
Abstract
Rationale
The endocannabinoid system has been implicated in the pathogenesis of major depression (MD) as well as in the mediation of antidepressant drug effects.
Objectives
To analyze CNR1 gene variants in MD and clinical response to citalopram (selective serotonin re-uptake inhibitors [SSRI]).
Methods
The role of CNR1 gene (rs806368, rs1049353, rs806371, rs806377 and rs1535255) was investigated in 319 outpatients with MD and 150 healthy individuals. A subsample of 155 depressive patients were treated with citalopram and evaluated for response (fourth week) and remission (12th week) by the 21-item Hamilton Depression Rating Scale (HDRS).
Results
We observed a higher frequency of rs806371 G carriers in MD patients with both presence of melancholia (p = 0.018) and psychotic symptoms (p = 0.007) than in controls. Haplotype frequency distributions between MD sample and controls showed a significant difference for Block 1 (rs806368–rs1049353–rs806371) (p = 0.008). This haplotype finding was consistent when we compared controls with MD subsample stratified by melancholia (p = 0.0009) and psychotic symptoms (p = 0.014). The TT homozygous of the rs806368 and rs806371 presented more risk of no Remission than the C carriers (p = 0.008 and 0.012, respectively). Haplotype frequency distributions according to Remission status showed a significant difference for Block 1 (p = 0.032). Also, we observed significant effect of time–sex–genotype interaction for the rs806368, showing that the C carrier men presented a better response to antidepressant treatment throughout the follow-up than TT homozygous men and women group (p = 0.026).
Conclusions
These results suggest an effect of CNR1 gene in the etiology of MD and clinical response to citalopram.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Major depression (MD) is a common disease caused by a complex interaction of a large number of genetic and non-genetic factors, each of them with a relatively small contribution to the disorder (Caspi et al. 2003). Treatment of MD is principally based on selective serotonin re-uptake inhibitors (SSRI) that enhanced serotonergic neurotransmission by blocking the serotonin transporter. However, clinical response to drug treatment in depression is a highly complex biological phenomenon in which several factors are involved, some of them genetic (Rasmussen-Torvik and McAlpine 2007; Klengel and Binder 2011; Uher 2011).
Recently, the endocannabinoid system has been implicated in the pathogenesis of depression and anxiety, the mediation of antidepressant drug effects in animal models and the neurobiology of emotion processing in healthy volunteers (Domschke et al. 2008). Physiological actions of endocannabinoid system in the central nervous system (CNS) are mediated by the activation of a specific cannabinoid receptor, the CB1 receptor (Matsuda et al. 1990). This receptor is coded by the CNR1 gene located on chromosome 6 (6q14–15). It is considered the most abundant G protein-coupled receptor expressed in the CNS of mammalian brain, being present in the limbic system and in the brain areas related to stress response, such as the central amygdala and the paraventricular nucleus (PVN) of the hypothalamus (Herkenham 1991). In addition, changes in the functional activity of the endocannabinoid system can cause altered activity in other neuromodulatory systems as well as imbalance in the primary GABA/glutamate control system (Rodriguez de Fonseca et al. 2005). Moreover, endocannabinoid system could activate the hypothalamic–pituitary–adrenal (HPA) axis (Weidenfeld et al. 1994), the neuroendocrine system involved in the responses to emotional stress.
Experimental data provide evidence that blocking the endocannabinoid system is a risk factor in the pathogenesis of depression as well as in anxiety disorders (Martin et al. 2002; Hill and Gorzalka 2005a). The administration of CB1 receptor agonist or endogenous cannabinoid re-uptake inhibitors results in antidepressant-like effects and increases efficacy of the antidepressant fluoxetine in experimental animal models (Gobbi et al. 2005; Hill and Gorzalka 2005a; Adamczyk et al. 2008).
In line with that, patients diagnosed with depression are found to have a reduced levels of circulating endocannabinoids (Hill et al. 2009). Moreover, a decreased in CB1 receptor density in grey matter glial cells was found in the post mortem brains of patients with MD (Koethe et al. 2007). Furthermore, an up-regulation of CB1 receptors was observed in the prefrontal cortex of subjects with MD who died by suicide (Hungund et al. 2004).
The involvement of CB1 receptors in regulating mood is further supported by evidences showing that the CB1 receptor antagonist, rimonabant, administered to humans for weight loss and obesity-related metabolic disorders has been shown to increase the risk of depressed mood disorder and anxiety along the treatment even though when the presence of depressed mood was an exclusion criteria in the study (Christensen et al. 2007). Moreover, a genetic study described the association between the polymorphism rs1049353 at the CNR1 gene and major depressive individuals when comparing with healthy controls showing an odds ratio (OR) of 2.46 for the contribution of the A allele to the probability of having MD (Monteleone et al. 2010).
Recent studies show the link between endocannabinoid system and antidepressant treatment. It has recently been suggested that the expression of CB1 receptor in the hippocampus and the hypothalamus is up regulated by chronic tricyclic antidepressant treatment (Hill et al. 2006). Furthermore, Domschke and colleagues (Domschke et al. 2008) found that individuals with G allele at rs1049353 had increased risk for antidepressant treatment resistance, particularly in females with comorbid anxiety. In contrast, we described that rs1049353 GG men presented better response along the follow-up than A carrier men or the women group (Mitjans et al. 2012).
According to these previous results, which seem to indicate a possible role of CNR1 gene (rs1049353 polymorphism) in both MD (Monteleone et al. 2010) and pharmacogenetics (Domschke et al. 2008; Mitjans et al. 2012), the aims of this study are therefore to investigate the role of several genetic variability at the CNR1 gene (rs806368, rs1049353, rs806371, rs806377 and rs1535255) as a risk factor for (a) MD and severity clinical features associated with the disease (b) response to citalopram (CIT) treatment.
Materials and methods
Total sample
The MD sample consisted of 319 depressive outpatients (227 females and 92 males; mean age 46.38 years, SD = 15.08 age of onset 38.29 years, SD = 14.92) from the Centre de Salut Mental of the Hospital Clinic de Barcelona. All patients suffered an active episode of MD diagnosed following the Diagnostic and Statistical Manual of Mental Disorders (DSM-IV) at the time of inclusion in the study. All cases were diagnosed using the Spanish version of the Structures Clinical Interview (SCID-I) (Spitzer et al. 1990). Detailed data about severity clinical features such as presence of melancholic features (n = 151 (50.3 %)), psychotic symptoms (n = 75 (25.1 %)) or previous suicide attempts (n = 55 (18.6 %)) were also collected (Arias et al. 2009). No patients with bipolar I or II disorder were included in this sample. Patients with drug abuse and dependence, mental retardation or with a medical disease that impairs evaluation have been excluded from the study.
A control sample consisting of 150 healthy individuals (71 females and 79 males; mean age 42.1 years, SD = 10.3) with no personal history of mental illness was included in the study. The Spanish version of the 28-item General Health Questionnaire (Goldberg and Hillier 1979) was used to assess their current mental condition.
All the individuals included in the study were of Spanish origin as stated through the birthplace of their four grandparents, thereby reducing the possibility of confounding genetic differences by population stratification (Freedman et al. 2004).
Ethnical approval was obtained from Spanish local research ethic committees. All patients and controls provided a complete written informed consent before inclusion in the study. All procedures were carried out according to the Declaration of Helsinki.
Pharmacogenetic subsample
A subsample of 155 patients out of the total depressive sample (120 females and 35 males) was followed for a pharmacogenetic study. All patients were treated with CIT and followed along 12 weeks by experienced psychiatrists. Patients were initially evaluated for the severity of their symptoms using 21-item Hamilton Depression Rating Scale (HDRS) (mean initial HDRS 24.72, SD = 4.74). A new HDRS was assessed to all patients every 4 weeks until completion of the follow-up at week 12. Clinical response to CIT treatment was considered when a decreased of at least 50 % in the baseline HDRS score was observed at the fourth week (Baumann et al. 1996). Remission for the index episode was considered when HDRS scores were equal or under 7 by the end of 12th week (Frank et al. 1991). Plasma levels of CIT were determined at sixth week using high-performance liquid chromatography (Olesen and Linnet 1996).
All patients were treated with CIT at standard therapeutic doses (mean initial dose 26.39 mg/day; range 20–40 mg/day). Before their inclusion in the study, a 2-week wash-out was carried out with those patients who were being treated with different drugs. In case it was necessary, low dose concomitant treatments with drug such as neuroleptics (10 % of the sample) or benzodiazepines at bedtime (55.4 % of the sample) were allowed. The presence and intensity of side effects was assessed by using the UKU scale (Lingjaerde et al. 1987) at the end of the fourth week of pharmacological treatment.
Ethnical approval was obtained from local research ethic committees. Patients provided written informed consent before inclusion in the study.
Genetic analysis
A total of five single nucleotide polymorphisms (SNPs) located at the CNR1 gene were selected according to previous literature: rs806368 (T/C), rs1049353 (G/A), rs806371 (T/G), rs806377 (T/C) and rs1535255 (T/G). Genomic DNA was extracted from blood samples using a standard phenol–chloroform extraction protocol. All the polymorphisms were successfully assayed using Sequenom MassArray technology (Tang et al. 1999).
Statistical analysis
The Hardy–Weinberg equilibrium for genotype frequencies in all samples was calculated using chi-square tests with EpiInfo v.3.5.1 (Dean et al. 1991).
Simple chi-square tests of independence were performed to confirm the presence or absence of allele or genotype associations. OR with 95 % confidence intervals (CI) were estimated for the effects of high-risk genotypes. The combined case–control study (MD vs. Controls) had an 80 % power (95 % CI) to detect OR equal or greater than 2.21 for disease according to the minimum allele frequencies of the different polymorphisms analyzed in our sample. In reference to the pharmacogenetic sample, the minimum detectable OR for no-response or no-remission will be equal or greater than 3.1 or 2.99, respectively (Cohen 1988). Bonferroni correction was conservatively applied for multiple analyses in single polymorphism analyses (p = 0.01 (=0.05/5 variations)).
Haploview 3.2 (Barret et al. 2005; Barrett et al. 2005) was used to generate a linkage disequilibrium map and to test for Hardy–Weinberg equilibrium in the haplotype analysis. The ‘R’ software (http://www.r-project.org) was used to calculate haplotype frequencies and to include covariates (see “Pharmacogenetic study” section) in the analysis for quantitative traits by the “haplo.stat” package (Schaid et al. 2002). Rare haplotypes less frequent than 1 % were excluded from the analyses. The global significance of the results for haplotype analyses was estimated using permutation (50,000 permutations) to confirm the asymptotic p values.
In the pharmacogenetic subsample the genetic variant effects on HDRS change scores over 12 weeks of CIT treatment was performed using analysis of variance (ANOVA) with repeated measures (genotype and gender as fixed factor, time point as a repeated measure and a number of covariates; see “Pharmacogenetic study” section). These analyses were processed using SPSS 17.00 (SPSS for Windows; SPSS Inc., Chicago, IL, USA).
Because the SNP rs1049353 (G/A) was analyzed in our previous study (Mitjans et al. 2012) according to response and remission status in the subsample of major depressive patients treated with CIT (n = 155), this polymorphism was only considered in the analyses that include the total sample of MD and controls. It has been also included when haplotype analyses were performed in all case–control design analyses.
Results
Total sample (major depression and control samples)
Genotype distribution of all SNPs was found to be in Hardy–Weinberg equilibrium in the control sample (rs806368 χ 2 = 0.35, df = 2, p = 0.839; rs1049353 χ 2 < 0.01, df = 2, p = 1; rs806371 χ 2 = 0.4, df = 2, p = 0.819; rs806377 χ 2 = 0.88, df = 2, p = 0.644; rs1535255 χ 2 = 0.1, df = 2, p = 0.95) as well as in the MD sample (rs806368 χ 2 = 0.85, df = 2, p = 0.652; rs1049353 χ 2 = 0.19, df = 2, p = 0.91; rs806371 χ 2 = 0.08, df = 2, p = 0.959; rs806377 χ 2 = 2.08, df = 2, p = 0.354; rs1535255 χ2 < 0.01, df = 2, p = 1). Genotypic and allelic frequencies in patients and controls are shown in Table 1.
Allele and genotype frequencies for the five SNPs analyzed did not significantly differ between MD patients and the control sample (see Table 1 for details).
Also, we did not find any significant difference when we compared the allele and genotype distribution according to clinical features (presence of melancholic features, psychotic symptoms and suicide attempts) in the major depressive sample (data not shown).
However, genotype and allele frequencies of the rs806371 significantly differed between the control sample and those patients that present MD with melancholia (n = 151) (genotype: χ 2 = 6.42, df = 2, p = 0.04; allele: χ 2 = 5.97, df = 1, p = 0.014). We observed a higher frequency of G carriers in patients with presence of melancholia than in healthy subjects (χ 2 = 5.59, df = 1, p = 0.018; OR = 1.83 95 % CI [1.07–3.15]). After multiple correction adjustment these results were no longer significant. Similar results were found when we compare genotype and allele frequencies between the control sample and depressive patients with psychotic symptoms (n = 75) (genotype: χ 2 = 8.56, df = 2, p = 0.01; allele: χ 2 = 7.89, df = 1, p = 0.004), showing G carriers presented increased risk of 2.22 for suffering MD with psychotic symptoms compared to healthy subjects (χ 2 = 6.96, df = 1, p = 0.008; OR = 2.22 95 % CI [1.17–4.22]).
Haplotype analysis has shown the existence of linkage disequilibrium among rs806368–rs1049353–rs806371 (Block 1: D′=0.907, r 2 = 0.557) and rs806377–rs1535255 (Block 2: D′=0.938, r 2 = 0.173) in the MD sample. As we detected the same results for the control sample (Block 1: D′=0.957; r 2 = 0.36; Block 2: D′=1.0, r 2 = 0.191), Fig. 1 shows linkage disequilibrium for the whole sample (MD + controls).

Linkage disequilibrium among markers in the whole Spanish sample (Block 1: D′=0.918, r 2 = 0.485; Block 2: D′=0.95, r 2 = 0.18)
Comparisons of the haplotype frequency distributions between MD and control samples showed a significant difference for Block 1 (Global-stat = 13.76, df = 4, simulated p = 0.0078), showing a lower haplotype frequency for the C–G–T haplotype (rs806368–rs1049353–rs806371) in the MD sample (7.5 %) than in the control sample (14.3 %) (simulated p = 0.001) (see Table 2 for haplotype frequencies details).
We did not find any significant difference when we compared the haplotype frequency distributions according to clinical features (presence of melancholic features, psychotic symptoms and suicide attempts) in the major depressive sample either for Block 1 or Block 2 (data not shown).
When patients were stratified according to clinical features and compared to the control subjects, we detect a significant association between the haplotype Block 1 and melancholia (Global-stat = 17.537, df = 4, simulated p = 0.0009) and psychotic symptoms (Global-stat = 12.003, df = 4, simulated p = 0.014) (see Table 2 for haplotype frequencies details).
Pharmacogenetic study
In the pharmacogenetic subsample, 95 patients (64.6 %) were considered Responders (Rp) and 52 (35.4 %) were classified as no-Responders (No-Rp) according to the decrease of their HDRS scores at fourth week. Considering the remission criteria at 12th week, 96 patients (65.3 %) were classified as Remitters (Rm) and 51 (34.7 %) as no-Remitters (No-Rm).
Concerning response at the fourth week, we did not observe any significant difference in genotype or allele distribution of any polymorphism between Rp/no-Rp (Table 3). However, we observed significant differences when we consider Remission status. There was significant differences for the rs806368 allele and genotype distribution according to Remission [genotype: χ 2 = 7.07, df = 2, p = 0.029; allele: χ 2 = 5.27, df = 1, p = 0.021], however these results did not survive multiple correction. Carriers analyses showed that TT homozygous presented almost 2.7 times more risk of no-remission than the C carriers (χ 2 = 6.94, df = 1, p = 0.008; OR = 2.64 95 % CI [1.20–5.89]). Furthermore, we observed the same significant difference for rs806371 (genotype: χ 2 = 6.18, df = 2, p = 0.045; allele: χ 2 = 5.74, df = 1, p = 0.016). The TT homozygous of the rs806371 presented 2.8 times more risk of no-remission than the G carriers (χ2 = 6.18, df = 1, p = 0.012; OR = 2.8 95 % CI [1.14–7.01]).
We considered the presence of suicide attempts and CIT levels at the sixth week as covariates in the response and remission haplotype analyses and in the longitudinal study because of their implication in response treatment. In a previous study with the same sample, the presence of suicide attempts was associated with no response at fourth week (χ 2 = 3.75, df = 1, p = 0.046) and no remission at 12th week (χ 2 = 10.204, df = 1, p = 0.002). Also, lower CIT levels at sixth week were associated with no response at fourth week (F = 4.72, df = 1, p = 0.032) (Mitjans et al. 2012).
Haplotype analyses for the two analyzed blocks did not yield a significant association with response at fourth week (Table 4). However, haplotype analysis showed a significant association between the haplotype Block 1 and remission (Global-stat = 10.503, df = 4, simulated p = 0.029) (see Table 4 for haplotype frequencies details).
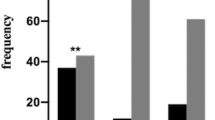
We performed a longitudinal study through a two-way repeated-measures ANOVA on HDRS scores to evaluate the effect of different polymorphisms on the 12-week clinical outcome of the patients treated with CIT. The longitudinal study showed no effects of the rs806371, rs806377 and rs1535255 polymorphisms on the 12-week clinical outcome. We found a significant effect of rs1049353 and rs806368. The rs1049353 effect had been reported in our previous study showing that individuals with GG genotype presented better response along the follow-up than A carriers (Mitjans et al. 2012). The longitudinal study of rs806368 showed that there was a significant decrease of HDRS scores over 12 weeks (F (2.76, 284.98) = 138.539, p < 0.001), a significant effects of time–sex interaction (F (2.76, 284.98) = 6.85, p < 0.001), time–genotype interaction (F (2.76, 284.98) = 4.987, p = 0.003) and a time–sex–genotype interaction (F (2.76, 284.98) = 3.233, p = 0.026). So, we observed significant effect of time–genotype interaction, showing that the C carriers presented a better response to antidepressant treatment throughout the follow up than TT homozygous. Stratification for gender revealed that this effect is originated by the subgroup of male patients (Fig. 2).

Genotype distribution of the CNR1-rs806368 polymorphism according to the different follow-ups based on CIT treatment. a rs806368-C allele carriers presented a better response to antidepressant treatment compared to rs806368-TT homozygous (F (2.76, 284.98) = 4.987, p = 0.003). b This effect is originated by the subgroup of male patients that showed a better outcome in response to CIT treatment (F (2.76, 284.98) = 3.233, p = 0.026). Tables show the Hamilton scores along the follow-up in relation to genotypes
Discussion
We have conducted an association study in which we have analyzed the genetic variability at CNR1 gene as a genetic risk factor for MD as well as for no response to clinical treatment with SSRIs.
When analysing the SNPs variability at the CNR1 gene (rs806368, rs1049353, rs806371, rs806377 and rs1535255), the results of the case–control association study did not show any genetic influence of this variability on the overall risk to suffer MD. However, the haplotype analysis showed that Block 1 C–G–T combination (rs806368–rs1049353–rs806371) is associated with an increased risk for MD. These results are in line with a previous study by Monteleone and colleagues (2010) that associated the CNR1 gene with depression.
Moreover, we grouped major depressive patients according to clinical features of severity such as the presence of melancholia, psychotic symptoms or suicide attempts and compared them to the control sample. Our results showed that patients with presence of melancholia or psychotic symptoms presented a higher frequency of rs806371 G carriers than healthy controls. Furthermore, the haplotype analysis showed an association between the haplotype Block 1 and melancholia and psychotic symptoms.
These results highlight the clinical and biological heterogeneity underlying the categorical diagnoses of MD which can easily overcome the power of genetic association studies (Winokur 1997). Categorical diagnostic tools are based on clusters of symptoms and characteristics of clinical course that maybe are not defining the different pathophysiological processes occurring in the disease. The definition of genetically relevant phenotypes in MD could help to increase the success of genetic studies (Hasler et al. 2004). Our results are in line with those defending that a stricter phenotype re-definition could increase power to detect more robust genetic effects (van der Sluis et al. 2010).
MD with melancholia has been identified as a valid subtype of MD that identifies a subset of more severe depressive patients with a particularly high genetic background (Kendler 1997). It has been shown that the genetic or pharmacological blockade of endocannabinoid system in animal models provoked similar symptomatology than melancholic depression (Hill and Gorzalka 2005b). One of the most reliable biological markers of melancholic depression is alterations in the HPA axis. Recent evidences show the role of the endocannabinoid system in regulation of the HPA axis activity (Di et al. 2003; Barna et al. 2004; Patel et al. 2004). Recent studies have shown that CB1 knockout mice present hypersecretion of corticotropin-releasing hormone (CRH) in the PVN (Cota et al. 2003), as well as elevated basal adrenocorticotropin (ACTH) and corticosterone (Barna et al. 2004). Consistent with the findings, glucocorticoid receptors (GR) antagonists have been found to be effective in very severe forms of depression, such as psychotic or endogenous forms of depression (Belanoff et al. 2001; Reus and Wolkowitz 2001). Our findings are in line with these evidences suggesting that severe forms of depression may have specific biological processes.
When we analyzed genetic variability in relation to clinical response or remission in the pharmacogenetic subsample, we did not observe any effect of the single different polymorphisms analyzed in response at fourth week to CIT treatment. However, we found significant effects of rs806368 and rs806371 polymorphisms on remission at 12th week. The TT homozygous of the rs806368 presented more risk of no Remission than the C carriers and TT homozygous of the rs806371 also presented more risk of no Remission than the G carriers. Although previous studies have shown the involvement of the rs806368 in substance use disorder or cannabis dependence (Zuo et al. 2007; Agrawal et al. 2009), no association study considering its role in clinical response has been published.
The results of our longitudinal study showed an influence of the rs806368 polymorphism on the response to treatment. G carrier men presented better response along the follow-up than TT homozygous men or the women group. Specifically, G carrier men had presented the highest HDRS scores at baseline and the lowest scores at the end of the study being the group with the greatest reduction in HDRS scores. According to that, haplotype analysis showed linkage disequilibrium between rs806368 and rs1049353 polymorphisms in our samples. It has recently reported that rs1049353 has an effect in antidepressant treatment response in MD (Domschke et al. 2008; Mitjans et al. 2012). Domschke and colleagues (2008) reported that the G allele of the rs1049353 confers an increased risk of resistance to antidepressant treatment, particularly in female patients with MD and high comorbid anxiety. In contrast, in a recent work, we described that men carrying the GG genotype presented better response along the follow-up than A carrier men or the women group (Mitjans et al. 2012). Although both studies show the involvement of this polymorphism in clinical response to antidepressant treatment, the results according to sex showed opposite directions.
Differential response mediated by gender remains still controversial (Serretti et al. 2008; Vermeiden et al. 2010; Carter et al. 2012). It might be hypothesized that gender differences in the response could be also reflecting the differences that are found in the aetiology of MD as physiological and epidemiological studies have shown (Biver et al. 1996; Weissman et al. 1996; Nishizawa et al. 1997; Kendler et al. 2001; Legato 2010; Lai 2011). Studies suggesting a role of estradiol in expression regulation of CB1 receptor mRNA (Gonzalez et al. 2000; Hill et al. 2007) could explain the differential response by gender found in this study. However, more research is still needed to better understand the gender specific contribution in antidepressant response. Pharmacogenetics could help to elucidate the role of CNS neurotransmission systems, such as the endocannabinoid system, in response to antidepressants. However, genetics will provide information for just a part of the complex puzzle of clinical response to psychodrugs. Other factors such environmental or clinical will be also necessary in order to understand the total phenotype. Nowadays, a test with widespread clinical use and adoption is still missing (Arranz and Kapur 2008).
All the analyzed polymorphisms are synonymous then not altering amino acid residues. Although synonymous SNPs have often been called silent or unable to affect functional changes, recent reports indicate that there are several mechanisms by which synonymous mutations could bring about such changes (Komar 2007; Sauna et al. 2007). These may have important implications in biology and in the diagnosis and treatment of human diseases. Alternatively, these polymorphisms might not constitute the actual causative variant, but rather reflect association of other polymorphisms in linkage disequilibrium with this locus.
Our study has several limitations. The relatively small size of our pharmacogenetic sample limits the power to detect small differences. However, this study has enough power to detect small–medium effect sizes. Moreover, the possible functional effects of the analyzed markers are still under investigation. We consider that multiple testing corrections are likely to be excessively exclusive in the context of the present study since the selection of the genetic polymorphisms, the sample size and the analyses performed had a directional hypothesis based on previous findings (Cardon and Bell 2001). However, as multiple testing based on Bonferroni’s procedures were taking into account; part of our results referred to the single SNP analyses (rs806368 and rs806371) did not survive the correction. Subsequent statistical analyses such as the genotype carrier’s analyses or the haplotype analyses, demonstrate the involvement of these polymorphisms in the risk for MD or response to antidepressant treatment.
In summary, CNR1 gene variants seem to be associated with the etiology of MD and specifically with the severity of MD showing that maybe a redefinition of the phenotype could help to a better understanding of the disease. Additionally, CB1 receptor gene seems to have an indirect effect on clinical response to CIT (SSRIs) basically in remission at the 12th week and along the follow-up.
Further studies focusing on other genes involved in the endocannabinoid system or other systems related to endocannabinoid system could help to elucidate the complex mechanism of aetiology of MD and clinical response to antidepressants.
References
Adamczyk P, Golda A, McCreary AC, Filip M, Przegalinski E (2008) Activation of endocannabinoid transmission induces antidepressant-like effects in rats. J Physiol Pharmacol 59:217–28
Agrawal A, Wetherill L, Dick DM, Xuei X, Hinrichs A, Hesselbrock V, Kramer J, Nurnberger JI Jr, Schuckit M, Bierut LJ, Edenberg HJ, Foroud T (2009) Evidence for association between polymorphisms in the cannabinoid receptor 1 (CNR1) gene and cannabis dependence. Am J Med Genet B Neuropsychiatr Genet 150B:736–40
Arias B, Serretti A, Mandelli L, Gasto C, Catalan R, Ronchi DD, Fananas L (2009) Dysbindin gene (DTNBP1) in major depression: association with clinical response to selective serotonin reuptake inhibitors. Pharmacogenet Genomics 19:121–128
Arranz MJ, Kapur S (2008) Pharmacogenetics in psychiatry: are we ready for widespread clinical use? Schizophr Bull 34:1130–1144
Barna I, Zelena D, Arszovszki AC, Ledent C (2004) The role of endogenous cannabinoids in the hypothalamo-pituitary-adrenal axis regulation: in vivo and in vitro studies in CB1 receptor knockout mice. Life Sci 75:2959–2970
Barret O, Carpenter TA, Clark JC, Ansorge RE, Fryer TD (2005) Monte Carlo simulation and scatter correction of the GE advance PET scanner with SimSET and Geant4. Phys Med Biol 50:4823–40
Barrett JC, Fry B, Maller J, Daly MJ (2005) Haploview: analysis and visualization of LD and haplotype maps. Bioinformatics 21:263–5
Baumann P, Nil R, Souche A, Montaldi S, Baettig D, Lambert S, Uehlinger C, Kasas A, Amey M, Jonzier-Perey M (1996) A double-blind, placebo-controlled study of citalopram with and without lithium in the treatment of therapy-resistant depressive patients: a clinical, pharmacokinetic, and pharmacogenetic investigation. J Clin Psychopharmacol 16:307–14
Belanoff JK, Flores BH, Kalezhan M, Sund B, Schatzberg AF (2001) Rapid reversal of psychotic depression using mifepristone. J Clin Psychopharmacol 21:516–21
Biver F, Lotstra F, Monclus M, Wikler D, Damhaut P, Mendlewicz J, Goldman S (1996) Sex difference in 5HT2 receptor in the living human brain. Neurosci Lett 204:25–8
Cardon LR, Bell JI (2001) Association study designs for complex diseases. Nat Rev Genet 2:91–9
Carter GC, Cantrell RA, Victoria Z, Haynes VS, Phillips G, Alatorre CI, Goetz I, Paczkowski R, Marangell LB (2012) Comprehensive review of factors implicated in the heterogeneity of response in depression. Depress Anxiety 29:340–54
Caspi A, Sugden K, Moffitt TE, Taylor A, Craig IW, Harrington H, McClay J, Mill J, Martin J, Braithwaite A, Poulton R (2003) Influence of life stress on depression: moderation by a polymorphism in the 5-HTT gene. Science 301:386–9
Cohen J (1988) Statistical power analysis for the behavioral sciences. New Jersey, Hillsdale, pp 8–14
Cota D, Marsicano G, Tschop M, Grubler Y, Flachskamm C, Schubert M, Auer D, Yassouridis A, Thone-Reineke C, Ortmann S, Tomassoni F, Cervino C, Nisoli E, Linthorst ACE, Pasquali R, Lutz B, Stalla GK, Pagotto U (2003) The endogenous cannabinoid system affects energy balance via central orexigenic drive and peripheral lipogenesis. J Clin Invest 112:423–431
Christensen R, Kristensen PK, Bartels EM, Blidda H, Astrup A (2007) Efficacy and safety of the weight-loss drug rimonabant: a meta-analysis of randomised trials. Lancet 370:1706–1713
Dean AG, Dean JA, Burton AH, Dicker RC (1991) Epi Info: a general-purpose microcomputer program for public health information systems. Am J Prev Med 7:178–82
Di S, Malcher-Lopes R, Halmos KC, Tasker JG (2003) Nongenomic glucocorticoid inhibition via endocannabinoid release in the hypothalamus: a fast feedback mechanism. J Neurosci 23:4850–7
Domschke K, Danniowski U, Ohrmann P, Lawford B, Bauer J, Kugel H, Heindel W, Young R, Morris P, Arolt V, Deckert J, Suslow T, Baune BT (2008) Cannabinoid receptor 1 (CNR1) gene: impact on antidepressant treatment response and emotion processing in Major Depression. Eur Neuropsychopharmacol 18:751–759
Frank E, Prien RF, Jarrett RB, Keller MB, Kupfer DJ, Lavori PW, Rush AJ, Weissman MM (1991) Conceptualization and rationale for consensus definitions of terms in major depressive disorder. Remission, recovery, relapse, and recurrence. Arch Gen Psychiatry 48:851–5
Freedman ML, Reich D, Penney KL, McDonald GJ, Mignault AA, Patterson N, Gabriel SB, Topol EJ, Smoller JW, Pato CN, Pato MT, Petryshen TL, Kolonel LN, Lander ES, Sklar P, Henderson B, Hirschhorn JN, Altshuler D (2004) Assessing the impact of population stratification on genetic association studies. Nat Genet 36:388–93
Gobbi G, Bambico FR, Mangieri R, Bortolato M, Campolongo P, Solinas M, Cassano T, Morgese MG, Debonnel G, Duranti A, Tontini A, Tarzia G, Mor M, Trezza V, Goldberg SR, Cuomo V, Piomelli D (2005) Antidepressant-like activity and modulation of brain monoaminergic transmission by blockade of anandamide hydrolysis. Proc Natl Acad Sci USA 102:18620–18625
Goldberg DP, Hillier VF (1979) Scaled version of the General Health Questionnaire. Psychol Med 9:139–145
Gonzalez S, Bisogno T, Wenger T, Manzanares J, Milone A, Berrendero F, Di Marzo V, Ramos JA, Fernandez-Ruiz JJ (2000) Sex steroid influence on cannabinoid CB1 receptor mRNA and endocannabinoid levels in the anterior pituitary gland. Biochem Biophys Res Commun 270:260–266
Hasler G, Drevets WC, Manji HK, Charney DS (2004) Discovering endophenotypes for major depression. Neuropsychopharmacology 29:1765–1781
Herkenham M (1991) Characterization and localization of cannabinoid receptors in brain: an in vitro technique using slide-mounted tissue sections. NIDA Res Monogr 112:129–45
Hill MN, Gorzalka BB (2005a) Pharmacological enhancement of cannabinoid CB1 receptor activity elicits an antidepressant-like response in the rat forced swim test. Eur Neuropsychopharmacol 15:593–599
Hill MN, Gorzalka BB (2005b) Is there a role for the endocannabinoid system in the etiology and treatment of melancholic depression? Behav Pharmacol 16:333–352
Hill MN, Ho WS, Sinopoli KJ, Viau V, Hillard CJ, Gorzalka BB (2006) Involvement of the endocannabinoid system in the ability of long-term tricyclic antidepressant treatment to suppress stress-induced activation of the hypothalamic–pituitary–adrenal axis. Neuropsychopharmacology 31:2591–9
Hill MN, Karacabeyli ES, Gorzalka BB (2007) Estrogen recruits the endocannabinoid system to modulate emotionality. Psychoneuroendocrinology 32:350–357
Hill MN, Miller GE, Carrier EJ, Gorzalka BB, Hillard CJ (2009) Circulating endocannabinoids and N-acyl ethanolamines are differentially regulated in major depression and following exposure to social stress. Psychoneuroendocrinology 34:1257–1262
Hungund BL, Vinod KY, Kassir SA, Basavarajappa BS, Yalamanchili R, Cooper TB, Mann JJ, Arango V (2004) Upregulation of CB1 receptors and agonist-stimulated S-35 GTP gamma S binding in the prefrontal cortex of depressed suicide victims. Mol Psychiatry 9:184–190
Kendler KS (1997) The diagnostic validity of melancholic major depression in a population-based sample of female twins. Arch Gen Psychiatry 54:299–304
Kendler KS, Gardner CO, Neale MC, Prescott CA (2001) Genetic risk factors for major depression in men and women: similar or different heritabilities and same or partly distinct genes? Psychol Med 31:605–16
Klengel T, Binder EB (2011) Using gene–environment interactions to target personalized treatment in mood disorder. Personalized Medicine 8:23–34
Koethe D, Llenos IC, Dulay JR, Hoyer C, Torrey EF, Leweke FM, Weis S (2007) Expression of CB1 cannabinoid receptor in the anterior cingulate cortex in schizophrenia, bipolar disorder, and major depression. J Neural Transm 114:1055–1063
Komar AA (2007) Genetics. SNPs, silent but not invisible. Science 315:466–7
Lai CH (2011) Major Depressive disorder gender differences in symptoms, life quality, and sexual function. J Clin Psychopharmacol 31:39–44
Legato MJ (2010) The skewed sex distribution in affective disorders—a diagnostic, social, or biological problem? Prog Brain Res 186:159–66
Lingjaerde O, Ahlfors UG, Bech P, Dencker SJ, Elgen K (1987) The UKU side effect rating scale. A new comprehensive rating scale for psychotropic drugs and a cross-sectional study of side effects in neuroleptic-treated patients. Acta Psychiatr Scand Suppl 334:1–100
Martin M, Ledent C, Parmentier M, Maldonado R, Valverde O (2002) Involvement of CB1 cannabinoid receptors in emotional behaviour. Psychopharmacology 159:379–387
Mitjans M, Gasto C, Catalan R, Fananas L, Arias B (2012) Genetic variability in the endocannabinoid system and 12-week clinical response to citalopram treatment: the role of the CNR1, CNR2 and FAAH genes. J Psychopharmacol 26:1391–8
Monteleone P, Bifulco M, Maina G, Tortorella A, Gazzerro P, Proto MC, Di Filippo C, Monteleone F, Canestrelli B, Buonerba G, Bogetto F, Maj M (2010) Investigation of CNR1 and FAAH endocannabinoid gene polymorphisms in bipolar disorder and major depression. Pharmacol Res 61:400–404
Nishizawa S, Benkelfat C, Young SN, Leyton M, Mzengeza S, de Montigny C, Blier P, Diksic M (1997) Differences between males and females in rates of serotonin synthesis in human brain. Proc Natl Acad Sci USA 94:5308–13
Olesen OV, Linnet K (1996) Simplified high-performance liquid chromatographic method for the determination of citalopram and desmethylcitalopram in serum without interference from commonly used psychotropic drugs and their metabolites. J Chromatogr B Biomed Appl 675:83–8
Patel S, Roelke CT, Rademacher DJ, Cullinan WE, Hillard CJ (2004) Endocannabinoid signaling negatively modulates stress-induced activation of the hypothalamic–pituitary–adrenal axis. Endocrinology 145:5431–8
Rasmussen-Torvik LJ, McAlpine DD (2007) Genetic screening for SSRI drug response among those with major depression: great promise and unseen perils. Depress Anxiety 24:350–7
Reus VI, Wolkowitz OM (2001) Antiglucocorticoid drugs in the treatment of depression. Expert Opin Investig Drugs 10:1789–1796
Rodriguez de Fonseca F, del Arco I, Bermudez-Silva FJ, Bilbao A, Cippitelli A, Navarro M (2005) The endocannabinoid system: physiology and pharmacology. Alcohol Alcohol 40:2–14
Sauna ZE, Kimchi-Sarfaty C, Ambudkar SV, Gottesman MM (2007) Silent polymorphisms speak: how they affect pharmacogenomics and the treatment of cancer. Cancer Res 67:9609–12
Schaid DJ, Rowland CM, Tines DE, Jacobson RM, Poland GA (2002) Score tests for association between traits and haplotypes when linkage phase is ambiguous. Am J Hum Genet 70:425–434
Serretti A, Kato M, Kennedy JL (2008) Pharmacogenetic studies in depression: a proposal for methodologic guidelines. Pharmacogenomics J 8:90–100
Spitzer RL WJ, Gibbon M, First MEB (1990) Structured clinical interview for Dsm-Iii-R. Washington, DC: American Psychiatric Press
Tang K, Fu DJ, Julien D, Braun A, Cantor CR, Koster H (1999) Chip-based genotyping by mass spectrometry. Proc Natl Acad Sci USA 96:10016–10020
Uher R (2011) Genes, environment, and individual differences in responding to treatment for depression. Harv Rev Psychiatry 19:109–24
van der Sluis S, Verhage M, Posthuma D, Dolan CV (2010) Phenotypic complexity, measurement bias, and poor phenotypic resolution contribute to the missing heritability problem in genetic association studies. PLoS One 5:e13929
Vermeiden M, van den Broek WW, Mulder PGH, Birkenhager TK (2010) Influence of gender and menopausal status on antidepressant treatment response in depressed inpatients. J Psychopharmacol 24:497–502
Weidenfeld J, Feldman S, Mechoulam R (1994) effect of the brain constituent anandamide, a cannabinoid receptor agonist, on the hypothalamo–pitutary–adrenal axis in the rat. Neuroendocrinology 59:110–112
Weissman MM, Bland RC, Canino GJ, Faravelli C, Greenwald S, Hwu HG, Joyce PR, Karam EG, Lee CK, Lellouch J, Lepine JP, Newman SC, Rubio-Stipec M, Wells JE, Wickramaratne PJ, Wittchen H, Yeh EK (1996) Cross-national epidemiology of major depression and bipolar disorder. JAMA 276:293–9
Winokur G (1997) All roads lead to depression: clinically homogeneous, etiologically heterogeneous. J Affect Disord 45:97–108
Zuo L, Kranzler HR, Luo X, Covault J, Gelernter J (2007) CNR1 variation modulates risk for drug and alcohol dependence. Biol Psychiatry 62:616–26
Acknowledgments
This study was supported through research projects funded by the Ministry of Science and Innovation (SAF2008-05674-C03-00/03, FIS07/0815 and IT2009-0016), the Institute of Health Carlos III, CIBER of Mental Health (CIBERSAM), the Comissionat per a Universitats I Recerca del DIUE of the Generalitat de Catalunya (2009SGR827) and the Ministero dell’Istruzione, dell’Università e della Ricerca, Italy (IT107CB8DC).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Mitjans, M., Serretti, A., Fabbri, C. et al. Screening genetic variability at the CNR1 gene in both major depression etiology and clinical response to citalopram treatment. Psychopharmacology 227, 509–519 (2013). https://doi.org/10.1007/s00213-013-2995-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00213-013-2995-y