
Abstract
Pyroptosis-mediated neuron death plays a crucial role in neurodegenerative diseases, such as Alzheimer’s disease (AD). However, the effect of 1,7-diphenyl-4-hepten-3-one (C1), a natural diarylheptanoid, on AD is unclear. Herein, we investigated the therapeutic effect of C1 on APP/PS1 mice and β-amyloid (Aβ)-induced HT22 cells. Our findings showed that C1 attenuated cognitive impairment and mitigated pathological damage in APP/PS1 mice. Furthermore, we found that C1 prevented oxidative stress damage and decreased the levels of pyroptosis-related proteins. In vitro experiments showed that C1 can improve the proliferation of Aβ-induced HT22 cells and decrease the levels of pyroptosis-related proteins in them. When Nrf2 was silenced, the positive effects of C1 in inhibiting pyroptosis were inhibited. Particularly, the production of pyroptosis-associated proteins, including NLRP3, GSDMD, and caspase-1, and the secretion of pro-inflammatory molecules, including IL-1 and IL-18, were increased. Altogether, these findings indicate that C1 can mitigate AD-like pathology via the inhibition of pyroptosis by activating the Nrf2 pathway. We believe that this study can provide alternative strategies for the prevention and treatment of AD.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Alzheimer’s disease (AD) is a neurodegenerative disorder that is characterized by progressive cognitive impairment and movement dysfunction. The primary pathological phenomena of AD include β-amyloid (Aβ) deposition and abnormal phosphorylation of Tau protein, which causes neuronal damage (Chen et al. 2021). Because individuals over the age of 65 are most susceptible to AD, it is a serious threat to public health due to the aging global population (Koutsodendris et al. 2022; Ahami Monfared et al. 2022). Currently, clinically available treatments for AD (e.g., donepezil, galantamine, and memantine) only improve short-term memory and cannot decrease disease progression (Ahami Monfared et al. 2022). Therefore, new therapies or treatment strategies should be urgently developed for the betterment of patients and society.
Pyroptosis is a novel mechanism of programmed cell death that depends on the activation of the caspase-1 pathway by the inflammasome (Wu et al. 2020). Inflammasomes, such as the nucleotide-binding domain, leucine-rich-containing family, and pyrin domain-containing-3 (NLRP3), promote the release of interleukin-18 (IL-18), interleukin-1β (IL-1β), and cleave the N-terminal sequence of gasdermin-D (GSDMD), thus inducing an inflammatory response, disrupting cell membrane integrity, and ultimately causing cell death (Zhou et al. 2021; He et al. 2021). Recently, pyroptosis-mediated neuroinflammation has garnered attention as a landmark event during the entire course of AD (Han et al. 2020). Inflammasomes are involved in the formation of Aβ, and IL-18 and IL-1β exacerbate the neuroinflammatory response. Furthermore, reducing pyroptosis in the brain can effectively rescue neuronal loss in the hippocampal and cortical regions (Han et al. 2020; Lyu et al. 2021). Although gene editing techniques have been used to regulate pyroptosis, its safety is not known. Therefore, a safe and effective strategy should be established (Xiao et al. 2021).
Mounting evidence suggests that nuclear factor erythroid 2-related factor 2 (Nrf2) plays a crucial role in regulating organismal pyroptosis (Cai et al. 2021). Nrf2 is the core transcription factor that antagonizes oxidative stress injury, and its binding to the antioxidant response element promotes the expression of downstream phase II detoxification enzymes and an array of protective proteins and decreases reactive oxygen species (ROS) production (Chen et al. 2022). Recent studies have shown that excess ROS and mitochondrial dysfunction are key in triggering NLRP3 inflammasome activation (Wang et al. 2019; Simon et al. 2021). Therefore, Nrf2 is a potential target for regulating pyroptosis. Previous studies have shown that dihydromyricetin reduces pyroptosis in vascular endothelial cells by activating Nrf2 to decrease ROS production (Zhang et al. 2016). This also indicates that natural products have great potential in antagonizing pyroptosis.
Alpinia oxyphylla Mique (AOM) is a plant still used in traditional Chinese medicine, which is also popular in other Asian countries (e.g., Japan and Korea). Previous studies have revealed that AOM exhibits several biological activities such as anti-inflammatory, antioxidant, and neuroprotective properties (Li et al. 2021; Zhang et al. 2018). AOM can ameliorate AD-induced learning memory decline and movement dysfunction by reducing Aβ production, inhibiting abnormal phosphorylation of Tau protein, and promoting hippocampal neurogenesis (Koo et al. 2004; Tang et al. 2015). However, it is unknown whether AOM and its main active substance can treat AD by inhibiting pyroptosis. Herein, the inhibitory effect of 1,7-diphenyl-4-hepten-3-one (C1), an active substance of AOM, on the pyroptosis of neurons was evaluated in vitro and in vivo. The findings of this study provide a promising treatment or prevention strategy for AD.
Materials and methods
Plant material, extraction, and isolation
The fruits of Alpinia oxyphylla Mique (50 kg) were collected from the Tunchang Town of Hainan Province, People’s Republic of China, in August 2022 and identified by Prof. Xiao-Zhong Chen from Heilongjiang University of Traditional Chinese Medicine. The detailed protocol for extraction and isolation is available in the supplementary materials.
Animals
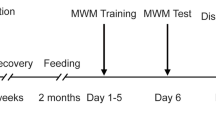
APP/PS1 mice (age: 6 months, male) were purchased from Nanjing Huimiaoxin Biotechnology Co., Ltd. (Nanjing, China). All mice were maintained in a designated animal care room (temperature: 25 ± 2 °C, 12-h light/dark photophase) with access to mouse food and water ad libitum. All experiments involving animals were approved by the Animal Care and Welfare Committee of Dalian Minzu University. The animal groupings and experimental procedures (Fig. S3) were detailed as follows: C57BL/6 J mice (n = 6) were included in the control group (WT group); APP/PS1 mice (n = 18) were assigned to the model (TG group), C1 low-dose (C1L group), and C1 high-dose group (C1H group). WT and TG group mice were administered 0.5% sodium carboxyl methylcellulose. C1L and C1H group mice were provided C1 (30 and 60 mg/kg) suspended in 0.5% sodium carboxyl methylcellulose. All experimental mice underwent intragastric administration for 4 weeks, once daily (Tang et al. 2015).
Cell culture
The HT22 cells (mouse hippocampal neurons) were obtained from Cyagen Biosciences (Suzhou, China). They were cultured in an H-DMEM medium supplemented with 10% fetal bovine serum and 1% penicillin/streptomycin at 37 °C.
Cell viability and LDH assay
The HT22 cells (5 × 104 cells/mL) were plated into 96-well plates (NEST Biotechnology) and pretreated with C1 (0.25, 0.5, 1, 2, and 4 μM) for 24 h. Then, the cells were exposed to Aβ (20 μM) for 24 h. All subsequent procedures were conducted in accordance with the manufacturer’s instructions. Cell counting kit-8 (CCK-8, MA0218) was purchased from Meilun Biological Technology (Dalian, China), and lactate dehydrogenase kit (LDH, KGT02448) was obtained from KeyGen Biological Technology (Nanjing, China).
CCK-F, Edu, and ROS staining assay
The HT22 cells (1 × 105 cells/mL) were plated into 6-well plates (NEST Biotechnology) and pretreated with C1 (1 and 4 μM) for 24 h. Then, the cells were exposed to Aβ (20 μM) for 24 h. All subsequent procedures were conducted in accordance with the manufacturer’s instructions. The cell fluorescence counting kit (CCK-F, C2013S), Edu cell proliferation kit (Edu, C0085L), and reactive oxygen species assay kit (ROS, S0033S) were purchased from Beyotime Biotechnology (Shanghai, China).
Morris water maze test
The Morris water maze test contains training trials and a probe test, which was performed to evaluate the learning and memory abilities of mice. In the training trial, the platform was placed in a fixed position in the pool and trained twice daily for 5 days, recording the escape latency (the time mice spent climbing onto the platform). In the probe trial (the sixth day), the platform was removed. The time residing in the target quadrant, the times of platform crossings, swimming distance, and speed were also monitored and recorded.
Histology evaluation
Murine brain tissues were immersed in 4% paraformaldehyde (PFA) for 24 h before dehydrating and embedding in paraffin. The processed tissues were sliced into 10 μM sections and subjected to hematoxylin-eosin (HE), Nissl, and IHC staining as detailed previously.
Immunofluorescence chemistry staining
The brain tissue sections were washed thrice with PBS and fixed in 4% PFA for 30 min. Subsequently, the sections were permeabilized by 0.5% Triton X-100 for 10 min and blocked with 5% bovine serum albumin (BSA, SW3015, Solarbio). After 30 min, rabbit polyclonal primary antibody against GFAP (bsm-52254R, Bioss) was used to incubate the sections at 4 °C overnight. Then, the sections were incubated with appropriate fluorescein secondary antibodies conjugated with Cy3 at room temperature for 2 h in the dark. Nuclei were stained with DAPI and captured under a fluorescence microscope.
MDA, SOD, and GSH assays
After C1 and Aβ treatments, the cell membranes were solubilized by freeze-thaw cycles. All subsequent procedures were performed in accordance with the manufacturer’s instructions. The malonaldehyde kit (MDA, BC0025) and L-glutathione kit (GSH, BC1175) were purchased from Beijing Solarbio Science & Technology Co., Ltd. (Beijing, China). A superoxide dismutase kit (SOD, KGT00150-2) was obtained from KeyGen Biological Technology (Nanjing, China).
ELISA
The murine hippocampal tissues were homogenized, and the supernatant was collected. The cell culture supernatant was collected after treatment. The levels of tumor necrosis factor-α (TNF-α; BDEL-0070, Biodragon), IL-1β (SEKM-0002, Solarbio), and IL-18 (BDEL-0652, Biodragon) were assessed using an ELISA kit, as prescribed by the manufacturer’s instructions.
Western blotting
Murine hippocampal tissues were collected, and the proteins were released using a whole protein extraction kit containing protease and phosphatase inhibitors. After determining the protein concentration by the BCA method, the samples were prepared and subjected to SDS-PAGE gel electrophoresis. The gels were excised horizontally by using the Prestained Color Protein Marker (P0079, Beyotime). Next, the proteins were transferred onto PVDF membranes and blocked with 5% BSA (SW3015, Solarbio) for 2 h. The proteins were labeled with primary antibodies as follows: rabbit polyclonal antibodies against Nrf2 (WL02135, Wanlei), NLRP3 (bs-6655R, Bioss), GSDMD (ab219800, Abcam), and caspase-1 (WL02996, Wanlei). Then, the proteins were incubated with a secondary antibody (1:1000) for 2 h in the dark. A Touch Imager was used to visualize the proteins. The optical density was analyzed by ImageJ software.
Statistical analysis
All data were expressed as the mean ± SEM and compared using one-way ANOVA and two-way ANOVA. The statistical plots were drawn by GraphPad Prism 9 software, and P < 0.05 was considered to indicate statistical significance.
Results
C1 protects Aβ-induced HT22 cells from death
We first investigated the neuroprotective effect of C1 in vitro. CCK-8 results showed that Aβ can decrease the viability of HT22 cells, but C1 (1, 2, and 4 μM) pretreatment can increase this. Meanwhile, C1 (1, 2, and 4 μM) can decrease the levels of LDH in the cell supernatant (Fig. 1A, B). After careful consideration, we used 1 μM and 4 μM as the subsequent doses of administration. Microscopically, we observed that HT22 cells regained their previous interwoven meshwork after the addition of C1 culture (Fig. 1C). Subsequently, we performed CCK-F staining, and the results indicated that the green fluorescence was significantly decreased in the Mod group than in the Con group, indicating that the Aβ-induced proliferation of HT22 cells was decreased. However, C1 reversed the aforementioned phenomenon (Fig. 1D, E). Similar conclusions were obtained in the Edu staining experiments (Fig. 1F, G). Altogether, those results indicate that C1 can protect Aβ-induced HT22 cells from death.

C1 protects Aβ-induced HT22 cells from death. A The HT22 cells were pretreated by C1 (0.25, 0.5, 1, 2, and 4 μM) for 24 h and then exposed to Aβ (20 μM) for 24 h. Cell viability was tested by CCK-8 assay. B LDH release was measured using a commercial kit. C Morphology of Aβ-induced HT22 cells. D Cell viability was assessed by CCK-F staining. E Quantitative analysis of MFI in D. F The proliferation of Aβ-induced HT22 cells were detected by Edu staining. Red fluorescence represents Edu+ cells which mean cells were proliferating, and blue fluorescence represents nucleus stained by DAPI. Red and blue fluorescence merge to form the last line of the image. G Quantitative analysis of Edu+ cells in F. Data are represented as mean ± SEM, n = 3–6, scale bar = 100 μM. ###P < 0.001 vs. Con group; *P < 0.05, **P < 0.01, ***P < 0.001 vs. Mod group
C1 inhibits oxidative stress and pyroptosis in Aβ-induced HT22 cells
Oxidative stress damage is a crucial cause of neuroinflammation. We examined the production of reactive oxygen species (ROS). The results showed that the green fluorescence of the ROS probe increased significantly after Aβ induction in HT22 cells, but a significant decrease in fluorescence was found after C1 treatment. This indicated that C1 can decrease the generation of ROS (Fig. 2D, E). Furthermore, other oxidative stress indicators such as MDA, SOD, and GSH were significantly improved after C1 intervention (Fig. 2A–C). We then detected the changes in pyroptosis-related protein levels in Aβ-induced HT22 cells using western blotting. The results indicated that NLRP3, GSDMD, and caspase-1 gene expression were increased in the Mod group cells, which was reversed in C1. However, the opposite trend was observed for Nrf2 (Fig. 2F–J). Meanwhile, the pyroptosis marker pro-inflammatory factors, IL-1β and IL-18, were also detected. ELISA results showed that C1 effectively downregulated their expression (Fig. 2K, L). Our results indicate that C1 decreased oxidative stress and pyroptosis in Aβ-induced HT22 cells.

C1 inhibits oxidative stress and pyroptosis in Aβ-induced HT22 cells. A–C The HT22 cells were pretreated by C1 (1 and 4 μM) for 24 h and then exposed to Aβ (20 μM) for 24 h. The levels of MDA, SOD, and GSH were tested by commercial kits. D Quantitative analysis of MFI in E. E The ROS content was detected using the DCFH-DA molecular probe. F The expression of Nrf2, NLRP3, GSDMD, and caspase-1 were assessed by western blot. G–J Quantitative analysis of protein levels in F. K, L The levels of IL-1β and IL-18 were measured by ELISA. Data are represented as mean ± SEM, n = 3–6, scale bar = 100 μM. ##P < 0.01, ###P < 0.001 vs. Con group; *P < 0.05, **P < 0.01, ***P < 0.001 vs. Mod group
C1 improves the learning and memory ability of APP/PS1 mice
Progressive memory loss is the major clinical manifestation of AD, and its improvement is the fundamental criterion for evaluating the efficacy of the drug intervention. Herein, the Morris water maze (MWM) test was performed to determine the effect of C1 on the learning and memory capacity of APP/PS1 mice. Our results showed that the TG group mice had a significantly longer escape latency compared with that of the WT group mice, and representative swim path maps were presented (Fig. 3A, B). Additionally, the number of platforms crossing and time in the target quadrant decreased considerably, and the swimming distance increased significantly in the TG group. However, the aforementioned phenomenon was reversed after the C1 intervention (Fig. 3C, D, F). These results indicate that C1 can effectively improve the learning and memory abilities of APP/PS1 mice.

C1 enhances the learning and memory ability of APP/PS1 mice. A–F After treatment with C1 (30 and 60 mg/kg) for 4 weeks, MWM test was carried out to evaluate the cognitive function of mice. A Swimming paths in last training trial. B Escape latency (the time of mice spend to climb onto the platform) in the training trial. C–F Number of platform crossings, swimming distance, swimming speed, and time in target quadrant in the probe trial. Data are represented as mean ± SEM, n = 6. ###P < 0.001 vs. WT group; *P < 0.05, **P < 0.01, ***P < 0.001 vs. TG group; &P < 0.05 vs. C1L group
C1 ameliorates pathological damage and attenuates Aβ1-42 production of APP/PS1 mice
The degree of Aβ plaque deposition and neuronal damage are crucial indicators to evaluate the progression of AD. Therefore, we performed neuropathological staining. Nissl staining indicated that the mice in the TG group had loosely arranged Nissl bodies at the cortex, with reduced coloration and a decreased number compared with those in the WT group. A significant improvement in the number of Nissl bodies was observed after the C1 intervention (Fig. 4A, B). Meanwhile, in the TG group, more extensive inflammatory infiltration and neurodegeneration involving neuronal atrophy, loosely arranged cells, and nuclear condensation were observed in HE staining, which was improved after C1 administration (Fig. 4C). The amount of Aβ in the cortex and hippocampus is closely associated with the learning and memory abilities of mice. We found that a wide distribution of Aβ was present in the brain of the TG group of mice, which was reduced after C1 intervention (Fig. 4D–F). Our results indicate that C1 can ameliorate pathological damage and attenuate Aβ1–2 production in APP/PS1 mice.

C1 ameliorates pathological damage and attenuates Aβ1–42 production of APP/PS1 mice. A After 4 weeks of C1 (30 and 60 mg/kg) administration, Nissl staining was performed to assess neurogenic damage of mice in different groups. B Quantitative analysis of Nissl bodies in A. C HE staining was carried out to evaluate pathological damage. D IHC staining was applied to assess the AOD of Aβ1–42 in cortex and hippocampus, respectively. E, F Quantitative analysis of AOD of Aβ1–42 in D. Data are represented as mean ± SEM, n = 3, scale bar = 100 μM. ###P < 0.001 vs. WT group; *P < 0.05, **P < 0.01, ***P < 0.001 vs. TG group
C1 inhibits oxidative stress and pyroptosis in the brain of APP/PS1 mice
Neuroinflammation is a crucial factor affecting AD progression; hence, we evaluated microglia and astrocyte activation in the present study. Microglia were marked by Iba-1, and the number of Iba-1 + cells was significantly increased in the TG group. However, the number of activated microglia in the brains of APP/PS1 mice was significantly decreased after C1 administration (Fig. 5A). IF staining showed a significantly increased level of the astrocyte marker GFAP in the TG group; however, C1 administration reversed this increase (Fig. 5B, C). Furthermore, C1 administration decreased the level of TNF-α, an important pro-inflammatory factor (Fig. 5D). Oxidative stress-mediated inflammatory responses are an essential component of neuroinflammation. The present results showed decreased SOD and GSH levels and increased MDA content in the TG group. However, the C1 administration reversed these changes (Fig. 5E-G). Pyroptosis plays a crucial role in AD progression. The western blot results showed increased NLRP3, GSDMD, and caspase-1 levels in the TG group, which decreased after C1 administration (Fig. 5H–L). IL-18 and IL-1β are the key markers of pyroptosis. We observed higher levels of these markers in brain tissues in the TG group, whereas their levels decreased after C1 administration (Fig. 5M, N). In summary, these results indicated that C1 inhibited oxidative stress and pyroptosis in the APP/PS1 mouse brain.

C1 inhibits oxidative stress and pyroptosis in the brain of APP/PS1 mice. A, B After 4 weeks of C1 (30 and 60 mg/kg) administration, IF staining was carried out to assess the number of Iba-1+and GFAP+ cells. C Quantitative analysis of GFAP+ cells in B. D The level of TNF-α was measured by ELISA. E–G The levels of MDA, GSH, and SOD were tested by commercial kits. H The protein expression of Nrf2, NLRP3, GSDMD, and caspase-1 were detected by western blot. I–L Quantitative analysis of protein levels in G. M, N The levels of IL-1β and IL-18 were evaluated by ELISA. Data are represented as mean ± SEM, n = 3–6, scale bar = 100 μM. ##P < 0.01, ###P < 0.001 vs. WT group; *P < 0.05, **P < 0.01, ***P < 0.001 vs. TG group
C1 reduces pyroptosis by activating the Nrf2 pathway in Aβ-induced HT22 cells
Nrf2, a core transcription factor, exerts antioxidant and anti-inflammatory effects on the body. Herein, siRNA was introduced to silence Nrf2. The ELISA results showed that C1 inhibited IL-1β, IL-18, and TNF-α secretion; however, this phenomenon was reversed by Nrf2 silencing (Fig. 6A–C). Furthermore, western blotting was performed to detect the levels of Nrf2 and pyroptosis-associated proteins, and the results showed that C1 effectively rescued the Aβ-induced decrease in Nrf2 levels and inhibited the increase in NLRP3, GSDMD, and caspase-1 levels. Similarly, Nrf2 silencing eliminated the positive effect of C1 (Fig. 6D–H). Overall, the results showed that C1 decreased pyroptosis by increasing Nrf2 levels.

C1 diminishes pyroptosis via activating Nrf2 pathway in Aβ-induced HT22 cells. A–C After 24 h of pretreatment with C1 (4 μM) and siRNA, HT22 cells were exposed to Aβ (20 μM) for 24 h. The levels of IL-1β, IL-18, and TNF-α were tested by ELISA. D The protein expression of Nrf2, NLRP3, GSDMD, and caspase-1 were evaluated by western blot. E–H Quantitative analysis of protein levels in D. Data are represented as mean ± SEM, n = 3–6. #P < 0.05, ##P < 0.01, ###P < 0.001 vs. NC siRNA-Con group; **P < 0.01, ***P < 0.001 vs. siRNA-Mod group; &P < 0.05, &&&P < 0.001 vs. NC siRNA-C1 group; ^P < 0.05, ^^P < 0.01, ^^^P < 0.001 vs. Nrf2 siRNA-Mod group
Discussion
Herein, we revealed the therapeutic effect of C1, a natural diarylheptanoid of AOM, on AD and its potential mechanisms in vitro and in vivo. We observed that C1 could mitigate cognitive impairment, pathological damage, and oxidative stress damage in APP/PS1 mice. Additionally, we found that C1 administration increased proliferation and inhibited inflammatory responses in Aβ-induced HT22 cells. To summarize, C1 administration mitigated AD-like pathology by inhibiting pyroptosis via activating the Nrf2 pathway. Given the lack of AD therapeutic agents as well as the good performance of C1, we believe that C1 may be a promising drug candidate for AD prevention and treatment.
Progressive memory loss is a critical clinical characteristic of AD that accurately and directly reflects disease progression (Chen et al. 2021). In the present study, we evaluated the effect of C1 by performing a water maze experiment, and the results revealed that C1 administration significantly improved learning and memory abilities and rescued cognitive impairment in APP/PS1 mice. Furthermore, Nissl staining showed that C1 protected the morphology and number of Nissl bodies. Previous studies have shown that A1, another natural diarylheptanoid of AOM, effectively protects neurons from Aβ damage (Tang et al. 2015). Moreover, AOM exerts good neuroprotective effects in vitro and in vivo (Zhang et al. 2018). Overall, these results show the potential of AOM and its major components in treating neurodegenerative diseases, especially AD.
Oxidative stress refers to oxidation-reduction balance disruption in an organism caused by excessive ROS accumulation (Forman and Zhang 2021). Current evidence suggests that oxidative stress injury occurs throughout the pathogenic course of AD (Wang et al. 2022). Furthermore, some scholars believe that oxidative stress is an important risk factor for inducing AD development (Plascencia-Villa and Perry 2021). Herein, we found that C1 administration increased SOD and GSH levels in APP/PS1 mice, as well as inhibited lipid peroxidation, which decreased MDA content. Similarly, C1 exerted antioxidant effects on Aβ-induced HT22 cells. In particular, C1 effectively reduced Aβ-induced ROS production. The above findings were consistent with previous findings (Koo et al. 2004). Thus, we can infer that AOM and its main compounds exhibit good antioxidant activity.
Neurogenesis in the hippocampal region is a vital way for an organism to rescue neuronal damage, and we have extensively worked on promoting hippocampal neurogenesis in the early stage (Wang et al. 2022). Owing to limitations such as the presence of the blood–brain barrier, we have studied a limited number of small natural molecules. Herein, we found that C1 effectively reduced Aβ-induced LDH release from HT22 cells and improved cell viability. Additionally, C1 promoted HT22 cell proliferation. Thus, we will further explore C1 applications in promoting hippocampal neurogenesis.
The inflammatory response mediated by glial cells, including microglia and astrocytes, has been studied widely. Previous evidence has shown that restoring microglial phagocytosis and inhibiting the astrocyte-induced inflammatory storm can effectively help reduce cognitive impairment in AD model mice (Cai et al. 2016; Smith et al. 2022). Herein, we observed that the levels of GFAP, a marker of astrocytes, decreased significantly compared with those in the TG group after C1 intervention. However, owing to the contradictory functions of astrocytes, the decreased levels of GFAP do not sufficiently indicate its usefulness in treating patients with AD. Astrocytes function mainly by secreting cytokines (Hasel and Liddelow 2021). Recent studies have shown that astrocytes are classified into A1 and A2 types according to the type of cytokines they secrete. Type A1 astrocytes mainly secrete pro-inflammatory factors such as TNF-α, whereas type A2 astrocytes mainly secrete neurotrophic factors such as BDNF (Lee et al. 2022; Hasel et al. 2021). Therefore, the detailed effects of AOM and its effective components on astrocytes need further exploration, which will be the direction of subsequent studies by our research group.
Pyroptosis-mediated neuroinflammation has garnered increasing attention in recent years. The production and release of IL-1β and IL-18 are the hallmark events of pyroptosis (Huang et al. 2021); hence, in the present study, we assessed the levels of these pro-inflammatory factors by ELISA and showed that C1 intervention decreased their secretion compared with that in the Mod group. In summary, we identified the therapeutic effect of C1 on AD, and the underlying mechanism might be related to pyroptosis inhibition. Furthermore, we observed that the levels of the pro-inflammatory factors IL-1β, IL-18, and TNF-α decreased after C1 administration in APP/PS1 mice. These results provided additional evidence that C1 inhibited inflammatory responses in the APP/PS1 mouse brain.
The antioxidant effects of Nrf2 have been widely studied by researchers, and we have previously demonstrated that Jujuboside A could inhibit H2O2-induced oxidative stress damage in human umbilical cord mesenchymal stem cells by activating the Nrf2/HO-1 signaling pathway (Chen et al. 2022). Additionally, a study has shown the involvement of Nrf2 immunomodulation and inflammatory responses (Guo et al. 2021). Hence, we silenced Nrf2 in HT22 cells, which reversed the positive effect of C1 to inhibit pyroptosis. Specifically, the levels of pyroptosis-related proteins such as NLRP3, GSDMD, and caspase-1 and pro-inflammatory factors such as IL-18 and IL-1β were increased. To summarize, we found that C1 inhibited pyroptosis by increasing Nrf2 levels. However, previous studies have shown the dual role of Nrf2. Nrf2 activation in cancer promotes the growth and proliferation of cancer cells, prevents their apoptosis, enhances the self-renewal ability of cancer stem cells, and improves the chemoresistance and radioresistance of cancer cells (Huang et al. 2022). Therefore, the manipulation of C1 to specifically activate Nrf2 is necessary. A recent study has shown that gene-edited Wnt7a specifically activates the Wnt pathway that plays a dual role in normal and tumor cells in brain endothelial cells to repair the blood–brain barrier (Martin et al. 2022). This finding may provide a useful basis for subsequent studies.
Overall, the present findings showed that C1, an effective component of AOM, exerts AD therapeutic effects by inhibiting oxidative stress injury and pyroptosis by increasing Nrf2 levels.
Conclusions
In conclusion, we found that C1 obtained from AOM mitigated AD-like pathology by inhibiting pyroptosis via activating the Nrf2 pathway. Moreover, a new diarylheptanoid and six known compounds extracted during the phytochemical investigation of AOM exhibited anti-AD activity. We hope that the present study contributes to the discovery of active substances in AOM and provides an alternative strategy for AD prevention and treatment.
Data availability
Data and materials are available in the paper.
Abbreviations
- AD:
-
Alzheimer’s disease
- Aβ:
-
β-Amyloid
- NLRP3:
-
NOD-like receptor thermal protein domain associated protein 3
- IL-18:
-
Interleukin-18
- IL-1β:
-
Interleukin-1β
- GSDMD:
-
Gasdermin-D
- Nrf2:
-
Nuclear factor erythroid 2-related factor 2
- ROS:
-
Reactive oxygen species
- PFA:
-
Paraformaldehyde
- BSA:
-
Bovine serum albumin
- TNF-α:
-
Tumor necrosis factor-α
- CCK-8:
-
Cell counting kit-8
- LDH:
-
Lactate dehydrogenase
- SOD:
-
Superoxide dismutase
- MDA:
-
Malonaldehyde
- GSH:
-
L-Glutathione
- AOM:
-
Alpinia oxyphylla Mique
- CCK-F:
-
Cell fluorescence counting kit-F
- Edu:
-
5-Ethynyl-2′-deoxyuridine
- Mod:
-
Molde group
- C1L:
-
C1 low-dose group
- C1H:
-
C1 high-dose group
- MFI:
-
Median fluorescence intensity
- AOD:
-
Average optical density
References
Ahami Monfared AA, Byrnes MJ, White LA, Zhang Q (2022) Alzheimer’s disease: epidemiology and clinical progression. Neurol Ther 11:553–569. https://doi.org/10.1007/s40120-022-00338-8
Cai J, Yi M, Tan Y, Li X, Li G, Zeng Z, Xiong W, Xiang B (2021) Natural product triptolide induces GSDME-mediated pyroptosis in head and neck cancer through suppressing mitochondrial hexokinase-ΙΙ. J Exp Clin Cancer Res 40:190. https://doi.org/10.1186/s13046-021-01995-7
Cai H, Liang Q, Ge G (2016) Gypenoside attenuates β amyloid-induced inflammation in N9 microglial cells via SOCS1 signaling. Neural Plast 6362707. https://doi.org/10.1155/2016/6362707
Chen JC, Xiao HH, Zhang Q, Kong L, Wang TM, Tian Y, Zhao YM, Li H, Tian JM, Wang C, Yang JX (2022) Jujuboside A inhibits oxidative stress damage and enhances immunomodulatory capacity of human umbilical cord mesenchymal stem cells through up-regulating IDO expression, Chin. J Nat Med 20:494–505. https://doi.org/10.1016/S1875-5364(22)60176-6
Chen X, Sun G, Tian E, Zhang M, Davtyan H, Beach TG, Reiman EM, Blurton-Jones M, Holtzman DM, Shi Y (2021) Modeling sporadic Alzheimer’s disease in human brain organoids under serum exposure. Adv Sci (Weinh) 8:e2101462T. https://doi.org/10.1002/advs.202101462
Forman HJ, Zhang H (2021) Targeting oxidative stress in disease: promise and limitations of antioxidant therapy. Nat Rev Drug Discov 20:689–709. https://doi.org/10.1038/s41573-021-00233-1
Guo C, Bi J, Li X, Lyu J, Liu X, Wu X, Liu J (2021) Immunomodulation effects of polyphenols from thinned peach treated by different drying methods on RAW264.7 cells through the NF-κB and Nrf2 pathways. Food Chem 340:127931. https://doi.org/10.1016/j.foodchem.2020.127931
Han C, Yang Y, Guan Q, Zhang X, Shen H, Sheng Y, Wang J, Zhou X, Li W, Guo L, Jiao Q (2020) New mechanism of nerve injury in Alzheimer’s disease: β-amyloid-induced neuronal pyroptosis. J Cell Mol Med 24:8078–8090. https://doi.org/10.1111/jcmm.15439
Hasel P, Liddelow SA (2021) Astrocytes. Curr Biol 31:326–327. https://doi.org/10.1016/j.cub.2021.01.056
Hasel P, Rose IVL, Sadick JS, Kim RD, Liddelow SA (2021) Neuroinflammatory astrocyte subtypes in the mouse brain. Nat Neurosci 24:1475–1487. https://doi.org/10.1038/s41593-021-00905-6
He B, Nie Q, Wang F, Han Y, Yang B, Sun M, Fan X, Ye Z, Liu P, Wen J (2021) Role of pyroptosis in atherosclerosis and its therapeutic implications. J Cell Physiol 236:7159–7175. https://doi.org/10.1002/jcp.30366
Huang Y, Xu W, Zhou R (2021) NLRP3 inflammasome activation and cell death. Cell Mol Immunol 18:2114–2127. https://doi.org/10.1038/s41423-021-00740-6
Huang W, Wen F, Gu P, Liu J, Xia Y, Li Y, Zhou J, Song S, Ruan S, Gu S, Chen X, Shu P (2022) The inhibitory effect and mechanism of Yi-qi-hua-yu-jie-du decoction on the drug resistance of gastric cancer stem cells based on ABC transporters. Chin Med 17:93. https://doi.org/10.1186/s13020-022-00647-y
Koo BS, Lee WC, Chang YC, Kim CH (2004) Protective effects of Alpinae oxyphyllae Fructus (Alpinia oxyphylla MIQ) water-extracts on neurons from ischemic damage and neuronal cell toxicity. Phytother Res 18:142–148. https://doi.org/10.1002/ptr.1382
Koutsodendris N, Nelson MR, Rao A, Huang Y (2022) Apolipoprotein E and Alzheimer’s disease: findings, hypotheses, and potential mechanisms. Annu Rev Pathol 17:73–99. https://doi.org/10.1146/annurev-pathmechdis-030421-112756
Lee HG, Wheeler MA, Quintana FJ (2022) Function and therapeutic value of astrocytes in neurological diseases. Nat Rev Nat Rev Drug Discov 21:339–358. https://doi.org/10.1038/s41573-022-00390-x
Li J, Du Q, Li N, Du S, Sun Z (2021) Alpiniae oxyphyllae Fructus and Alzheimer’s disease: an update and current perspective on this traditional Chinese medicine. Biomed Pharmacother 135:111167. https://doi.org/10.1016/j.biopha.2020.111167
Lyu Z, Li Q, Yu Z, Chan Y, Fu L, Li Y, Zhang C (2021) Yi-Zhi-Fang-Dai Formula exerts neuroprotective effects against pyroptosis and blood-brain barrier-glymphatic dysfunctions to prevent amyloid-beta acute accumulation after cerebral ischemia and reperfusion in rats. Front Pharmacol 12:791059. https://doi.org/10.3389/fphar.2021.791059
Martin M, Vermeiren S, Bostaille N, … Vanhollebeke B (2022) Engineered Wnt ligands enable blood-brain barrier repair in neurological disorders. Science 375:6582 https://doi.org/10.1126/science.abm4459
Plascencia-Villa G, Perry G (2021) Preventive and therapeutic strategies in Alzheimer’s disease: focus on oxidative stress, redox metals, and ferroptosis. Antioxid Redox Signal 34:591–610. https://doi.org/10.1089/ars.2020.8134
Simon MS, Schiweck C, Arteaga-Henríquez G, … Drexhage HA (2021) Monocyte mitochondrial dysfunction, inflammaging, and inflammatory pyroptosis in major depression. Prog Neuropsychopharmacol Biol Psychiatry 111:110391. https://doi.org/10.1016/j.pnpbp.2021.110391
Smith AM, Davey K, Tsartsalis S, Khozoie C, Fancy N, Tang SS, Liaptsi E, Weinert M, McGarry A, Muirhead RCJ, Gentleman S, Owen DR, Matthews PM (2022) Diverse human astrocyte and microglial transcriptional responses to Alzheimer’s pathology. Acta Neuropathol 143:75–91. https://doi.org/10.1007/s00401-021-02372-6
Tang G, Dong X, Huang X, Huang XJ, Liu H, Wang Y, Ye WC, Shi L (2015) A natural diarylheptanoid promotes neuronal differentiation via activating ERK and PI3K-Akt dependent pathways. Neuroscience 303:389–401. https://doi.org/10.1016/j.neuroscience.2015.07.019
Wang Y, Shi P, Chen Q, Huang Z, Zou D, Zhang J, Gao X, Lin Z (2019) Mitochondrial ROS promote macrophage pyroptosis by inducing GSDMD oxidation. J Mol Cell Biol 11:1069–1082. https://doi.org/10.1093/jmcb/mjz020
Wang C, Chen JC, Xiao HH, Kong L, Zhao YM, Tian Y, Li H, Tian JM, Cui L, Wen CM, Shi YJ, Yang JX, Shang DJ (2022) Jujuboside A promotes proliferation and neuronal differentiation of APPswe-overexpressing neural stem cells by activating Wnt/β-catenin signaling pathway. Neurosci Lett 772:136473. https://doi.org/10.1016/j.neulet.2022.136473
Wu P, Chen J, Chen J, Tao J, Wu S, Xu G, Wang Z, Wei D, Yin W (2020) Trimethylamine N-oxide promotes apoE-/- mice atherosclerosis by inducing vascular endothelial cell pyroptosis via the SDHB/ROS pathway. J Cell Physiol 235:6582–6591. https://doi.org/10.1002/jcp.29518
Xiao Y, Zhang T, Ma X, Yang QC, Yang LL, Yang SC, Liang M, Xu Z, Sun ZJ (2021) Microenvironment-responsive prodrug-induced pyroptosis boosts cancer immunotherapy. Adv Sci (Weinh) 8:e2101840. https://doi.org/10.1002/advs.202101840
Zhang T, Hu Q, Shi L, Qin L, Zhang Q, Mi M (2016) Equol attenuates atherosclerosis in apolipoprotein E-deficient mice by inhibiting endoplasmic reticulum stress via activation of Nrf2 in endothelial cells. PLoS ONE 11:e0167020. https://doi.org/10.1371/journal.pone.0167020
Zhang Q, Zheng Y, Hu X, Hu X, Lv W, Lv D, Chen J, Wu M, Song Q, Shentu J (2018) Ethnopharmacological uses, phytochemistry, biological activities, and therapeutic applications of Alpinia oxyphylla Miquel: a review. J Ethnopharmacol 224:149–168. https://doi.org/10.1016/j.jep.2018.05.002
Zhou Y, Zhou H, Hua L, Hou C, Jia Q, Chen J, Zhang S, Wang Y, He S, Jia E (2021) Verification of ferroptosis and pyroptosis and identification of PTGS2 as the hub gene in human coronary artery atherosclerosis. Free Radic Biol Med 171:55–68. https://doi.org/10.1016/j.freeradbiomed.2021.05.009
Funding
The authors appreciate the support from the Natural Science Foundation of Liaoning Province of China (grant no. 20170540201) and the Key Research and Development Plan of Anhui Province in 2022 (grant no. 2022e07020065).
Author information
Authors and Affiliations
Contributions
Yu-Sheng Shi: conceptualization, methodology, software, and writing—original draft. Yan Zhang: writing—original draft, data curation, and investigation. Xiao Luo: validation and resources. Hong-Kai Yang: validation and resources. Yong-Sheng He: methodology, writing—review and editing, and funding acquisition. The authors declare that all data were generated in-house and that no paper mill was used.
Corresponding author
Ethics declarations
Ethical approval
The animals were acclimated for 1 week before experimentation. This study was conducted following the guidelines of the Ethics Committee of Dalian Minzu University (Dalian, China) (Approval AEWC-20210303-1).
Competing interests
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Shi, YS., Zhang, Y., Luo, X. et al. 1,7-diphenyl-4-hepten-3-one mitigates Alzheimer’s-like pathology by inhibiting pyroptosis via activating the Nrf2 pathway. Naunyn-Schmiedeberg's Arch Pharmacol 397, 3065–3075 (2024). https://doi.org/10.1007/s00210-023-02765-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00210-023-02765-2