 1992; Pliakas et al. 2001) that are mimicked by elevating CREB levels in the NAc using viral-mediated gene transfer (Barrot et al. 2002; Pliakas et al. 2001). In contrast, decreasing CREB activity in the NAc through expression of a dominant-negative form of CREB leads to antidepressant-like effects in rodents (Newton et al. 2002; Pliakas et al. 2001). Notably, these changes in behavior due to increases or decreases in CREB activity were shown to be mediated largely by CREB-induced changes in dynorphin expression. Dynorphin is a target of CREB-induced gene expression in vitro (Cole et al. 1995; Douglass et al. 1994; Turgeon et al. 1997) and manipulating CREB levels changes dynorphin expression in vivo (Carlezon et al. 1998; Pliakas et al. 2001). Administration of norBNI attenuates the behavioral effects of elevated CREB levels within the NAc (Carlezon et al. 1998; Pliakas et al. 2001), whereas blockade of endogenous dynorphin actions through direct injection of norBNI into the NAc is sufficient to produce antidepressant-like effects (Newton et al. 2002). It is postulated that some features of depression are the result of dynorphin control of mesocorticolimbic DA function, either by actions at KORs on VTA cell bodies or terminals that project to the NAc (Nestler and Carlezon 2006). Given the high co-morbidity of depressive and anxiety disorders (Kaufman and Charney 2000; Kessler 2000), KOR signaling and control of DA function may underlie the pathophysiology of both. The question of whether these effects are mediated within the NAc itself, or the result of alterations in NAc-to-VTA feedback that subsequently affect neural activity in regions that receive VTA input, remains open.
The AMY is another target of VTA dopamine neurons, and is the brain region most often considered to be the epicenter of fear responsiveness. Much preclinical work has elucidated AMY cellular and molecular mechanisms in fear as reviewed elsewhere (Davis 1997; Davis and Shi 2000). Recent evidence indicates that fear conditioning induces plasticity in KOR systems leading to upregulation of KOR mRNA in the basolateral nucleus of the AMY (BLA) suggesting that KOR signaling in this region may mediate the expression of conditioned fear. Indeed, microinfusions of KOR antagonist into the BLA reduces conditioned fear responses and produces anxiolytic-like effects in the EPM (Knoll et al. 2011). Induction of stress-like states through central administration of CRF induces avoidance of the open arms of an EPM, an effect that is abolished with prior norBNI treatment or Pdyn gene disruption (Bruchas et al. 2009). In agreement with fear conditioning studies, the BLA is critical for this anxiogenic effect, because direct injection of norBNI into this region is sufficient to block CRF-induced decreases in open arm time (Bruchas et al. 2009). Microinjections of KOR antagonist into the AMY also attenuate the stress-related effects of withdrawal from nicotine (Smith et al. 2012). Although the AMY is clearly involved in the expression of fear and anxiety behaviors, it is embedded within a circuit of highly interconnected brain structures that are known to be involved in processes that reflect motivation and emotion. Recent work suggests that KORs are expressed on the terminals of AMY inputs to the BNST (Li et al. 2012), a brain area strongly implicated in anxiety behavior (Walker et al. 2003). It is increasingly evident that structures with amygdalar afferent and/or efferent projections contribute to normal and pathologic anxiety. A deeper understanding of how these interconnected regions function in isolation as well as in circuits may enable new insights into the neurobiology of stress and anxiety responses as well as the pathophysiology of psychiatric disorders.
In studies of stress-induced aversion and potentiation of drug reward, the DRN is implicated in an elegant mechanism that explains how KORs expressed on terminals of axon projections from the DRN to the NAc are involved in stress-induced responses (Land et al. 2009; Schindler et al. 2012). KOR-dependent activation of p38 MAPK by stress in DRN serotonergic neurons is necessary and sufficient to induce a negative affective state (Bruchas et al. 2007a, 2011; Land et al. 2009). These effects are hypothesized to result from decreased serotonergic tone considering that KOR activation in DRN slice preparations induces p38 MAPK-dependent activation of GIRKs and presynaptic inhibition of excitatory neurotransmission resulting in decreased serotonergic neuron excitability and increased serotonin uptake in nerve terminals (Bruchas et al. 2011; Lemos et al. 2012).
Although the role that KORs within these regions play on stress and anxiety-related behavior has not been thoroughly examined, future studies using the recently generated floxed KOR mouse (Van’t Veer et al. 2013) in combination with promoter-driven Cre viral vectors have the potential to elucidate the function of KORs within particular cell populations and brain regions and lead to a more comprehensive understanding of interactions between stress and KOR systems.
Systemic administration of KOR antagonists to prevent stress-related illness
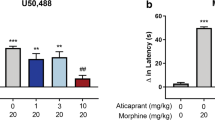
The idea of preventing psychiatric illness may seem fanciful or provocative, in part because our understanding of the brain and the pathophysiology of neuropsychiatric disorders remains incomplete. However, if stress can cause psychiatric illness and KOR antagonists can block stress, the concept of prevention becomes feasible. The discovery that systemic (or central) administration of KOR antagonists can block the effects of stress has at least some of its basis in the unusual pharmacodynamics of the prototypical KOR antagonist norBNI: slow onset, lack of initial selectivity for KORs, and exceptionally long duration of action. Early work indicated that norBNI initially blocks all opioid receptors non-selectively and requires 4–24 h to reach maximal kappa-selective antagonism (Endoh et al. 1992) and that a single injection produces behavioral effects that persist for weeks (Bruchas et al. 2007b; Horan et al. 1992; Jones and Holtzman 1992). Indeed, we have found that a single injection of norBNI blocks the depressive-like effects of the highly selective KOR agonist salvinorin A for at least 84 days, at which time the experiments were terminated (Potter et al. 2011). The mechanisms by which norBNI and other KOR antagonists (JDTic) produce long-lasting effects are not understood, but may involve a process known as biased agonism (or ligand-directed signaling) (see Carroll and Carlezon 2013). These unique properties made it necessary to use experimental designs in which KOR antagonists were administered far in advance of exposure to stress, to ensure selective KOR antagonism at the time of initial stress exposure (Pliakas et al. 2001), since the long-lasting effects of the drug made concerns about time course irrelevant. The interpretation of these original studies, which demonstrated that norBNI produced antidepressant-like effects, were focused on the similarities between the effects of KOR antagonists and standard antidepressant drugs in the FST rather than the fact that the data also raised the possibility of anti-stress actions. Subsequent studies in which the effects of KOR antagonists were evaluated in the EPM and FPS test also used this experimental design—administration of the drug before exposure to stress (Knoll et al. 2007). These studies provided clear evidence that these agents had acute anxiolytic-like effects and could reduce the persistent behavioral consequences of fear conditioning. To the extent that fear conditioning can accurately model key aspects of PTSD (Mahan and Ressler 2012) the ability of KOR antagonists to reduce FPS may reflect an ability to prevent stress-related neuroadaptations that can cause psychiatric illness. It is important to note that some studies showing anxiolytic or antidepressant effects of KOR antagonists use pretreatment times and/or drug doses capable of antagonizing other opioid receptors (Endoh et al. 1992; Thomas et al. 2004), suggesting these behavioral effects may occur through non-KOR receptors. Because delta-opioid receptor-deficient mice and those treated with delta receptor antagonist show prodepressant- and anxiogenic-like effects (Filliol et al. 2000; Perrine et al. 2006), it seems unlikely that the effects of KOR antagonists on depressive and anxiety behavior in these studies are due to blockade of delta receptor function. However, mu opioid receptor knockout mice or mice treated with mu antagonists demonstrate antidepressant- and anxiolytic-like behaviors (Filliol et al. 2000; Komatsu et al. 2011; Yoo et al. 2004), which could potentially underlie KOR antagonist effects in some instances, although the preponderance of data are collected at time points of selective KOR antagonism. In addition, non-selective opioid antagonists produce anhedonia in the ICSS test (West and Wise 1988), suggesting that blockade of mu and/or delta receptors can induce a prominent sign of depressive illness. The combination of acute antidepressant-like and anxiolytic-like effects distinguishes KOR antagonists from standard antidepressant drugs, which tend to have acute anxiogenic effects (Knoll et al. 2007). The ability of KOR antagonists to block the cognitive-disrupting effects of CRF (Van’t Veer et al. 2012), as well as other behaviors that characterize PTSD (e.g., persistent hyperarousal; Van’t Veer et al. 2011), provides converging evidence for the anti-stress effects of these agents.
Preclinical studies that provide support for the concept that systemic administration of KOR antagonists might be useful for mitigating stress effects and thus preventing the development of stress-related psychiatric illness are summarized in Table 1. Clearly, the ability of KOR antagonists to block reinstatement of addiction-like behaviors does not qualify as prevention of addiction, per se. However, addiction and psychiatric illness are often co-morbid, and it is not always clear which condition precedes which (Kessler 1997). As such, KOR antagonist-induced reductions in addictive behaviors may serve to prevent psychiatric illnesses that are secondary consequences of addiction.
Summary
It is often easier and less costly to prevent illness than to treat it. Familiar examples of broad efforts to prevent disease include campaigns to decrease smoking, promote exercise, and outlaw harmful foods. Vaccines have been developed to prevent debilitating diseases ranging from polio to, more recently, influenza. The best-selling prescription medication of all time (atorvastatin [Lipitor™]) treats a risk factor for disease (high cholesterol) rather than a disease itself. The idea of preventing psychiatric illness, however, can seem fanciful. Although there have been advances in early diagnosis and intervention to mitigate conditions such as bipolar disorder, schizophrenia, and attention-deficit hyperactivity disorder (Andersen 2003; McNamara et al. 2012; Sonuga-Barke et al. 2011), as well as increasing efforts to identify the biological basis of resilience (Russo et al. 2012), there are still no widely accepted methods of actually preventing psychiatric illness. One strategy is to attenuate the effects of stress, a major cause of new illness and a precipitating factor in existing illness. While it is certainly true that stress can be unpredictable in the context of everyday life, there is often adequate lead-time preceding exposure to some of the most severe, debilitating, and costly forms of stress (e.g., those encountered during a combat mission or while responding to a disaster). In animal models, KOR antagonists appear to have a general effect of mitigating the perception and/or consequences of stress, which may account for their ability to produce such a wide variety of beneficial effects. Perhaps most importantly, these agents produce combined antidepressant and anxiolytic effects, whereas standard antidepressants initially produce anxiogenic effects that, in humans, may contribute to problems with tolerability and adherence. Together, these results indicate that KOR antagonists may be useful in humans to prevent the development and expression of stress-induced illnesses such as anxiety, depressive disorders, and addiction. While there are numerous gaps in our knowledge with regard to the mechanisms of their beneficial effects (e.g., how KOR antagonists might block stress, the brain areas in which their effects are mediated) as well as their pharmacodynamics (e.g., why the effects of the prototypical antagonists are so persistent despite little apparent structural overlap, if shorter-acting agents would also be effective), there is an increasing appreciation that this class of agents may fill a novel and unique therapeutic niche.
References
Abercrombie ED, Keefe KA, DiFrischia DS, Zigmond MJ (1989) Differential effect of stress on in vivo dopamine release in striatum, nucleus accumbens, and medial frontal cortex. J Neurochem 52:1655–1658
Adell A, Trullas R, Gelpi E (1988) Time course of changes in serotonin and noradrenaline in rat brain after predictable or unpredictable shock. Brain Res 459:54–59
Ahima RS, Harlan RE (1990) Charting of type II glucocorticoid receptor-like immunoreactivity in the rat central nervous system. Neuroscience 39:579–604
Aisa B, Tordera R, Lasheras B, Del Rio J, Ramirez MJ (2008) Effects of maternal separation on hypothalamic–pituitary–adrenal responses, cognition and vulnerability to stress in adult female rats. Neuroscience 154:1218–1226
Aldrich JV, Patkar KA, McLaughlin JP (2009) Zyklophin, a systemically active selective kappa opioid receptor peptide antagonist with short duration of action. Proc Natl Acad Sci U S A 106:18396–18401
Allen CP, Zhou Y, Leri F (2013) Effect of food restriction on cocaine locomotor sensitization in Sprague–Dawley rats: role of kappa opioid receptors. Psychopharmacology 226:571–578
Almeida OF, Nikolarakis KE, Herz A (1986) Regulation of hypothalamic beta-endorphin and dynorphin release by corticotropin-releasing factor (CRF). NIDA Res Monogr 75:401–402
Andersen SL (2003) Trajectories of brain development: point of vulnerability or window of opportunity? Neurosci Biobehav Rev 27:3–18
Antoni FA (1986) Hypothalamic control of adrenocorticotropin secretion: advances since the discovery of 41-residue corticotropin-releasing factor. Endocr Rev 7:351–378
Arato M, Banki CM, Bissette G, Nemeroff CB (1989) Elevated CSF CRF in suicide victims. Biol Psychiatry 25:355–359
Attali B, Saya D, Vogel Z (1989) Kappa-opiate agonists inhibit adenylate cyclase and produce heterologous desensitization in rat spinal cord. J Neurochem 52:360–369
Autelitano DJ, Blum M, Lopingco M, Allen RG, Roberts JL (1990) Corticotropin-releasing factor differentially regulates anterior and intermediate pituitary lobe proopiomelanocortin gene transcription, nuclear precursor RNA and mature mRNA in vivo. Neuroendocrinology 51:123–130
Avgustinovich DF, Kovalenko IL, Kudryavtseva NN (2005) A model of anxious depression: persistence of behavioral pathology. Neurosci Behav Physiol 35:917–924
Baker DG, West SA, Nicholson WE, Ekhator NN, Kasckow JW, Hill KK, Bruce AB, Orth DN, Geracioti TD Jr (1999) Serial CSF corticotropin-releasing hormone levels and adrenocortical activity in combat veterans with posttraumatic stress disorder. Am J Psychiatry 156:585–588
Bals-Kubik R, Ableitner A, Herz A, Shippenberg TS (1993) Neuroanatomical sites mediating the motivational effects of opioids as mapped by the conditioned place preference paradigm in rats. J Pharmacol Exp Ther 264:489–495
Barrot M, Olivier JD, Perrotti LI, DiLeone RJ, Berton O, Eisch AJ, Impey S, Storm DR, Neve RL, Yin JC, Zachariou V, Nestler EJ (2002) CREB activity in the nucleus accumbens shell controls gating of behavioral responses to emotional stimuli. Proc Natl Acad Sci U S A 99:11435–11440
Basso AM, Spina M, Rivier J, Vale W, Koob GF (1999) Corticotropin-releasing factor antagonist attenuates the "anxiogenic-like" effect in the defensive burying paradigm but not in the elevated plus-maze following chronic cocaine in rats. Psychopharmacology 145:21–30
Beardsley PM, Howard JL, Shelton KL, Carroll FI (2005) Differential effects of the novel kappa opioid receptor antagonist, JDTic, on reinstatement of cocaine-seeking induced by footshock stressors vs cocaine primes and its antidepressant-like effects in rats. Psychopharmacology 183:118–126
Beardsley PM, Pollard GT, Howard JL, Carroll FI (2010) Effectiveness of analogs of the kappa opioid receptor antagonist (3R)-7-hydroxy-N-((1S)-1-{[(3R,4R)-4-(3-hydroxyphenyl)-3,4-dimethyl-1-piperidinyl]methyl}-2-methylpropyl)-1,2,3,4-tetrahydro-3-isoquinolinecarboxamide (JDTic) to reduce U50,488-induced diuresis and stress-induced cocaine reinstatement in rats. Psychopharmacology 210:189–198
Beaulieu S, Di Paolo T, Cote J, Barden N (1987) Participation of the central amygdaloid nucleus in the response of adrenocorticotropin secretion to immobilization stress: opposing roles of the noradrenergic and dopaminergic systems. Neuroendocrinology 45:37–46
Beck LH, Bransome ED Jr, Mirsky AF, Rosvold HE, Sarason I (1956) A continuous performance test of brain damage. J Consult Psychol 20:343–350
Belcheva MM, Clark AL, Haas PD, Serna JS, Hahn JW, Kiss A, Coscia CJ (2005) Mu and kappa opioid receptors activate ERK/MAPK via different protein kinase C isoforms and secondary messengers in astrocytes. J Biol Chem 280:27662–27669
Belcheva MM, Vogel Z, Ignatova E, Avidor-Reiss T, Zippel R, Levy R, Young EC, Barg J, Coscia CJ (1998) Opioid modulation of extracellular signal-regulated protein kinase activity is ras-dependent and involves Gbetagamma subunits. J Neurochem 70:635–645
Berton O, McClung CA, Dileone RJ, Krishnan V, Renthal W, Russo SJ, Graham D, Tsankova NM, Bolanos CA, Rios M, Monteggia LM, Self DW, Nestler EJ (2006) Essential role of BDNF in the mesolimbic dopamine pathway in social defeat stress. Science 311:864–868
Bilkei-Gorzo A, Racz I, Michel K, Mauer D, Zimmer A, Klingmuller D (2008) Control of hormonal stress reactivity by the endogenous opioid system. Psychoneuroendocrinology 33:425–436
Bodkin JA, Zornberg GL, Lukas SE, Cole JO (1995) Buprenorphine treatment of refractory depression. J Clin Psychopharmacol 15:49–57
Bohn LM, Belcheva MM, Coscia CJ (2000) Mitogenic signaling via endogenous kappa-opioid receptors in C6 glioma cells: evidence for the involvement of protein kinase C and the mitogen-activated protein kinase signaling cascade. J Neurochem 74:564–573
Bremner JD, Licinio J, Darnell A, Krystal JH, Owens MJ, Southwick SM, Nemeroff CB, Charney DS (1997) Elevated CSF corticotropin-releasing factor concentrations in posttraumatic stress disorder. Am J Psychiatry 154:624–629
Britton DR, Koob GF, Rivier J, Vale W (1982) Intraventricular corticotropin-releasing factor enhances behavioral effects of novelty. Life Sci 31:363–367
Britton KT, Lee G, Vale W, Rivier J, Koob GF (1986) Corticotropin releasing factor (CRF) receptor antagonist blocks activating and 'anxiogenic' actions of CRF in the rat. Brain Res 369:303–306
Brown PJ, Wolfe J (1994) Substance abuse and post-traumatic stress disorder comorbidity. Drug Alcohol Depend 35:51–59
Bruchas MR, Chavkin C (2010) Kinase cascades and ligand-directed signaling at the kappa opioid receptor. Psychopharmacology 210:137–147
Bruchas MR, Land BB, Aita M, Xu M, Barot SK, Li S, Chavkin C (2007a) Stress-induced p38 mitogen-activated protein kinase activation mediates kappa-opioid-dependent dysphoria. J Neurosci 27:11614–11623
Bruchas MR, Land BB, Lemos JC, Chavkin C (2009) CRF1-R activation of the dynorphin/kappa opioid system in the mouse basolateral amygdala mediates anxiety-like behavior. PLoS One 4:e8528
Bruchas MR, Macey TA, Lowe JD, Chavkin C (2006) Kappa opioid receptor activation of p38 MAPK is GRK3- and arrestin-dependent in neurons and astrocytes. J Biol Chem 281:18081–18089
Bruchas MR, Schindler AG, Shankar H, Messinger DI, Miyatake M, Land BB, Lemos JC, Hagan CE, Neumaier JF, Quintana A, Palmiter RD, Chavkin C (2011) Selective p38alpha MAPK deletion in serotonergic neurons produces stress resilience in models of depression and addiction. Neuron 71:498–511
Bruchas MR, Yang T, Schreiber S, Defino M, Kwan SC, Li S, Chavkin C (2007b) Long-acting kappa opioid antagonists disrupt receptor signaling and produce noncompetitive effects by activating c-Jun N-terminal kinase. J Biol Chem 282:29803–29811
Bruijnzeel AW (2009) kappa-Opioid receptor signaling and brain reward function. Brain Res Rev 62:127–146
Buckingham JC, Cooper TA (1986) Pharmacological characterization of opioid receptors influencing the secretion of corticotrophin releasing factor in the rat. Neuroendocrinology 44:36–40
Buydens-Branchey L, Noumair D, Branchey M (1990) Duration and intensity of combat exposure and posttraumatic stress disorder in Vietnam veterans. J Nerv Ment Dis 178:582–587
Calogero AE, Scaccianoce S, Burrello N, Nicolai R, Muscolo LA, Kling MA, Angelucci L, D’Agata R (1996) The kappa-opioid receptor agonist MR-2034 stimulates the rat hypothalamic–pituitary–adrenal axis: studies in vivo and in vitro. J Neuroendocrinol 8:579–585
Campeau S, Liberzon I, Morilak D, Ressler K (2011) Stress modulation of cognitive and affective processes. Stress 14:503–519
Canteras NS, Simerly RB, Swanson LW (1995) Organization of projections from the medial nucleus of the amygdala: a PHAL study in the rat. J Comp Neurol 360:213–245
Carey AN, Borozny K, Aldrich JV, McLaughlin JP (2007) Reinstatement of cocaine place-conditioning prevented by the peptide kappa-opioid receptor antagonist arodyn. Eur J Pharmacol 569:84–89
Carey AN, Lyons AM, Shay CF, Dunton O, McLaughlin JP (2009) Endogenous kappa opioid activation mediates stress-induced deficits in learning and memory. J Neurosci 29:4293–4300
Carlezon WA Jr, Beguin C, DiNieri JA, Baumann MH, Richards MR, Todtenkopf MS, Rothman RB, Ma Z, Lee DY, Cohen BM (2006) Depressive-like effects of the kappa-opioid receptor agonist salvinorin A on behavior and neurochemistry in rats. J Pharmacol Exp Ther 316:440–447
Carlezon WA Jr, Duman RS, Nestler EJ (2005) The many faces of CREB. Trends Neurosci 28:436–445
Carlezon WA Jr, Thomas MJ (2009) Biological substrates of reward and aversion: a nucleus accumbens activity hypothesis. Neuropharmacology 56(Suppl 1):122–132
Carlezon WA Jr, Thome J, Olson VG, Lane-Ladd SB, Brodkin ES, Hiroi N, Duman RS, Neve RL, Nestler EJ (1998) Regulation of cocaine reward by CREB. Science 282:2272–2275
Carr GV, Bangasser DA, Bethea T, Young M, Valentino RJ, Lucki I (2010) Antidepressant-like effects of kappa-opioid receptor antagonists in Wistar Kyoto rats. Neuropsychopharmacol 35:752–763
Carr GV, Lucki I (2010) Comparison of the kappa-opioid receptor antagonist DIPPA in tests of anxiety-like behavior between Wistar Kyoto and Sprague Dawley rats. Psychopharmacology 210:295–302
Carrasco GA, Van de Kar LD (2003) Neuroendocrine pharmacology of stress. Eur J Pharmacol 463:235–272
Carroll FI, Carlezon WA Jr (2013) Development of kappa opioid receptor antagonists. J Med Chem 56:2178–2195
Chartoff E, Sawyer A, Rachlin A, Potter D, Pliakas A, Carlezon WA (2012) Blockade of kappa opioid receptors attenuates the development of depressive-like behaviors induced by cocaine withdrawal in rats. Neuropharmacology 62:167–176
Chaudhury D, Walsh JJ, Friedman AK, Juarez B, Ku SM, Koo JW, Ferguson D, Tsai HC, Pomeranz L, Christoffel DJ, Nectow AR, Ekstrand M, Domingos A, Mazei-Robison MS, Mouzon E, Lobo MK, Neve RL, Friedman JM, Russo SJ, Deisseroth K, Nestler EJ, Han MH (2013) Rapid regulation of depression-related behaviours by control of midbrain dopamine neurons. Nature 493:532–536
Chavkin C, James IF, Goldstein A (1982) Dynorphin is a specific endogenous ligand of the kappa opioid receptor. Science 215:413–415
Chrousos GP, Gold PW (1992) The concepts of stress and stress system disorders. Overview of physical and behavioral homeostasis. JAMA: J Am Med Assoc 267:1244–1252
Cintra A, Zoli M, Rosen L, Agnati LF, Okret S, Wikstrom AC, Gustaffsson JA, Fuxe K (1994) Mapping and computer assisted morphometry and microdensitometry of glucocorticoid receptor immunoreactive neurons and glial cells in the rat central nervous system. Neuroscience 62:843–897
Cole RL, Konradi C, Douglass J, Hyman SE (1995) Neuronal adaptation to amphetamine and dopamine: molecular mechanisms of prodynorphin gene regulation in rat striatum. Neuron 14:813–823
Colombo PJ, Martinez JL Jr, Bennett EL, Rosenzweig MR (1992) Kappa opioid receptor activity modulates memory for peck-avoidance training in the 2-day-old chick. Psychopharmacology 108:235–240
Crabbe JC, Wahlsten D, Dudek BC (1999) Genetics of mouse behavior: interactions with laboratory environment. Science 284:1670–1672
Crane JW, Ebner K, Day TA (2003) Medial prefrontal cortex suppression of the hypothalamic–pituitary–adrenal axis response to a physical stressor, systemic delivery of interleukin-1beta. Eur J Neurosci 17:1473–1481
Cullinan WE, Herman JP, Watson SJ (1993) Ventral subicular interaction with the hypothalamic paraventricular nucleus: evidence for a relay in the bed nucleus of the stria terminalis. J Comp Neurol 332:1–20
Dalman FC, O’Malley KL (1999) kappa-Opioid tolerance and dependence in cultures of dopaminergic midbrain neurons. J Neurosci 19:5750–5757
Daumas S, Betourne A, Halley H, Wolfer DP, Lipp HP, Lassalle JM, Frances B (2007) Transient activation of the CA3 Kappa opioid system in the dorsal hippocampus modulates complex memory processing in mice. Neurobiol Learn Mem 88:94–103
Dautzenberg FM, Hauger RL (2002) The CRF peptide family and their receptors: yet more partners discovered. Trends Pharmacol Sci 23:71–77
Davis M (1997) Neurobiology of fear responses: the role of the amygdala. J Neuropsychiatry Clin Neurosci 9:382–402
Davis M, Shi C (2000) The amygdala. Curr Biol 10:R131
de Kloet CS, Vermetten E, Geuze E, Lentjes EG, Heijnen CJ, Stalla GK, Westenberg HG (2008) Elevated plasma corticotrophin-releasing hormone levels in veterans with posttraumatic stress disorder. Progress Brain Res 167:287–291
De Souza EB (1995) Corticotropin-releasing factor receptors: physiology, pharmacology, biochemistry and role in central nervous system and immune disorders. Psychoneuroendocrinology 20:789–819
De Souza EB, Insel TR, Perrin MH, Rivier J, Vale WW, Kuhar MJ (1985) Corticotropin-releasing factor receptors are widely distributed within the rat central nervous system: an autoradiographic study. J Neurosci 5:3189–3203
Deak T, Nguyen KT, Ehrlich AL, Watkins LR, Spencer RL, Maier SF, Licinio J, Wong ML, Chrousos GP, Webster E, Gold PW (1999) The impact of the nonpeptide corticotropin-releasing hormone antagonist antalarmin on behavioral and endocrine responses to stress. Endocrinology 140:79–86
Detke MJ, Rickels M, Lucki I (1995) Active behaviors in the rat forced swimming test differentially produced by serotonergic and noradrenergic antidepressants. Psychopharmacology 121:66–72
Di Chiara G, Imperato A (1988) Opposite effects of mu and kappa opiate agonists on dopamine release in the nucleus accumbens and in the dorsal caudate of freely moving rats. J Pharmacol Exp Ther 244:1067–1080
Dinieri JA, Nemeth CL, Parsegian A, Carle T, Gurevich VV, Gurevich E, Neve RL, Nestler EJ, Carlezon WA Jr (2009) Altered sensitivity to rewarding and aversive drugs in mice with inducible disruption of cAMP response element-binding protein function within the nucleus accumbens. J Neurosci 29:1855–1859
Dong HW, Petrovich GD, Watts AG, Swanson LW (2001) Basic organization of projections from the oval and fusiform nuclei of the bed nuclei of the stria terminalis in adult rat brain. J Comp Neurol 436:430–455
Dong HW, Swanson LW (2004) Projections from bed nuclei of the stria terminalis, posterior division: implications for cerebral hemisphere regulation of defensive and reproductive behaviors. J Comp Neurol 471:396–433
Douglass J, McKinzie AA, Pollock KM (1994) Identification of multiple DNA elements regulating basal and protein kinase A-induced transcriptional expression of the rat prodynorphin gene. Mol Endocrinol 8:333–344
Dunn AJ, File SE (1987) Corticotropin-releasing factor has an anxiogenic action in the social interaction test. Horm Behav 21:193–202
Eans SO, Ganno ML, Reilley KJ, Patkar KA, Senadheera SN, Aldrich JV, McLaughlin JP (2013) The macrocyclic tetrapeptide [D-Trp]CJ-15,208 produces short-acting kappa opioid receptor antagonism in the CNS after oral administration. Br J Pharmacol 169:426–436
Endoh T, Matsuura H, Tanaka C, Nagase H (1992) Nor-binaltorphimine: a potent and selective kappa-opioid receptor antagonist with long-lasting activity in vivo. Arch Int Pharmacodyn Ther 316:30–42
Fallon JH, Leslie FM (1986) Distribution of dynorphin and enkephalin peptides in the rat brain. J Comp Neurol 249:293–336
Filliol D, Ghozland S, Chluba J, Martin M, Matthes HW, Simonin F, Befort K, Gaveriaux-Ruff C, Dierich A, LeMeur M, Valverde O, Maldonado R, Kieffer BL (2000) Mice deficient for delta- and mu-opioid receptors exhibit opposing alterations of emotional responses. Nat Genet 25:195–200
Foa EB, Zinbarg R, Rothbaum BO (1992) Uncontrollability and unpredictability in post-traumatic stress disorder: an animal model. Psychol Bull 112:218–238
Ford CP, Beckstead MJ, Williams JT (2007) Kappa opioid inhibition of somatodendritic dopamine inhibitory postsynaptic currents. J Neurophysiol 97:883–891
Ford CP, Mark GP, Williams JT (2006) Properties and opioid inhibition of mesolimbic dopamine neurons vary according to target location. J Neurosci 26:2788–2797
Fox HC, Bergquist KL, Hong KI, Sinha R (2007) Stress-induced and alcohol cue-induced craving in recently abstinent alcohol-dependent individuals. Alcohol Clin Exp Res 31:395–403
Fuxe K, Wikstrom AC, Okret S, Agnati LF, Harfstrand A, Yu ZY, Granholm L, Zoli M, Vale W, Gustafsson JA (1985) Mapping of glucocorticoid receptor immunoreactive neurons in the rat tel- and diencephalon using a monoclonal antibody against rat liver glucocorticoid receptor. Endocrinology 117:1803–1812
George O, Ghozland S, Azar MR, Cottone P, Zorrilla EP, Parsons LH, O’Dell LE, Richardson HN, Koob GF (2007) CRF–CRF1 system activation mediates withdrawal-induced increases in nicotine self-administration in nicotine-dependent rats. Proc Natl Acad Sci U S A 104:17198–17203
Goeders NE, Guerin GF (2000) Effects of the CRH receptor antagonist CP-154,526 on intravenous cocaine self-administration in rats. Neuropsychopharmacol 23:577–586
Gray TS, Carney ME, Magnuson DJ (1989) Direct projections from the central amygdaloid nucleus to the hypothalamic paraventricular nucleus: possible role in stress-induced adrenocorticotropin release. Neuroendocrinology 50:433–446
Graziane NM, Polter AM, Briand LA, Pierce RC, Kauer JA (2013) Kappa opioid receptors regulate stress-induced cocaine seeking and synaptic plasticity. Neuron 77:942–954
Greenwell TN, Funk CK, Cottone P, Richardson HN, Chen SA, Rice KC, Zorrilla EP, Koob GF (2009) Corticotropin-releasing factor-1 receptor antagonists decrease heroin self-administration in long- but not short-access rats. Addict Biol 14:130–143
Griebel G, Simiand J, Steinberg R, Jung M, Gully D, Roger P, Geslin M, Scatton B, Maffrand JP, Soubrie P (2002) 4-(2-Chloro-4-methoxy-5-methylphenyl)-N-[(1S)-2-cyclopropyl-1-(3-fluoro-4-methylphenyl)ethyl]5-methyl-N-(2-propynyl)-1,3-thiazol-2-amine hydrochloride (SSR125543A), a potent and selective corticotrophin-releasing factor(1) receptor antagonist. II. Characterization in rodent models of stress-related disorders. J Pharmacol Exp Ther 301:333–345
Grilli M, Neri E, Zappettini S, Massa F, Bisio A, Romussi G, Marchi M, Pittaluga A (2009) Salvinorin A exerts opposite presynaptic controls on neurotransmitter exocytosis from mouse brain nerve terminals. Neuropharmacology 57:523–530
Gross RA, Moises HC, Uhler MD, Macdonald RL (1990) Dynorphin A and cAMP-dependent protein kinase independently regulate neuronal calcium currents. Proc Natl Acad Sci U S A 87:7025–7029
Habib KE, Weld KP, Rice KC, Pushkas J, Champoux M, Listwak S, Webster EL, Atkinson AJ, Schulkin J, Contoreggi C, Chrousos GP, McCann SM, Suomi SJ, Higley JD, Gold PW (2000) Oral administration of a corticotropin-releasing hormone receptor antagonist significantly attenuates behavioral, neuroendocrine, and autonomic responses to stress in primates. Proc Natl Acad Sci U S A 97:6079–6084
Hauger RL, Risbrough V, Oakley RH, Olivares-Reyes JA, Dautzenberg FM (2009) Role of CRF receptor signaling in stress vulnerability, anxiety, and depression. Ann N Y Acad Sci 1179:120–143
Hayes AG, Stewart BR (1985) Effect of mu and kappa opioid receptor agonists on rat plasma corticosterone levels. Eur J Pharmacol 116:75–79
Heinrichs SC, Menzaghi F, Schulteis G, Koob GF, Stinus L (1995) Suppression of corticotropin-releasing factor in the amygdala attenuates aversive consequences of morphine withdrawal. Behav Pharmacol 6:74–80
Henry DJ, Grandy DK, Lester HA, Davidson N, Chavkin C (1995) Kappa-opioid receptors couple to inwardly rectifying potassium channels when coexpressed by Xenopus oocytes. Mol Pharmacol 47:551–557
Herman JP, Ostrander MM, Mueller NK, Figueiredo H (2005) Limbic system mechanisms of stress regulation: hypothalamo-pituitary-adrenocortical axis. Prog Neuro-Psychopharmacol Biol Psychiatry 29:1201–1213
Herman JP, Schafer MK, Young EA, Thompson R, Douglass J, Akil H, Watson SJ (1989) Evidence for hippocampal regulation of neuroendocrine neurons of the hypothalamo-pituitary-adrenocortical axis. J Neurosci 9:3072–3082
Hiramatsu M, Hoshino T (2004) Involvement of kappa-opioid receptors and sigma receptors in memory function demonstrated using an antisense strategy. Brain Res 1030:247–255
Hiramatsu M, Hoshino T (2005) Improvement of memory impairment by (+)- and (−)-pentazocine via sigma, but not kappa opioid receptors. Brain Res 1057:72–80
Hiramatsu M, Hyodo T, Kameyama T (1996) U-50488H, a selective kappa-opioid receptor agonist, improves carbon monoxide-induced delayed amnesia in mice. Eur J Pharmacol 315:119–125
Hjelmstad GO, Fields HL (2003) Kappa opioid receptor activation in the nucleus accumbens inhibits glutamate and GABA release through different mechanisms. J Neurophysiol 89:2389–2395
Horan P, Taylor J, Yamamura HI, Porreca F (1992) Extremely long-lasting antagonistic actions of nor-binaltorphimine (nor-BNI) in the mouse tail-flick test. J Pharmacol Exp Ther 260:1237–1243
Hurd YL (1996) Differential messenger RNA expression of prodynorphin and proenkephalin in the human brain. Neuroscience 72:767–783
Imperato A, Cabib S, Puglisi-Allegra S (1993) Repeated stressful experiences differently affect the time-dependent responses of the mesolimbic dopamine system to the stressor. Brain Res 601:333–336
Iyengar S, Kim HS, Wood PL (1986) Kappa opiate agonists modulate the hypothalamic–pituitary–adrenocortical axis in the rat. J Pharmacol Exp Ther 238:429–436
Jackson KJ, Carroll FI, Negus SS, Damaj MI (2010) Effect of the selective kappa-opioid receptor antagonist JDTic on nicotine antinociception, reward, and withdrawal in the mouse. Psychopharmacology 210:285–294
Jackson KJ, McLaughlin JP, Carroll FI, Damaj MI (2013) Effects of the kappa opioid receptor antagonist, norbinaltorphimine, on stress and drug-induced reinstatement of nicotine-conditioned place preference in mice. Psychopharmacology 226:763–768
James IF, Fischli W, Goldstein A (1984) Opioid receptor selectivity of dynorphin gene products. J Pharmacol Exp Ther 228:88–93
Jamot L, Matthes HW, Simonin F, Kieffer BL, Roder JC (2003) Differential involvement of the mu and kappa opioid receptors in spatial learning. Genes Brain Behav 2:80–92
Janis IL, Mann L (1977) Decision making: a psychological analysis of conflict, choice, and commitment. Free Press, New York
Jones DN, Holtzman SG (1992) Long term kappa-opioid receptor blockade following nor-binaltorphimine. Eur J Pharmacol 215:345–348
Kalin NH (1990) Behavioral and endocrine studies of corticotropin-releasing hormone in primates. In: De Souza EB, Nemeroff CB (eds) Corticotropin-releasing factor: band and clinical studies of a neuropeptide. CRC, Boca Raton, pp 275–289
Kam AY, Chan AS, Wong YH (2004) Kappa-opioid receptor signals through Src and focal adhesion kinase to stimulate c-Jun N-terminal kinases in transfected COS-7 cells and human monocytic THP-1 cells. J Pharmacol Exp Ther 310:301–310
Kasckow JW, Baker D, Geracioti TD Jr (2001) Corticotropin-releasing hormone in depression and post-traumatic stress disorder. Peptides 22:845–851
Kaufman J, Charney D (2000) Comorbidity of mood and anxiety disorders. Depression Anxiety 12(Suppl 1):69–76
Keay KA, Bandler R (2001) Parallel circuits mediating distinct emotional coping reactions to different types of stress. Neurosci Biobehav Rev 25:669–678
Keeney AJ, Hogg S (1999) Behavioural consequences of repeated social defeat in the mouse: preliminary evaluation of a potential animal model of depression. Behav Pharmacol 10:753–764
Keinan G (1987) Decision making under stress: scanning of alternatives under controllable and uncontrollable threats. J Personal Soc Psychol 52:639–644
Kendler KS, Karkowski LM, Prescott CA (1999) Causal relationship between stressful life events and the onset of major depression. Am J Psychiatry 156:837–841
Kessler RC (1997) The effects of stressful life events on depression. Annu Rev Psychol 48:191–214
Kessler RC (2000) The epidemiology of pure and comorbid generalized anxiety disorder: a review and evaluation of recent research. Acta Psychiatr Scandinavica Suppl 7–13
Knoll AT, Carlezon WA Jr (2010) Dynorphin, stress, and depression. Brain Res 1314:56–73
Knoll AT, Meloni EG, Thomas JB, Carroll FI, Carlezon WA Jr (2007) Anxiolytic-like effects of kappa-opioid receptor antagonists in models of unlearned and learned fear in rats. J Pharmacol Exp Ther 323:838–845
Knoll AT, Muschamp JW, Sillivan SE, Ferguson D, Dietz DM, Meloni EG, Carroll FI, Nestler EJ, Konradi C, Carlezon WA Jr (2011) Kappa opioid receptor signaling in the basolateral amygdala regulates conditioned fear and anxiety in rats. Biol Psychiatry 70:425–433
Komatsu H, Ohara A, Sasaki K, Abe H, Hattori H, Hall FS, Uhl GR, Sora I (2011) Decreased response to social defeat stress in mu-opioid-receptor knockout mice. Pharmacol Biochem Behav 99:676–682
Konkoy CS, Childers SR (1989) Dynorphin-selective inhibition of adenylyl cyclase in guinea pig cerebellum membranes. Mol Pharmacol 36:627–633
Konkoy CS, Childers SR (1993) Relationship between kappa 1 opioid receptor binding and inhibition of adenylyl cyclase in guinea pig brain membranes. Biochem Pharmacol 45:207–216
Koob G, Kreek MJ (2007) Stress, dysregulation of drug reward pathways, and the transition to drug dependence. Am J Psychiatry 164:1149–1159
Koob GF (1999) Corticotropin-releasing factor, norepinephrine, and stress. Biol Psychiatry 46:1167–1180
Koob GF, Heinrichs SC, Pich EM, Menzaghi F, Baldwin H, Miczek K, Britton KT (1993) The role of corticotropin-releasing factor in behavioural responses to stress. CIBA Found Symp 172:277–289, discussion 290–5
Koob GF, Volkow ND (2010) Neurocircuitry of addiction. Neuropsychopharmacol 35:217–238
Kuzmin A, Madjid N, Terenius L, Ogren SO, Bakalkin G (2006) Big dynorphin, a prodynorphin-derived peptide produces NMDA receptor-mediated effects on memory, anxiolytic-like and locomotor behavior in mice. Neuropsychopharmacol 31:1928–1937
Lammel S, Lim BK, Ran C, Huang KW, Betley MJ, Tye KM, Deisseroth K, Malenka RC (2012) Input-specific control of reward and aversion in the ventral tegmental area. Nature 491:212–217
Land BB, Bruchas MR, Lemos JC, Xu M, Melief EJ, Chavkin C (2008) The dysphoric component of stress is encoded by activation of the dynorphin kappa-opioid system. J Neurosci 28:407–414
Land BB, Bruchas MR, Schattauer S, Giardino WJ, Aita M, Messinger D, Hnasko TS, Palmiter RD, Chavkin C (2009) Activation of the kappa opioid receptor in the dorsal raphe nucleus mediates the aversive effects of stress and reinstates drug seeking. Proc Natl Acad Sci U S A 106:19168–19173
Law PY, Wong YH, Loh HH (2000) Molecular mechanisms and regulation of opioid receptor signaling. Annu Rev Pharmacol Toxicol 40:389–430
Lawrence DM, Bidlack JM (1993) The kappa opioid receptor expressed on the mouse R1.1 thymoma cell line is coupled to adenylyl cyclase through a pertussis toxin-sensitive guanine nucleotide-binding regulatory protein. J Pharmacol Exp Ther 266:1678–1683
Lemos JC, Roth CA, Messinger DI, Gill HK, Phillips PE, Chavkin C (2012) Repeated stress dysregulates kappa-opioid receptor signaling in the dorsal raphe through a p38alpha MAPK-dependent mechanism. J Neurosci 32:12325–12336
Li C, Pleil KE, Stamatakis AM, Busan S, Vong L, Lowell BB, Stuber GD, Kash TL (2012) Presynaptic inhibition of gamma-aminobutyric acid release in the bed nucleus of the stria terminalis by kappa opioid receptor signaling. Biol Psychiatry 71:725–732
Liang KC, Melia KR, Miserendino MJ, Falls WA, Campeau S, Davis M (1992) Corticotropin-releasing factor: long-lasting facilitation of the acoustic startle reflex. J Neurosci 12:2303–2312
Lin S, Boey D, Lee N, Schwarzer C, Sainsbury A, Herzog H (2006) Distribution of prodynorphin mRNA and its interaction with the NPY system in the mouse brain. Neuropeptides 40:115–123
Logrip ML, Zorrilla EP, Koob GF (2012) Stress modulation of drug self-administration: implications for addiction comorbidity with post-traumatic stress disorder. Neuropharmacology 62:552–564
Mague SD, Pliakas AM, Todtenkopf MS, Tomasiewicz HC, Zhang Y, Stevens WC Jr, Jones RM, Portoghese PS, Carlezon WA Jr (2003) Antidepressant-like effects of kappa-opioid receptor antagonists in the forced swim test in rats. J Pharmacol Exp Ther 305:323–330
Mahan AL, Ressler KJ (2012) Fear conditioning, synaptic plasticity and the amygdala: implications for posttraumatic stress disorder. Trends Neurosci 35:24–35
Majzoub J (2006) Corticotropin-releasing hormone physiology. Eur J Endocrinol 155:S71–S76
Mansour A, Khachaturian H, Lewis ME, Akil H, Watson SJ (1987) Autoradiographic differentiation of mu, delta, and kappa opioid receptors in the rat forebrain and midbrain. J Neurosci 7:2445–2464
Mansour A, Khachaturian H, Lewis ME, Akil H, Watson SJ (1988) Anatomy of CNS opioid receptors. Trends Neurosci 11:308–314
Marchant NJ, Li X, Shaham Y (2013) Recent developments in animal models of drug relapse. Curr Opin Neurobiol S0959–4388(13)00019–6. doi:10.1016/j.conb.2013.01.003
Margolis EB, Hjelmstad GO, Bonci A, Fields HL (2003) Kappa-opioid agonists directly inhibit midbrain dopaminergic neurons. J Neurosci 23:9981–9986
Margolis EB, Hjelmstad GO, Bonci A, Fields HL (2005) Both kappa and mu opioid agonists inhibit glutamatergic input to ventral tegmental area neurons. J Neurophysiol 93:3086–3093
Margolis EB, Lock H, Chefer VI, Shippenberg TS, Hjelmstad GO, Fields HL (2006) Kappa opioids selectively control dopaminergic neurons projecting to the prefrontal cortex. Proc Natl Acad Sci U S A 103:2938–2942
Margolis EB, Mitchell JM, Ishikawa J, Hjelmstad GO, Fields HL (2008) Midbrain dopamine neurons: projection target determines action potential duration and dopamine D(2) receptor inhibition. J Neurosci 28:8908–8913
McLaughlin JP, Land BB, Li S, Pintar JE, Chavkin C (2006a) Prior activation of kappa opioid receptors by U50,488 mimics repeated forced swim stress to potentiate cocaine place preference conditioning. Neuropsychopharmacol 31:787–794
McLaughlin JP, Li S, Valdez J, Chavkin TA, Chavkin C (2006b) Social defeat stress-induced behavioral responses are mediated by the endogenous kappa opioid system. Neuropsychopharmacol 31:1241–1248
McLaughlin JP, Marton-Popovici M, Chavkin C (2003a) Kappa opioid receptor antagonism and prodynorphin gene disruption block stress-induced behavioral responses. J Neurosci 23:5674–5683
McLaughlin JP, Xu M, Mackie K, Chavkin C (2003b) Phosphorylation of a carboxyl-terminal serine within the kappa-opioid receptor produces desensitization and internalization. J Biol Chem 278:34631–34640
McNamara RK, Strawn JR, Chang KD, DelBello MP (2012) Interventions for youth at high risk for bipolar disorder and schizophrenia. Child Adolesc Psychiatr Clin N Am 21:739–751
Meng F, Xie GX, Thompson RC, Mansour A, Goldstein A, Watson SJ, Akil H (1993) Cloning and pharmacological characterization of a rat kappa opioid receptor. Proc Natl Acad Sci U S A 90:9954–9958
Merchenthaler I (1984) Corticotropin releasing factor (CRF)-like immunoreactivity in the rat central nervous system. Extrahypothalamic distribution. Peptides 5(Suppl 1):53–69
Merchenthaler I, Maderdrut JL, Cianchetta P, Shughrue P, Bronstein D (1997) In situ hybridization histochemical localization of prodynorphin messenger RNA in the central nervous system of the rat. J Comp Neurol 384:211–232
Miczek KA, Covington HE 3rd, Nikulina EM Jr, Hammer RP (2004) Aggression and defeat: persistent effects on cocaine self-administration and gene expression in peptidergic and aminergic mesocorticolimbic circuits. Neurosci Biobehav Rev 27:787–802
Miczek KA, Yap JJ, Covington HE 3rd (2008) Social stress, therapeutics and drug abuse: preclinical models of escalated and depressed intake. Pharmacol Ther 120:102–128
Millan MA, Jacobowitz DM, Hauger RL, Catt KJ, Aguilera G (1986) Distribution of corticotropin-releasing factor receptors in primate brain. Proc Natl Acad Sci U S A 83:1921–1925
Moreau JL, Jenck F, Martin JR, Mortas P, Haefely WE (1992) Antidepressant treatment prevents chronic unpredictable mild stress-induced anhedonia as assessed by ventral tegmentum self-stimulation behavior in rats. Eur Neuropsychopharmacol J Eur Coll Neuropsychopharmacol 2:43–49
Morimoto M, Morita N, Ozawa H, Yokoyama K, Kawata M (1996) Distribution of glucocorticoid receptor immunoreactivity and mRNA in the rat brain: an immunohistochemical and in situ hybridization study. Neurosci Res 26:235–269
Morris BJ, Haarmann I, Kempter B, Hollt V, Herz A (1986) Localization of prodynorphin messenger RNA in rat brain by in situ hybridization using a synthetic oligonucleotide probe. Neurosci Lett 69:104–108
Morris SE, Cuthbert BN (2012) Research domain criteria: cognitive systems, neural circuits, and dimensions of behavior. Dialogues Clin Neurosci 14:29–37
Munck A, Guyre PM, Holbrook NJ (1984) Physiological functions of glucocorticoids in stress and their relation to pharmacological actions. Endocr Rev 5:25–44
Muschamp JW, Carlezon WA, Jr (2013) Roles of nucleus accumbens CREB and dynorphin in dysregulation of motivation. Cold Spring Harbor Perspect Med 3: a012005
Muschamp JW, Van’t Veer A, Parsegian A, Gallo MS, Chen M, Neve RL, Meloni EG, Carlezon WA Jr (2011) Activation of CREB in the nucleus accumbens shell produces anhedonia and resistance to extinction of fear in rats. J Neurosci 31:3095–3103
Nemeroff CB (1992) New vistas in neuropeptide research in neuropsychiatry: focus on corticotropin-releasing factor. Neuropsychopharmacol 6:69–75
Nemeroff CB, Widerlov E, Bissette G, Walleus H, Karlsson I, Eklund K, Kilts CD, Loosen PT, Vale W (1984) Elevated concentrations of CSF corticotropin-releasing factor-like immunoreactivity in depressed patients. Science 226:1342–1344
Nestler EJ, Carlezon WA Jr (2006) The mesolimbic dopamine reward circuit in depression. Biol Psychiatry 59:1151–1159
Newton SS, Thome J, Wallace TL, Shirayama Y, Schlesinger L, Sakai N, Chen J, Neve R, Nestler EJ, Duman RS (2002) Inhibition of cAMP response element-binding protein or dynorphin in the nucleus accumbens produces an antidepressant-like effect. J Neurosci 22:10883–10890
Nguyen XV, Masse J, Kumar A, Vijitruth R, Kulik C, Liu M, Choi DY, Foster TC, Usynin I, Bakalkin G, Bing G (2005) Prodynorphin knockout mice demonstrate diminished age-associated impairment in spatial water maze performance. Behav Brain Res 161:254–262
Nikolarakis K, Pfeiffer A, Stalla GK, Herz A (1987) The role of CRF in the release of ACTH by opiate agonists and antagonists in rats. Brain Res 421:373–376
Nikolarakis KE, Almeida OF, Herz A (1986) Stimulation of hypothalamic beta-endorphin and dynorphin release by corticotropin-releasing factor (in vitro). Brain Res 399:152–155
Nikoshkov A, Hurd YL, Yakovleva T, Bazov I, Marinova Z, Cebers G, Pasikova N, Gharibyan A, Terenius L, Bakalkin G (2005) Prodynorphin transcripts and proteins differentially expressed and regulated in the adult human brain. FASEB J 19:1543–1545
Owens MJ, Vargas MA, Nemeroff CB (1993) The effects of alprazolam on corticotropin-releasing factor neurons in the rat brain: implications for a role for CRF in the pathogenesis of anxiety disorders. J Psychiatr Res 27(Suppl 1):209–220
Paris JJ, Reilley KJ, McLaughlin JP (2011) Kappa opioid receptor-mediated disruption of novel object recognition: relevance for psychostimulant treatment. J Addict Res Ther S4
Pascoe JE, Williams KL, Mukhopadhyay P, Rice KC, Woods JH, Ko MC (2008) Effects of mu, kappa, and delta opioid receptor agonists on the function of hypothalamic–pituitary–adrenal axis in monkeys. Psychoneuroendocrinology 33:478–486
Pascucci T, Ventura R, Latagliata EC, Cabib S, Puglisi-Allegra S (2007) The medial prefrontal cortex determines the accumbens dopamine response to stress through the opposing influences of norepinephrine and dopamine. Cereb Cortex 17:2796–2804
Peng J, Sarkar S, Chang SL (2012) Opioid receptor expression in human brain and peripheral tissues using absolute quantitative real-time RT-PCR. Drug Alcohol Depend 124:223–228
Perrine SA, Hoshaw BA, Unterwald EM (2006) Delta opioid receptor ligands modulate anxiety-like behaviors in the rat. Br J Pharmacol 147:864–872
Pezze MA, Feldon J (2004) Mesolimbic dopaminergic pathways in fear conditioning. Prog Neurobiol 74:301–320
Pfeiffer A, Brantl V, Herz A, Emrich HM (1986) Psychotomimesis mediated by kappa opiate receptors. Science 233:774–776
Piazza PV, Le Moal M (1998) The role of stress in drug self-administration. Trends Pharmacol Sci 19:67–74
Pickel VM, Chan J, Sesack SR (1993) Cellular substrates for interactions between dynorphin terminals and dopamine dendrites in rat ventral tegmental area and substantia nigra. Brain Res 602:275–289
Pine DS, Cohen P, Johnson JG, Brook JS (2002) Adolescent life events as predictors of adult depression. J Affect Disord 68:49–57
Pittenger C, Duman RS (2008) Stress, depression, and neuroplasticity: a convergence of mechanisms. Neuropsychopharmacol 33:88–109
Platt JE, Stone EA (1982) Chronic restraint stress elicits a positive antidepressant response on the forced swim test. Eur J Pharmacol 82:179–181
Pliakas AM, Carlson RR, Neve RL, Konradi C, Nestler EJ, Carlezon WA Jr (2001) Altered responsiveness to cocaine and increased immobility in the forced swim test associated with elevated cAMP response element-binding protein expression in nucleus accumbens. J Neurosci 21:7397–7403
Porsolt RD, Le Pichon M, Jalfre M (1977) Depression: a new animal model sensitive to antidepressant treatments. Nature 266:730–732
Potter DN, Damez-Werno D, Carlezon WA Jr, Cohen BM, Chartoff EH (2011) Repeated exposure to the kappa-opioid receptor agonist salvinorin A modulates extracellular signal-regulated kinase and reward sensitivity. Biol Psychiatry 70:744–753
Putnam FW (2013) The role of abusive states of being in interrogation. J Trauma Dissociation 14:147–158
Rassnick S, Heinrichs SC, Britton KT, Koob GF (1993) Microinjection of a corticotropin-releasing factor antagonist into the central nucleus of the amygdala reverses anxiogenic-like effects of ethanol withdrawal. Brain Res 605:25–32
Redila VA, Chavkin C (2008) Stress-induced reinstatement of cocaine seeking is mediated by the kappa opioid system. Psychopharmacology 200:59–70
Reul JM, de Kloet ER (1985) Two receptor systems for corticosterone in rat brain: microdistribution and differential occupation. Endocrinology 117:2505–2511
Robbins TW (2002) The 5-choice serial reaction time task: behavioural pharmacology and functional neurochemistry. Psychopharmacology 163:362–380
Ronken E, Van Muiswinkel FL, Mulder AH, Schoffelmeer AN (1993) Opioid receptor-mediated inhibition of evoked catecholamine release from cultured neurons of rat ventral mesencephalon and locus coeruleus. Eur J Pharmacol 230:349–355
Rusin KI, Giovannucci DR, Stuenkel EL, Moises HC (1997) Kappa-opioid receptor activation modulates Ca2+ currents and secretion in isolated neuroendocrine nerve terminals. J Neurosci 17:6565–6574
Russo SJ, Murrough JW, Han MH, Charney DS, Nestler EJ (2012) Neurobiology of resilience. Nat Neurosci 15:1475–1484
Sakanaka M, Shibasaki T, Lederis K (1987) Corticotropin releasing factor-like immunoreactivity in the rat brain as revealed by a modified cobalt-glucose oxidase-diaminobenzidine method. J Comp Neurol 260:256–298
Salamone JD (1994) The involvement of nucleus accumbens dopamine in appetitive and aversive motivation. Behav Brain Res 61:117–133
Sapolsky RM (1996) Stress, glucocorticoids, and damage to the nervous system: the current state of confusion. Stress 1:1–19
Sarnyai Z, Biro E, Gardi J, Vecsernyes M, Julesz J, Telegdy G (1995) Brain corticotropin-releasing factor mediates 'anxiety-like' behavior induced by cocaine withdrawal in rats. Brain Res 675:89–97
Schindler AG, Li S, Chavkin C (2010) Behavioral stress may increase the rewarding valence of cocaine-associated cues through a dynorphin/kappa-opioid receptor-mediated mechanism without affecting associative learning or memory retrieval mechanisms. Neuropsychopharmacol 35:1932–1942
Schindler AG, Messinger DI, Smith JS, Shankar H, Gustin RM, Schattauer SS, Lemos JC, Chavkin NW, Hagan CE, Neumaier JF, Chavkin C (2012) Stress produces aversion and potentiates cocaine reward by releasing endogenous dynorphins in the ventral striatum to locally stimulate serotonin reuptake. J Neurosci 32:17582–17596
Schwaber JS, Kapp BS, Higgins GA, Rapp PR (1982) Amygdaloid and basal forebrain direct connections with the nucleus of the solitary tract and the dorsal motor nucleus. J Neurosci 2:1424–1438
Schwarzer C (2009) 30 years of dynorphins—new insights on their functions in neuropsychiatric diseases. Pharmacol Ther 123:353–370
Shaham Y, Singer JE, Schaeffer MH (1992) Stability/instability of cognitive strategies across tasks determine whether stress will affect judgmental processes. J Appl Soc Psychol 22:691–713
Shaham Y, Stewart J (1995) Stress reinstates heroin-seeking in drug-free animals: an effect mimicking heroin, not withdrawal. Psychopharmacology 119:334–341
Shirayama Y, Ishida H, Iwata M, Hazama GI, Kawahara R, Duman RS (2004) Stress increases dynorphin immunoreactivity in limbic brain regions and dynorphin antagonism produces antidepressant-like effects. J Neurochem 90:1258–1268
Simmons ML, Chavkin C (1996) k-Opioid receptor activation of a dendrotoxin-sensitive potassium channel mediates presynaptic inhibition of mossy fiber neurotransmitter release. Mol Pharmacol 50:80–85
Simonin F, Gaveriaux-Ruff C, Befort K, Matthes H, Lannes B, Micheletti G, Mattei MG, Charron G, Bloch B, Kieffer B (1995) kappa-Opioid receptor in humans: cDNA and genomic cloning, chromosomal assignment, functional expression, pharmacology, and expression pattern in the central nervous system. Proc Natl Acad Sci U S A 92:7006–7010
Simonin F, Valverde O, Smadja C, Slowe S, Kitchen I, Dierich A, Le Meur M, Roques BP, Maldonado R, Kieffer BL (1998) Disruption of the kappa-opioid receptor gene in mice enhances sensitivity to chemical visceral pain, impairs pharmacological actions of the selective kappa-agonist U-50,488H and attenuates morphine withdrawal. EMBO J 17:886–897
Sirinathsinghji DJ, Nikolarakis KE, Herz A (1989) Corticotropin-releasing factor stimulates the release of methionine-enkephalin and dynorphin from the neostriatum and globus pallidus of the rat: in vitro and in vivo studies. Brain Res 490:276–291
Slattery DA, Uschold N, Magoni M, Bar J, Popoli M, Neumann ID, Reber SO (2012) Behavioural consequences of two chronic psychosocial stress paradigms: anxiety without depression. Psychoneuroendocrinology 37:702–714
Smith GW, Aubry JM, Dellu F, Contarino A, Bilezikjian LM, Gold LH, Chen R, Marchuk Y, Hauser C, Bentley CA, Sawchenko PE, Koob GF, Vale W, Lee KF (1998) Corticotropin releasing factor receptor 1-deficient mice display decreased anxiety, impaired stress response, and aberrant neuroendocrine development. Neuron 20:1093–1102
Smith JS, Schindler AG, Martinelli E, Gustin RM, Bruchas MR, Chavkin C (2012) Stress-induced activation of the dynorphin/kappa-opioid receptor system in the amygdala potentiates nicotine conditioned place preference. J Neurosci 32:1488–1495
Song ZH, Takemori AE (1992) Stimulation by corticotropin-releasing factor of the release of immunoreactive dynorphin A from mouse spinal cords in vitro. Eur J Pharmacol 222:27–32
Sonuga-Barke EJ, Koerting J, Smith E, McCann DC, Thompson M (2011) Early detection and intervention for attention-deficit/hyperactivity disorder. Expert Rev Neurother 11:557–563
Spanagel R, Herz A, Shippenberg TS (1992) Opposing tonically active endogenous opioid systems modulate the mesolimbic dopaminergic pathway. Proc Natl Acad Sci U S A 89:2046–2050
Specio SE, Wee S, O’Dell LE, Boutrel B, Zorrilla EP, Koob GF (2008) CRF(1) receptor antagonists attenuate escalated cocaine self-administration in rats. Psychopharmacology 196:473–482
Sperling RE, Gomes SM, Sypek EI, Carey AN, McLaughlin JP (2010) Endogenous kappa-opioid mediation of stress-induced potentiation of ethanol-conditioned place preference and self-administration. Psychopharmacology 210:199–209
Spiess J, Rivier J, Rivier C, Vale W (1981) Primary structure of corticotropin-releasing factor from ovine hypothalamus. Proc Natl Acad Sci U S A 78:6517–6521
Stenzel-Poore MP, Heinrichs SC, Rivest S, Koob GF, Vale WW (1994) Overproduction of corticotropin-releasing factor in transgenic mice: a genetic model of anxiogenic behavior. J Neurosci 14:2579–2584
Strome EM, Wheler GH, Higley JD, Loriaux DL, Suomi SJ, Doudet DJ (2002) Intracerebroventricular corticotropin-releasing factor increases limbic glucose metabolism and has social context-dependent behavioral effects in nonhuman primates. Proc Natl Acad Sci U S A 99:15749–15754
Swanson LW (1982) The projections of the ventral tegmental area and adjacent regions: a combined fluorescent retrograde tracer and immunofluorescence study in the rat. Brain Res Bull 9:321–353
Swanson LW, Sawchenko PE, Rivier J, Vale WW (1983) Organization of ovine corticotropin-releasing factor immunoreactive cells and fibers in the rat brain: an immunohistochemical study. Neuroendocrinology 36:165–186
Swanson LW, Simmons DM (1989) Differential steroid hormone and neural influences on peptide mRNA levels in CRH cells of the paraventricular nucleus: a hybridization histochemical study in the rat. J Comp Neurol 285:413–435
Swiergiel AH, Leskov IL, Dunn AJ (2008) Effects of chronic and acute stressors and CRF on depression-like behavior in mice. Behav Brain Res 186:32–40
Takahashi M, Senda T, Tokuyama S, Kaneto H (1990) Further evidence for the implication of a kappa-opioid receptor mechanism in the production of psychological stress-induced analgesia. Japan J Pharmacol 53:487–494
Tallent M, Dichter MA, Bell GI, Reisine T (1994) The cloned kappa opioid receptor couples to an N-type calcium current in undifferentiated PC-12 cells. Neuroscience 63:1033–1040
Tejeda HA, Counotte DS, Oh E, Ramamoorthy S, Schultz-Kuszak KN, Backman CM, Chefer V, O’Donnell P, Shippenberg TS (2013) Prefrontal cortical kappa-opioid receptor modulation of local neurotransmission and conditioned place aversion. Neuropsychopharmacology. doi:10.1038/npp.2013.76
Thierry AM, Tassin JP, Blanc G, Glowinski J (1976) Selective activation of mesocortical DA system by stress. Nature 263:242–244
Thomas JB, Fix SE, Rothman RB, Mascarella SW, Dersch CM, Cantrell BE, Zimmerman DM, Carroll FI (2004) Importance of phenolic address groups in opioid kappa receptor selective antagonists. J Med Chem 47:1070–1073
Timpl P, Spanagel R, Sillaber I, Kresse A, Reul JM, Stalla GK, Blanquet V, Steckler T, Holsboer F, Wurst W (1998) Impaired stress response and reduced anxiety in mice lacking a functional corticotropin-releasing hormone receptor 1. Nat Genet 19:162–166
Todtenkopf MS, Marcus JF, Portoghese PS, Carlezon WA Jr (2004) Effects of kappa-opioid receptor ligands on intracranial self-stimulation in rats. Psychopharmacology 172:463–470
Tomasiewicz HC, Todtenkopf MS, Chartoff EH, Cohen BM, Carlezon WA Jr (2008) The kappa-opioid agonist U69,593 blocks cocaine-induced enhancement of brain stimulation reward. Biol Psychiatry 64:982–988
Tsigos C, Chrousos GP (2002) Hypothalamic-pituitary-adrenal axis, neuroendocrine factors and stress. J Psychosom Res 53:865–871
Turgeon SM, Pollack AE, Fink JS (1997) Enhanced CREB phosphorylation and changes in c-Fos and FRA expression in striatum accompany amphetamine sensitization. Brain Res 749:120–126
Tye KM, Mirzabekov JJ, Warden MR, Ferenczi EA, Tsai HC, Finkelstein J, Kim SY, Adhikari A, Thompson KR, Andalman AS, Gunaydin LA, Witten IB, Deisseroth K (2013) Dopamine neurons modulate neural encoding and expression of depression-related behaviour. Nature 493:537–541
Ur E, Wright DM, Bouloux PM, Grossman A (1997) The effects of spiradoline (U-62066E), a kappa-opioid receptor agonist, on neuroendocrine function in man. Br J Pharmacol 120:781–784
Valdez GR, Harshberger E (2012) Kappa opioid regulation of anxiety-like behavior during acute ethanol withdrawal. Pharmacol Biochem Behav 102:44–47
Vale W, Spiess J, Rivier C, Rivier J (1981) Characterization of a 41-residue ovine hypothalamic peptide that stimulates secretion of corticotropin and beta-endorphin. Science 213:1394–1397
Valenti O, Lodge DJ, Grace AA (2011) Aversive stimuli alter ventral tegmental area dopamine neuron activity via a common action in the ventral hippocampus. J Neurosci 31:4280–4289
Van’t Veer A, Bechtholt AJ, Onvani S, Potter D, Wang Y, Liu-Chen LY, Schutz G, Chartoff EH, Rudolph U, Cohen BM, Carlezon WA Jr (2013) Ablation of kappa-opioid receptors from brain dopamine neurons has anxiolytic-like effects and enhances cocaine-induced plasticity. Neuropsychopharmacol 38:1585–1597
Van’t Veer A, Carroll FI, Carlezon WA Jr (2011) Antagonism of kappa-opioid receptors reduces corticotropin-releasing factor induced effects. Society for Neuroscience, Washington, D.C, Program Number 791.10
Van’t Veer A, Yano JM, Carroll FI, Cohen BM, Carlezon WA Jr (2012) Corticotropin-releasing factor (CRF)-induced disruption of attention in rats is blocked by the kappa-opioid receptor antagonist JDTic. Neuropsychopharmacol 37:2809–2816
van Gaalen MM, Stenzel-Poore MP, Holsboer F, Steckler T (2002) Effects of transgenic overproduction of CRH on anxiety-like behaviour. Eur J Neurosci 15:2007–2015
Van Pett K, Viau V, Bittencourt JC, Chan RK, Li HY, Arias C, Prins GS, Perrin M, Vale W, Sawchenko PE (2000) Distribution of mRNAs encoding CRF receptors in brain and pituitary of rat and mouse. J Comp Neurol 428:191–212
Viengchareun S, Le Menuet D, Martinerie L, Munier M, Pascual-Le Tallec L, Lombes M (2007) The mineralocorticoid receptor: insights into its molecular and (patho)physiological biology. Nuclear Receptor Signal 5: e012
Volkow ND, Li TK (2004) Drug addiction: the neurobiology of behaviour gone awry. Nat Rev Neurosci 5:963–970
Walker DL, Toufexis DJ, Davis M (2003) Role of the bed nucleus of the stria terminalis versus the amygdala in fear, stress, and anxiety. Eur J Pharmacol 463:199–216
Wee S, Koob GF (2010) The role of the dynorphin-kappa opioid system in the reinforcing effects of drugs of abuse. Psychopharmacology 210:121–135
Werling LL, Frattali A, Portoghese PS, Takemori AE, Cox BM (1988) Kappa receptor regulation of dopamine release from striatum and cortex of rats and guinea pigs. J Pharmacol Exp Ther 246:282–286
Widerlov E, Bissette G, Nemeroff CB (1988) Monoamine metabolites, corticotropin releasing factor and somatostatin as CSF markers in depressed patients. J Affect Disord 14:99–107
West TE, Wise RA (1988) Effects of naltrexone on nucleus accumbens, lateral hypothalamic and ventral tegmental self-stimulation rate-frequency functions. Brain research 462:126–33
Wiley MD, Poveromo LB, Antapasis J, Herrera CM, Bolanos Guzman CA (2009) Kappa-opioid system regulates the long-lasting behavioral adaptations induced by early-life exposure to methylphenidate. Neuropsychopharmacol 34:1339–1350
Wise RA, Bozarth MA (1987) A psychomotor stimulant theory of addiction. Psychol Rev 94:469–492
Wittmann W, Schunk E, Rosskothen I, Gaburro S, Singewald N, Herzog H, Schwarzer C (2009) Prodynorphin-derived peptides are critical modulators of anxiety and regulate neurochemistry and corticosterone. Neuropsychopharmacol 34:775–785
Xu Y, Day TA, Buller KM (1999) The central amygdala modulates hypothalamic–pituitary–adrenal axis responses to systemic interleukin-1beta administration. Neuroscience 94:175–183
Yoo JH, Lee SY, Loh HH, Ho IK, Jang CG (2004) Altered emotional behaviors and the expression of 5-HT1A and M1 muscarinic receptors in micro-opioid receptor knockout mice. Synapse 54:72–82
Zacharko RM, Anisman H (1991) Stressor-induced anhedonia in the mesocorticolimbic system. Neurosci Biobehav Rev 15:391–405
Zhu J, Chen C, Xue JC, Kunapuli S, DeRiel JK, Liu-Chen LY (1995) Cloning of a human kappa opioid receptor from the brain. Life Sci 56:PL201–PL207
Zhu J, Luo LY, Li JG, Chen C, Liu-Chen LY (1997) Activation of the cloned human kappa opioid receptor by agonists enhances [35S]GTPgammaS binding to membranes: determination of potencies and efficacies of ligands. J Pharmacol Exp Ther 282:676–684
Ziegler DR, Herman JP (2002) Neurocircuitry of stress integration: anatomical pathways regulating the hypothalamo–pituitary–adrenocortical axis of the rat. Integr Comp Biol 42:541–551
Zorrilla EP, Heilig M, de Wit H, Shaham Y (2013) Behavioral, biological, and chemical perspectives on targeting CRF(1) receptor antagonists to treat alcoholism. Drug Alcohol Depend 128:175–186
Zorrilla EP, Koob GF (2010) Progress in corticotropin-releasing factor-1 antagonist development. Drug Discov Today 15:371–383
Acknowledgments
This review represents an update on work described in the 2005 Jacob P. Waletzky Memorial Lecture, entitled “Experience-dependent alterations in the function of brain reward systems: the role of CREB” (WAC). The generosity of Dr. Jeremy Waletzky and his family is gratefully acknowledged.
Funding and disclosures
This research was supported by a National Defense Science and Engineering Graduate Fellowship (to AVV) and MH062366 (to WAC). Dr. Carlezon has a US patent covering the use of kappa antagonists in the treatment of depressive disorders (Assignee: McLean Hospital). In the last 3 years, Dr. Carlezon has received compensation for professional services from The American College of Neuropsychopharmacology and Concert Pharmaceuticals.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Van’t Veer, A., Carlezon, W.A. Role of kappa-opioid receptors in stress and anxiety-related behavior. Psychopharmacology 229, 435–452 (2013). https://doi.org/10.1007/s00213-013-3195-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00213-013-3195-5