
Abstract
Glibenclamide (GLB), most widely used in the treatment of type II diabetes mellitus, inhibits K+ATP channel in pancreatic-β cells and releases insulin, while thioacetamide (TAA) is a well-known hepatotoxicant and most recommended for the induction of acute and chronic liver disease. The purpose of this study was to evaluate the hepatoprotective potential of GLB against TAA-induced hepatic damage in Wistar rats. TAA (200 mg/kg, ip, twice weekly) and GLB (1.25, 2.5, and 5 mg/kg/day, po) were administered for 6 consecutive weeks. Different biochemical, DNA damage, histopathological, TEM, immunohistochemical, and western blotting parameters were evaluated. GLB treatment has no effects on the TAA-induced significant decrease in body and liver weights. TAA treatment significantly increased liver index and treatment with GLB has no effect the same. TAA treatment altered the liver morphology, whereas treatment with GLB normalized the alteration in morphology. Further, significant increase in oxidative stress, apoptosis, and DNA damage was found in TAA-treated animals and GLB treatment significantly reduced these effects. TAA-induced plasma transaminases and serum ALP levels were significantly restored by GLB. Furthermore, histopathological findings showed the presence of lymphocyte infiltration, collagen deposition, bridging fibrosis, degeneration of portal triad, and necrosis in TAA-treated animals and GLB intervention significantly reduced the same. TEM images revealed that GLB significantly normalized the hepatic stellate cell morphology as well as restored the number of lipid droplets. GLB treatment significantly downregulated the expressions of TGF-β1, α-SMA, NLRP3, ASC, caspase-1, and IL-1β, and upregulated MMP-2 and catalase against TAA-induced liver damage. The outcomes of the present study confirmed that GLB ameliorated the liver damage induced by TAA.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Global burden of chronic liver diseases and its complications are responsible for more than two million deaths annually. Cirrhosis is the top ten cause of death in lower middle-income countries, ranks 13th in overall disorders, and 4th among chronic diseases in terms of life expectancy (Rowe 2017; WHO 2015). In 2010, there were an estimated 188,575 cirrhosis deaths in India, accounting for almost one-fifth (18.3%) of the global liver cirrhosis deaths (Mokdad et al. 2014). In fact, approximately 45% of all deaths in western countries took place because of fibrosis (Wynn 2008). Until today, no US-FDA-approved drugs available for the treatment of fibrosis. Fibrosis is the defense mechanism of liver tissue in response to injury and inflammation. In the initial stage of liver injury, the quiescent hepatic stellate cells (HSCs) get activated and triggered the formation of extracellular matrix (ECM), which results the deposition of collagen. An imbalance between the collagen formation and breakdown leads to the deposition of excess collagen and subsequently triggers new angiogenesis. The excess deposition of collagen in the extracellular space of hepatic cells causes the obstruction of blood flow and results in the hardening of hepatic tissue (Bataller and Brenner 2005; Friedman 2004). Biotransformation of thioacetamide (TAA) by microsomal enzyme CYP2E1 results highly reactive TAA di-sulfoxide, which facilitates the formation of adducts with hepatic macromolecules. This leads to oxidative stress-induced cellular damage, apoptosis, and necrosis of hepatocytes. TAA induces periportal (zone 1) and pericentral (zone 3) damages as compared to other standard hepatotoxins such as carbon tetra chloride and diethylnitrosamine (Liu et al. 2013). Intraperitoneal TAA administration in rat induced hepatic encephalopathy, metabolic acidosis, increased levels of transaminases, abnormal coagulopathy, and centrilobular necrosis. These are the main features of clinical chronic liver disease and TAA can precisely replicate the initiation and progression of human liver disease in experimental animal model (Lee et al. 2011). It has also been well reported that thioacetamide administration in laboratory Wistar rats is the most recommended for the induction of liver damage in experimental animal model (Koblihova et al. 2014). Glibenclamide (GLB) is the most widely used long-acting sulfonylurea drug for the treatment of T2DM and acts on pancreatic beta cells by inhibiting ATP-sensitive K+ channels. It has been identified as NLRP3 inflammasome inhibitor in cultured monocytes, bone marrow-derived macrophages, and LPS-challenged endotoxemic mice (Lamkanfi et al. 2009). GLB improved the kidney and heart structure as well as functions in chronic kidney disease of male Wistar rats, which might be due to the inhibition of inflammation through NLRP3 inflammasome activity (Diwan et al. 2014). In another study, GLB acts as a NLRP3 inflammasome inhibitor and attenuated myocardial injury in diabetic mice (Cai et al. 2014). GLB blocked the NLRP3 inflammasome activation and physiological dysfunction in cyclophosphamide-induced bladder inflammation in female Sprague-Dawley rats (Hughes et al. 2014). NLRP3 inflammasome and ASC adapter knockout mice had reduced hepatic fibrosis, when induced with carbon tetrachloride and TAA (Watanabe et al. 2009). Low-dose delayed treatment with GLB demonstrated significant beneficial effects and displayed highly favorable therapeutic window in three different rat models of ischemic stroke (Simard et al. 2009b). It has been successfully reported that GLB inhibited NLRP3 and reduced the severity of experimental acute pancreatitis in genetically obese mice (York et al. 2014). Recent meta-analysis study showed that GLB was safe and effective for use in gestational diabetes when compared with insulin (Song et al. 2017). Further, GLB stimulates the fluid and electrolyte secretion in cystic fibrosis transmembrane conductance regulator defective mice, which makes sulfonylurea as the model class of compounds to design drugs useful in the treatment of cystic fibrosis in liver (Spirli et al. 2005). However, GLB at the dose of 20 mg/kg (po) administered in the 1st, 5th, 10th, and 15th day of gestation showed light brown precipitate in the central vein of the liver and no serious anatomical and histological changes reported in the liver of these fetuses and this might be due to the possible passage of the drug through the blood placental barrier (Aguillar-Gomes et al. 2014).
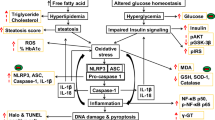
Chronic inflammation has been associated with progressive hepatic fibrosis and the subsequent development of cirrhosis (Czaja 2014). Inflammation contributes to the pathogenesis of acute and chronic liver diseases and the activation of inflammasome has been identified as the major contributing factor for hepatocyte damage, HSC activation, and amplification of liver inflammation (Szabo and Petrasek 2015). A paper published in Nature Scientific Report in 2013 reported that GLB also reduced the pro-inflammatory cytokine production by neutrophils of diabetic patients in response to bacterial infection (Kewcharoenwong et al. 2013). It is interesting to note that GLB reduced pro-inflammatory cytokines in an ex vivo model of human endotoxinemia under hypoxemic condition (Schmid et al. 2011). GLB acts as an anti-inflammatory agent by reducing the secretion of IL-1β and the bacterial dissemination to distant organs in streptozotocin-induced diabetic mice (Koh et al. 2013). Further, GLB reduced inflammation, vasogenic edema, and caspase-3 activation in mild-to-moderate subarachnoid hemorrhage rat model (Simard et al. 2009a). So far, there is no direct report on the effects of GLB on hepatic fibrosis and associated damage induced by thioacetamide in rat model. Recent clinical trial conducted as “Glyburide Advantage in Malignant Edema and Stroke Pilot (GAMES-PILOT)” revealed better outcomes in brain swelling or vasogenic edema in ischemic stroke patients (Kimberly et al. 2014; Sheth et al. 2014). Further, phase II clinical trial study reported that intravenous GLB is well tolerated in patients with risk for cerebral edema (Sheth et al. 2016). Another clinical trial reported that GLB can effectively and safely control the glycemic index in women with gestational diabetes mellitus in comparison to insulin (Mirzamoradi et al. 2015). In addition, sulfonylurea therapy benefits neurological and psychomotor functions in patients with neonatal diabetes owing to potassium channel mutations (Beltrand et al. 2016). In the present investigation, GLB was selected to evaluate its possible hepatoprotective effects at different molecular targets (Fig. 1), because it is one of the widely used drugs in the treatment of T2DM since 1966 and possesses both anti-inflammatory and anti-oxidant properties as reported in several literatures.

Pathways depict TAA-induced possible hepatic damage and subsequent protection with GLB at different molecular targets (showed in  shape). Dashed arrow represents either decrease or increase in respective expression/level
shape). Dashed arrow represents either decrease or increase in respective expression/level
Materials and method
Animals
Healthy male Wistar rats (250 ± 20 g) were procured from the Central Animal Facility (CAF), NIPER SAS Nagar. Rats were housed in polypropylene cages. Animals were divided into six groups (n = 7) at random. The room temperature controlled in between 20 and 26 °C, relative humidity between 50 and 70%, and 12-h light/dark cycle were maintained for the animals during the experiment. The experimental protocol was approved by the Institutional Animal Ethics Committee (IAEC) against approval number IAEC/16/54. Rats were fed with normal pellet diet and purified water ad libitum and the environmental conditions such as temperature and humidity were maintained throughout the experiment. The animals were well acclimatized for at least 1 week prior to the commencement of experiments.
Drugs and materials
GLB (CAS no. 10238-21-8), TAA (CAS no. 62-55-5), carboxy methyl cellulose sodium salt (sodium CMC) (CAS no. 9004-32-4), bovine serum albumin (CAS no. 9048-46-8), hematoxylin and eosin (H&E), trizma (CAS no. 77-86-1), glycine (CAS no. 56-40-6), and other commonly use reagents were purchased from Sigma-Aldrich Chemicals (St. Louis, MO, USA). Dimethyl sulfoxide (DMSO), normal melting point agarose, low-melting point agarose, Triton X-100, ethylenediaminetetraacetic acid (EDTA), and Hank’s balanced salt solution were obtained from Hi-Media Laboratories Ltd. (Mumbai, Maharashtra, India).
Rationality of dose selection
Kim et al. reported that thioacetamide is activated to its toxic metabolite(s) by CYP2E1 and not by CYP 2B in rats and mice (Kim et al. 2014). Further, Kim and Park reported that GLB has no effect on liver microsomal enzyme CYP2E1 activity (Kim and Park 2003). Hence, it can be deduced that GLB did not alter the metabolism of TAA in in vivo test system. Diwan et al. reported the improvement in kidney and heart structure and function in male Wistar rats aged in between 9 and 10 weeks administered with 10 mg/kg/day GLB in powdered food for 8 weeks, which significantly inhibited the expression of NLRP3 in kidney (Diwan et al. 2014). In another study of myocardial injury induced by lipopolysaccharides and streptozotocin in diabetic male C57BL/6 mice treated with GLB 20 mg/kg/day orally for 14 days, it showed significant inhibition of NLRP3 and other pro-inflammatory cytokines (Cai et al. 2014). Previous study on the inflammation of urinary bladder in adult female Sprague-Dawley rats treated with GLB (2.5 mg/kg in 10% ethanol, po) twice daily reported that GLB completely blocked cyclophosphamide-induced activation of caspase-1 and the production of IL-1β (Hughes et al. 2014). Based on the above information, three doses of GLB (1.25, 2.5, and 5 mg/kg/day for 6 weeks, po) were selected for the present investigation. TAA at the dose of 200 mg/kg, ip, twice in a week for 6 consecutive weeks was administered as reported in different studies for the successful induction of experimental rat model of hepatic fibrosis (El-Mihi et al. 2017; Tsai et al. 2010). TAA in the present investigation is used as an experimental model compound.
Experimental design
The animals were randomly divided into six groups (n = 7). Subsequent treatments were as follows: Group-1, animals were administered with normal saline, ip + 0.5% sodium carboxy methyl cellulose (CMC), po and served as control (CON); Group-2, animals were treated with GLB 5 mg/kg/day, po and served as GLB control (GLB 5); Group-3, animals were treated with TAA (200 mg/kg, ip) on every third day and served as TAA control (TAA); Group-4, animals were treated with TAA (200 mg/kg, ip, on every 3rd day) + GLB 1.25 mg/kg/day, po and served as TAA + GLB 1.25; Group-5, animals were treated with TAA (200 mg/kg, ip, on every 3rd day) + GLB 2.5 mg/kg/day, po and served as TAA + GLB 2.5; Group-6, animals treated with TAA (200 mg/kg, ip, on every 3rd day) + GLB 5 mg/kg/day, po and served as TAA + GLB 5 for 6 consecutive weeks. The study plan is diagrammatically described in Fig. 2a. In the present experiment, 0.5% sodium CMC was used to suspend glibenclamide and used normal saline to dissolve the thioacetamide. It is well reported in the literature that majority of the chemicals which are not soluble in water were given as suspension using CMC. Further, it is reported that CMC is well tolerated in rats when administered as 5% in water by oral gavage for 1 year (Gad et al. 2006). It is very well reported that GLB is an inflammasome (NLRP3) inhibitor. We thought it is imperative to use GLB to reduce TAA-induced inflammasome activation, thereby reducing fibrosis. Hence, the present experimental model was designed.

a Schematic diagram illustrates the experimental design in Wistar rats. Hepatic damage was induced by TAA (200 mg/kg, ip) on every third day (twice weekly) for 6 consecutive weeks, so the total study period was 42 days and the animals were sacrificed on day 43. GLB was administered daily at the doses of 1.25, 2.5, and 5 mg/kg, po for 6 consecutive weeks. b Representative photomicrographs depict liver morphology from different groups of rat at the time of necropsy. Control (CON) and GLB per se (GLB 5)-treated groups showed normal liver morphology. As thioacetamide per se (TAA)-treated group showed profuse nodular appearance in all the liver lobes, GLB at all the three (TAA + GLB 1.25, 2.5, and 5) doses reduced the nodular appearance uniformly in all the liver lobes. Effect of GLB treatment on the c change in body weight, d liver weight, e alanine transaminase (ALT), f aspartate transaminase (AST), g alkaline phosphatase (ALP), h γ-glutamyl transferase (GT), i lipid peroxidation (MDA), and j reactive oxygen species (DCFDA method) levels in TAA-induced liver damage. All the values are expressed as mean ± SEM (n = 4–7), ***p < 0.001, **p < 0.01, *p < 0.05, “a” vs. control, “c” vs. TAA control
Evaluation of biochemical parameters
Measurement of lipid peroxidation
Liver was dissected out, washed thoroughly in chilled PBS, and immediately blotted dry and weighed. Piece of liver was homogenized in phosphate buffer (pH 7.4, containing 3 mM EDTA) and centrifuged at 10,000 rpm. Supernatants (100 μL) were incubated with 100 μL 8.1% SDS, 750 μL 20% acetic acid, and 750 μL 0.8% thiobarbituric acid at 95 °C for 1 h until pink color developed. The product absorbance was taken at 532 nm along with standard prepared with known concentrations of malondialdehyde (MDA). Results were expressed as micromoles of MDA per milligram of protein (Khan and Jena 2014b).
Measurement of reactive oxygen species level
Level of reactive oxygen species (ROS) was measured using 2,7-dichlrofluorescein diacetate (DCFDA), which is converted to highly fluorescent DCF by cellular peroxides (including hydrogen peroxide). The assay was performed as described by Socci et al. Small piece of liver was homogenized in Tris-sucrose buffer (pH 7.4). Five microliters of supernatants was mixed with 5 μL of 5 μM DCFDA and added 990 μL of water. After 30 min, the product absorbance was taken at 485/525 nm. Result was expressed as fluorescence units per milligram of protein (Socci et al. 1999).
Measurement of superoxide dismutase level
Native polyacrylamide gel electrophoresis of supernatant from liver tissue homogenate was performed in 8% separating gel with 4% stacking gel. The gels were stained for superoxide dismutase (SOD) activity by the negative staining procedure. The gels were immersed in 1 mM nitro blue tetrazolium in distilled water and incubated for 20 min in the dark. Subsequently, gels were placed for 10 min in 38 mM phosphate buffer (pH 7.8) containing 28 mM tetramethyl ethylene diamine and 28 mM riboflavin in a bright place. Areas in the gel showing SOD activity remained achromatic (bands), whereas the remainder of the gel was stained violet (Beauchamp and Fridovich 1971).
Measurement of protein level
Supernatants (5 μL) of the liver tissue homogenate were incubated with solution D (2% sod. carbonate, 0.4% sod. hydroxide, 0.04% sod. tartrate, and 0.02% copper sulfate) for 10 min at 37 °C. Resultant solution was treated with equal volume of Folin’s reagent for 30 min at 37 °C. Blue color was developed. The product absorbance was taken at 660 nm along with standard prepared with known concentration of bovine serum albumin (Khan and Jena 2014b).
Evaluation of parameters in plasma and serum
Blood was collected in tubes and kept for half an hour at room temperature for serum; subsequently, tubes were centrifuged at 3000 rpm for 10 min at 4 °C. In another tube, blood was collected in tubes coated with EDTA for plasma and centrifuged at 7000 rpm for 10 min at 4 °C. The blood plasma was evaluated for alanine aminotransferase (ALT), aspartate aminotransferase (AST), and γ-glutamyl transferase (γ-GT), and serum was evaluated for alkaline phosphatase (ALP) by ERBA CHEM 7 semi auto-analyser (ERBA Diagnostics, Mannheim, Germany). Plasma glucose level was evaluated by GOD-POD kit (ACCUREX, Mumbai, India) method.
Quantification of DNA damage and cell death in liver tissue
The extent of DNA damage was analyzed by comet assay. Small piece of liver was taken out and chopped into single cell suspension in the solution containing Hank’s balanced salt solution, dimethyl sulfoxide, and EDTA. The comet assay was performed in the liver as described by Khan and Jena (2014a). Briefly, base slides were prepared by the coating of 1% normal melting agarose and dried overnight at room temperature. Every step (from tissue collection to scoring) was performed in the absence of light to avoid photo-induced DNA damage. The suspended cells were mixed with 0.5% low-melting point agarose and spread over a glass slide. The slides were kept at 4 °C for 16 h in lysis buffer [10 mM Tris (pH 10.0), 2.5 M NaCl, 100 mM EDTA, freshly prepared 10% DMSO, 1% Triton X-100]. Subsequently, slides were washed in chilled distilled water for the removal of detergents and salts. Slides were submerged in chilled running buffer [1 mM EDTA and 300 mM NaOH (pH 13)] for 20 min in horizontal electrophoresis tank [(model, CSLCOM20), Cleaver Scientific Ltd., Warwickshire, UK] for unwinding of DNA. Further, electrophoresis was performed at 300 mA and 38 V (0.90 V/cm) for 30 min. The slides were treated with neutralizing buffer containing 0.4 M Tris (pH 7.5) for 15 min and stained with SYBR Green I (1:10,000) for 1 h and cover slips were spread over the slides. The DNA damage was quantified using an AXIO imager M1 fluorescence microscope (Carl Zeiss, Altlussheim, Germany) and the pictures were captured with image analysis software (Comet Imager V.2.0.0). Arbitrarily, 50 comets per slide were captured and quantified for DNA damage. Duplicate slides were analyzed from each animal. DNA damage parameters included the following: % tail DNA (% DNA), tail moment (TM), tail length (TL), and tail moment olive (TMO). The comets were chosen for analyses that have uniform background, not any comets overlapping or touching other comets. Areas with disturbed agarose bed, edges of slides, and comets with irregular shapes were not taken into consideration for quantification. Cell death (apoptosis) was assessed by using TUNEL assay (Calbiochem, USA) commercial kit according to the manufacturer protocol.
Evaluation of cellular damage
Histological evaluation and quantification
Formalin-fixed liver tissue was washed dehydrated and embedded in paraffin. Sections (5 μm) were stained with hematoxylin and eosin (H&E). Other special stains for collagen deposition such as Masson’s trichrome (MT) (HT15-1KT, Sigma-Aldrich, USA), Picro Sirius Red (PSR), and PSR with fast green FCF (PSRF) were performed and %fibrotic area was quantified using ImageJ software (1.44p, NIH, USA). To check out the status of glycogen stores, periodic acid-Schiff (PAS) stain was performed (Marumo et al. 2010).
Immunohistochemical analysis and quantification
Immunohistochemistry of paraffin-embedded tissue were performed by using Novolink polymer detection system kit (RE7140-K, Leica Biosystems, UK). Briefly, 5-μM-thick liver sections were deparaffinized and hydrated and washed with TBS. Antigen retrieval were done by heating in citrate buffer at 95 °C for 30 min. Further, the rabbit polyclonal antibodies of anti-NLRP3 (sc-66846), anti-ASC (sc-22514), anti-caspase-1 (sc-514), anti-IL-1β (sc-7884), anti-TGF-β1 (sc-146), and mouse monoclonal antibody of anti-α-SMA (sc-53015) were incubated overnight. Subsequently, sections were incubated with HRP-conjugated Novolink polymer. Finally, slides were incubated with 3,3′-diaminobenzidine as a chromogen. Slides were counterstained with hematoxylin, dehydrated with gradient alcohols and xylene, mounted using DPX, and quantified using ImageJ software (Maremanda et al. 2016).
Detection of HSC activation by transmission electron microscopy
The liver tissue was cut into 1 × 1 × 1 mm3 blocks and fixed with 2.5% glutaraldehyde in the phosphate buffer. Samples were washed using phosphate buffer. Then samples were fixed with 1.0% osmium tetroxide for overnight, dehydrated with alcohol, and substituted with Spur’s embedding media. Ultrathin sections were taken using ultramicrotome, stained with uranyl acetate, and observed for morphological changes in HSC with the help of transmission electron microscope (model TF-20; FEI, Hillsboro, OR, USA).
Quantification of protein content by western blot analysis
Approximately, 100 mg of the liver tissue was homogenized in 1 mL radioimmunoprecipitation assay buffer containing protease inhibitor. Then, homogenized samples were sonicated for 10, 5, and 5 s. The samples were centrifuged at 10,000 rpm for 10 min at 4 °C and protein concentration was determined in supernatant. Samples were separated by 8–15% sodium dodecyl sulfate-polyacrylamide gel electrophoresis and transferred onto PVDF membrane. Three percent BSA in TBS was used as blocking agent then primary rabbit polyclonal antibody of anti-MMP-2 (sc-10736), anti-catalase (sc-50508), and mouse monoclonal anti-tubulin used in 1:1000 dilution in TBS, then incubation with HRP-conjugated secondary antibodies anti-mouse and anti-rabbit (Santa Cruz, CA, USA). Fluorescent signal was detected using enhanced chemiluminescence solution and X-ray films. Densitometric analyses of immunoblots were performed using ImageJ software (Maremanda et al. 2014).
Statistical analyses
Results were expressed as mean ± SEM for each group. Statistical analyses were performed using graph pad prism 6 (Statistical Software, California corporation). For multiple comparisons, ANOVA was used and post hoc analysis was performed with Tukey’s test. p values ≤ 0.05 were considered significant.
Results
Effect of GLB on hepatic morphology, body, and liver weight
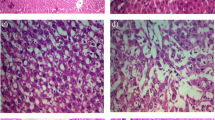
Control and GLB 5 per se treated groups have normal liver morphology; however, profuse superficial nodules were observed in TAA treatment groups. Treatment with GLB reduced the nodular appearance uniformly in all lobes (Fig. 2b). TAA treatment significantly decreased the body weight and significantly increased the liver weight while GLB treatment has no effect on the body weight (Fig. 2c, Supplementary Figs. 1 & 2) as well as the liver weight (Fig. 2d). TAA treatment increased the liver to body weight ratio (liver index), while GLB treatment has no effect on this parameter (Supplementary Fig. 3).
Effect of GLB on biochemical parameters
TAA per se treatment significantly increased the plasma ALT and AST levels and serum ALP level; significant protection for ALT was observed for the groups TAA + GLB 1.25 and TAA + GLB 2.5, and for AST and ALP, significant protection was observed only for TAA + GLB 1.25 when compared to TAA-treated group (Fig. 2e–g). Significant increase in the γ-GT levels was found in per se TAA-treated group and GLB treatment has no effect on the same (Fig. 2h). TAA treatment significantly increased the lipid peroxidation in the liver, and this was reflected by the increase in the MDA (malondialdehyde) and ROS level in comparison to control group. Treatment with GLB at low and mid doses decreased the MDA level in a significant manner and decreased ROS at low dose (Fig. 2i, j). Co-treatment of TAA and GLB (low and mid doses) significantly decreased the plasma glucose level (Supplementary Fig. 4). TAA treatment significantly reduced the SOD level while GLB treatment at low dose significantly restored the same (Supplementary Fig. 5).
Evaluation of cell apoptosis and DNA damage
TAA-treated group induced significant TUNEL-positive cells in comparison to control, and treatment with GLB decreased the TUNEL-positive cells in a significant way in comparison to TAA group (Fig. 3). Further, TAA treatment increased the comet assay parameters such as TL, TM, TMO, and % DNA, while GLB treatment significantly restored these comet assay parameters (Fig. 4).

Representative photomicrographs from different groups showing the TUNEL-positive cells (green signal indicated by arrows in FITC panels) in liver tissue sections. Effect of GLB treatment on apoptosis in TAA-induced liver fibrosis evaluated by TUNEL assay. All the values are expressed as mean ± SEM (n = 3), ***p < 0.001 and **p < 0.01, *p < 0.05, “a” vs. control, “c” vs. TAA

Representative photomicrographs from different groups showing the comets in single cell suspension of liver tissue in agarose gel electrophoresis. Effect of GLB treatment on % DNA, tail length, tail moment, and tail moment olive in TAA-induced liver fibrosis evaluated by comet assay. All the values are expressed as mean ± SEM (n = 3), ***p < 0.001 and **p < 0.01, “a” vs. control, “c” vs. TAA
Evaluation of cellular damage
Effect of GLB on histological alterations
Liver sections were processed for H&E staining to evaluate the alterations at tissue and cellular level. The infiltration of inflammatory lymphocytes, excessive ECM protein (collagen deposition) formation, and necrotic hepatocytes were observed (Fig. 5) and GLB treatment significantly ameliorated these effects. The extent of hepatic fibrosis especially collagen deposition (portal, septal, and sinusoidal) was determined by special staining such as MT, PSR, and PSRF staining. In MT staining, the collagen taken up blue stain against red background was quantified (Supplementary Fig. 6). Both PSR and PSRF that have red stain (for collagen) against yellow and green backgrounds, respectively, were quantified (Fig. 6, Supplementary Fig. 7). GLB significantly reduced the collagen deposition as observed in MT, PSR, and PSRF staining. PAS staining was performed as an indicator of glycogen storage. Hepatocytes with glycogen content have undergone the PAS reaction and showed magenta color. TAA treatment altered the glycogen storage as less hepatocytes have taken the magenta color, while treatment with GLB restored the magenta color (indicating restoration of glycogen) in the hepatocytes (Supplementary Fig. 8).

Representative photomicrographs from different groups showing the H&E staining of liver tissue sections. TAA group exhibited the extreme fibrosis as the portal zone showed a severe degree of infiltration of inflammatory lymphocytes in the area indicated by arrows. The hepatic artery and vein are present, but the corresponding interlobular bile duct is not clearly visible. Portal triad is not clearly visible. Higher magnifications of the indicated areas were further inserted in the respective panel

Representative photomicrographs from different groups showing the PSR staining of liver tissue sections. Effect of GLB treatment on TAA-induced hepatic fibrosis. As in TAA group, the presence of fibrosis (collagen stained red against yellow background also indicated by black arrows) that reaches from a portal area (or lobule) to another portal area (or lobule) known as bridging fibrosis. Green and red arrows showed the (peri-) sinusoidal and septal fibrosis, respectively. In the third panel (TAA), magnified area showed the portal fibrosis. Asterisk indicated the extent of fibrosis between and among the lobule has occurred. GLB treatment significantly reduced the all different forms of fibrosis. Higher magnifications of the indicated areas were further inserted in the respective panel. All the values are expressed as mean ± SEM (n = 4–7). ***p < 0.001, **p < 0.01, *p < 0.05, “a” vs. control, “c” vs. TAA. HA hepatic artery, BD bile duct, CV central vein
Effect of GLB on α-SMA, TGF-β1, NLRP3, ASC, caspase-1, and IL-1β expression
TGF-β1 and α-SMA are well-established markers to evaluate the hepatic fibrogenesis as well as progression of fibrosis. Immunohistochemistry was performed to validate our observation. TAA-treated group showed the enhance expression of α-SMA and TGF-β1, while GLB treatment significantly normalized these expressions (Fig. 7a, b). TAA treatment induced the NLRP3 inflammasome, its adapter ASC, and its downstream caspase-1 and IL-1β expression in TAA-treated group, while treatment with GLB restored the level of same (Fig. 8a–d).

Representative photomicrographs from different groups showed a α-SMA and b TGF-β1 expressions in the liver tissue and the effects of GLB treatment on TAA-induced hepatic fibrosis. Immunopositive area (brown against blue background) is indicated by black arrows. Higher magnifications of the indicated areas were further inserted in the respective panel. All the values are expressed as mean ± SEM (n = 3). ***p < 0.001, **p < 0.01, *p < 0.05, “a” vs. control, “c” vs. TAA


Representative photomicrographs from different groups showed a NLRP3, b ASC, c caspase-1, and d IL-1β expressions in the liver tissue and effect of GLB treatment on TAA-induced hepatic fibrosis. Immunopositive area (brown against blue background) is indicated by black arrows. Higher magnifications of the indicated areas were further inserted in the respective panel. All the values are expressed as mean ± SEM (n = 3). ***p < 0.001, **p < 0.01, “a” vs. control, “c” vs. TAA
Effect of GLB on hepatic stellate cells
Hepatic injury induced by TAA activated the quiescent state of HSC. TAA treatment activated HSC as well as significantly decreased the number of lipid droplets around the nucleus as visualized by TEM imaging. The comparison of HSC morphology was performed with control group. Activated HSC showed myofibroblast-like morphology as observed in TAA-treated group, when compared with control group. The treatment with GLB significantly restored the HSC morphology as well as the number of lipid droplets, when compared to TAA group (Fig. 9).

Representative photomicrographs from different groups showed morphology of HSC nucleus in liver as observed by transmission electron microscopy (TEM). CON and GLB 5 showed normal HSC (area enclosed in black dashed line). Black arrows represent the presence of lipid droplets around the nucleus of HSC. TAA group showed activated HSC with an irregular shape (as indicated by white arrows). Intervention of GLB against TAA with all the doses (TAA + GLB 1.25, 2.5, and 5 mg/kg) restored the changes in morphology as well as restored the number of lipid droplets
Effect of GLB on MMP-2 and catalase
TAA treatment significantly reduced the expressions of MMP-2 and catalase while GLB treatment significantly restored the expressions of same (Fig. 10a, b).

Representative photomicrographs from different groups showed a MMP-2 and b catalase expressions in the liver tissue and effect of GLB treatment on TAA-induced liver damage. All the values are expressed as mean ± SEM (n = 3), ***p < 0.001, **p < 0.01, *p < 0.05, “a” vs. control, “c” vs. TAA
Discussion
Liver fibrosis is a common pathophysiological disorder and initially manifested by the presence of inflammatory response, deposition of ECM, activation of HSC, and scar formation (Ouyang et al. 2013). The liver is one of the most vital organs that maintain the inner workings of the body. The liver serves as the first line of defense against and metabolizes/detoxifies the beneficial/toxic agents that are absorbed from the stomach and intestine. Hepatic lobule is a building block of the liver parenchyma. Lobule contains millions of functional units of liver known as hepatocytes. In addition to oxygen, hepatocytes require a lot of energy to perform their many and varied functions, and that hepatocytes contain abundant mitochondria, which results more production of ROS on the occurrence of any abnormal condition. Out of the total volume of the liver, 70–85% is occupied by parenchymal hepatocytes, and non-parenchymal cells (hepatic stellate cells) constitute only 6.5% of its volume (Kmiec 2001). Parenchymal cells (hepatocytes) are primary cells subjected to oxidative stress-induced injury in the liver. The mitochondria, microsomes, and peroxisomes in parenchymal cells can produce ROS. Moreover, Kupffer, hepatic stellate, and endothelial cells are potentially more exposed or sensitive to oxidative stress-related molecules (Li et al. 2015). The present experiment reported that TAA-induced oxidative stress decreased antioxidant status and increased inflammation-mediated liver damage. TAA treatment led to decrease the feed intake which resulted in decreased final body weight and body weight gain. In addition, TAA treatment increased in liver weight, liver index, and profuse nodular appearance as compared to control animal. Further, treatment with GLB improved the nodular appearance. TAA-induced significant increase in the liver weight might be due to collagen deposition, new angiogenesis, and increase in other fibrotic components. Study conducted by Hao et al. reported that TAA treatment for 8 weeks induced significant decrease in final body weight and significant increase in the liver weight index. Increased hepatic oxidative stress was confirmed by decreased antioxidant enzymes like SOD and catalase as well as increased MDA and ROS levels. GLB treatment at low dose significantly ameliorated these changes and improved the liver antioxidant status. In the present study, significant protection for ALT was observed for the groups TAA + GLB 1.25 and TAA + GLB 2.5, and for AST and ALP, significant protection was observed only for TAA + GLB 1.25 when compared to TAA-treated group. TAA per se treatment significantly elevated the γ-GT level and treatment with GLB has no effect on the same. Assessment of serum transaminases were long way considered to be an important factor to assess human liver abnormalities. Koblihova et al. reported that TAA-exposed Wistar rats have significant increase in plasma ALT and AST levels (Koblihova et al. 2014). Al-Attar also reported mice treated with TAA significantly increased the plasma ALT, AST, ALP, and γ-GT levels (Al-Attar 2012). Histomorphometric findings showed the presence of lymphocyte (inflammatory cells) infiltration, collagen deposition, bridging fibrosis, degeneration of portal triad, and necrosis in the TAA-treated animals, which clearly indicated the development of hepatic fibrosis and treatment with GLB significantly reduced the same. Further, MT, PSR, and PSRF staining revealed treatment with GLB significantly reduced the collagen deposition in the portal and central vein areas as compared to per se TAA treatment. In the present study, the HSC activation was revealed by TEM image analyses and the results indicated that TAA treatment significantly altered the morphology of the HSC and drastically decreased the number of lipid droplets around the HSC nucleus. GLB treatment restored the TAA-induced morphological changes in the HSC as well as restored the lipid droplets. It has also been reported that liver injury activates the HSC and plays a vital role in the development of hepatic fibrosis (Moreira 2007). Normal HSC nucleus looks regular in shape; once activated, it looks like elongated/irregular in shape (Soliman and Selim 2012). Further, the significant decrease of lipid droplets is another indication for the activation of HSC other than shape (Lee et al. 2010; Zhang et al. 2017b). Previous studies reported that, in response to injury, normal HSCs activate into myofibroblasts, which are characterized by an increased proliferation and high contractility with enhanced expression of pericellular matrix protein such as α-SMA (Puche et al. 2013). Activated HSCs not only secrete inflammatory, proliferative, and fibrogenic cytokine TGF-β1, but also express type I and II TGF-β receptors and the urokinase-type plasminogen activator, which activates latent TGF-β1 (Mann and Smart 2002). Immunohistochemistry was performed for α-SMA and TGF-β1. Per se TAA treatment increased the expression of α-SMA and TGF-1, while GLB treatment significantly reduced the overexpression of the same as revealed by DAB-positive cells around the central vein and the sinusoidal space as compared to control.
It has been reported that different stimuli including pathogens are involved in inflammasome activation and its components have been demonstrated in a wide range of fibrotic diseases including liver (Ouyang et al. 2013). As reported earlier, the inflammasomes majorly contribute to the biology of fibrogenesis as well as the progression of fibrosis (Wan et al. 2016). It is also reported that NLRP3 inflammasome complexes are present in hepatocytes, hepatic stellate cells, and Kupffer cells and mediated the inflammatory reaction in response to various stress conditions (Dixon et al. 2013; Meng et al. 2016). NLRP3 activation leads to hepatocyte pyroptosis, liver inflammation, and subsequently fibrosis (Szabo and Petrasek 2015; Wree et al. 2014). It is noteworthy to mention that NLRP3 and ASC knockout mice did not develop hepatic fibrosis when injected with carbon tetrachloride and TAA (Watanabe et al. 2009). Present investigation observed with marked activation of NLRP3 inflammasome complex, its adapter ASC, and its downstream caspase-1 and IL-1β tissue expression were significantly increased in TAA per se treated group, while treatment with GLB significantly normalized the expression of same. Recently, it has already been reported that NLRP3 inflammasome inhibitor decreased the hepatic inflammation and fibrosis in methionine/choline-deficient diet model as well as ameliorated kidney fibrosis in adenine crystal-induced experimental mice model (Ludwig-Portugall et al. 2016; Mridha et al. 2017). Anti-inflammatory and protective effects of GLB have been reported in diverse conditions such as respiration, digestion, urology, cardiology, CNS, melioidosis, ischemia-reperfusion (IR) injury, and septic shock (Zhang et al. 2017a). However, the most common problem with GLB is the wide variations of dose (from 0.15 to 500 mg/kg) as reported in different studies and summarized in Supplementary Table 1. In the present study, comet and TUNEL assay results revealed that TAA treatment significantly increased % DNA, TM, TMO, TL, and % TUNEL-positive cells, while GLB treatment reduced the same. TAA has already been reported as centrilobular hepatotoxicant and induces DNA damage, apoptosis, and necrosis in rat liver (Ledda-Columbano et al. 1991; Uno et al. 2015). Additionally, the expressions of MMP-2 and catalase were evaluated by western blot analyses, and it was observed that TAA significantly decreased the expression of MMP-2 and catalase as compared to control, whereas GLB treatment restored the same. Zhu et al. reported that low-dose GLB induces MMP-2 gelatinolytic activity in high glucose-stimulated rat primary mesangial cells (Zhu et al. 2012). It has already been reported that MMP-2 digests type I, II, and III collagens, thereby reducing the formation of ECM (Van Doren 2015). Earlier literatures reported that catalase is an antioxidant enzyme and serves as the defense against oxidative and other stresses in alloxan-induced diabetic rats (Chukwunonso Obi et al. 2016). Studies in both humans and experimental animal models indicate the protective role of GLB in the liver. GLB prevents the pathological liver damage due to severe hyperglycemia. It raises the insulin levels not only by augmenting the secretion of insulin but also selectively decreasing the catabolism of insulin by the endothelial cell of liver (Mulder et al. 1991; Sokolovska et al. 2012). It is clearly imperative from the present study that the lower dose of GLB is more effective than the higher doses in protecting the liver. Here, it is worthy to mention that the low dose of GLB (which is 40 to 400 times less than the dose usually needed to induce hypoglycemia in rat) has strong beneficial effects in reducing edema, mortality, and lesion volume as well as high therapeutic windows in ischemic stroke model of rat (Simard et al. 2009b). The dose in the present study was selected on the basis that at least one dose should be in the clinical dose range. The low dose 1.25 mg/kg and mid dose 2.5 mg/kg showed more prominent protective effects in the majority of parameters as compared to high dose 5 mg/kg. Similar results were also reported by other investigations such as Simard et al. that reported that low dose of GLB (infusion dose 75 ng/h, loading doses 3 and 33 μg/kg) reduced total lesion volume by 53% in the thromboembolic MCAO model at 6 h (Simard et al. 2009b). Zhu et al. reported that low dose of glibenclamide (0.01 and 1 μM) induced collagen IV catabolism in high glucose-stimulated rat primary mesangial cells (Zhu et al. 2012). However, previous studies have reported the conflicting evidences about the pro- or anti-apoptotic role of sulfonyl ureas, including GLB, which can be attributed due to a variety of mechanisms (Ansar and Ansari 2006; Kwon et al. 2013; Sawada et al. 2008). Recently, it has been reported that the non-alcoholic fatty liver diseases are in between 70 and 80% in patients with metabolic syndrome and T2DM (Masarone et al. 2017). As GLB is one of the important drugs of choice for T2DM, so it is worthy to evaluate its further hepatoprotective effects with appropriate dose ranges employing different experimental markers in both in vitro and in vivo models for future clinical applications particularly in humans with long-term hepatic disorders.
Abbreviations
- ALP:
-
Alkaline phosphatase
- ALT:
-
Alanine transaminase
- ASC:
-
Apoptosis-associated speck-like protein containing a caspase-recruiting domain
- AST:
-
Aspartate transaminase
- ATP:
-
Adenosine triphosphate
- CYP2E1:
-
Cytochrome P450 2E1
- DAB:
-
3,3′-Diaminobenzidine
- DMSO:
-
Dimethyl sulfoxide
- ECM:
-
Extracellular matrix
- GLB:
-
Glibenclamide
- HSC:
-
Hepatic stellate cell
- IL:
-
Interleukin
- IL-1β:
-
Interleukin-1 beta
- ip:
-
Intraperitoneal
- MT:
-
Masson’s trichrome
- MMP-2:
-
Matrix metalloproteinase-2
- NLRP3:
-
NACHT, LRR, and PYD domains-containing protein 3
- TMO:
-
Tail moment olive
- PAS:
-
Periodic acid-Schiff base
- po:
-
Per os (oral administration)
- PSR:
-
Picro Sirius Red
- PSRF:
-
PSR with fast green FCF
- SOD:
-
Superoxide dismutase
- T2DM:
-
Type II diabetes mellitus
- TAA:
-
Thioacetamide
- TEM:
-
Transmission electron microscopy
- TGF-β1:
-
Transforming growth factor beta1
- TL:
-
Tail length
- TM:
-
Tail moment
- TUNEL:
-
Terminal deoxynucleotidyl transferase dUTP nick end labeling assay
- US-FDA:
-
United States Food and Drug Administration
- WHO:
-
World Health Organization
- α-SMA:
-
Alpha-smooth muscle actin
- γ-GT:
-
γ-Glutamyl transferase
References
Aguillar-Gomes L, Lopes C, Barbieri D, Rocha T, Randazzo-Moura P (2014) Toxic effects of glibenclamide in fetuses of normoglycemic rats: an alternative therapy for gestational diabetes mellitus. Open Vet J 4:59–64
Al-Attar AM (2012) Attenuating effect of ginkgo biloba leaves extract on liver fibrosis induced by thioacetamide in mice. J Biomed Biotechnol 2012:9–9. https://doi.org/10.1155/2012/761450
Ansar MM, Ansari M (2006) Nitric oxide involvement in pancreatic beta cell apoptosis by glibenclamide. Nitric oxide : biology and chemistry 14:39–44. https://doi.org/10.1016/j.niox.2005.09.002
Bataller R, Brenner DA (2005) Liver fibrosis. J Clin Invest 115:209–218. https://doi.org/10.1172/jci200524282
Beauchamp C, Fridovich I (1971) Superoxide dismutase: improved assays and an assay applicable to acrylamide gels. Anal Biochem 44:276–287
Beltrand J, Elie C, Busiah K, Fournier E, Boddaert N, Bahi-Buisson N, Vera M, Bui-Quoc E, Ingster-Moati I, Berdugo M, Simon A, Gozalo C, Djerada Z, Flechtner I, Treluyer JM, Scharfmann R, Cave H, Vaivre-Douret L, Polak M (2016) Erratum. Sulfonylurea therapy benefits neurological and psychomotor functions in patients with neonatal diabetes owing to potassium channel mutations. Diabetes Care 2015;38:2033-2041. Diabetes Care 39: 175 https://doi.org/10.2337/dc16-er01
Cai J, Lu S, Yao Z, Deng YP, Zhang LD, Yu JW, Ren GF, Shen FM, Jiang GJ (2014) Glibenclamide attenuates myocardial injury by lipopolysaccharides in streptozotocin-induced diabetic mice. Cardiovasc Diabetol 13:106. https://doi.org/10.1186/s12933-014-0106-y
Chukwunonso Obi B, Chinwuba Okoye T, Okpashi VE, Nonye Igwe C, Olisah Alumanah E (2016) Comparative study of the antioxidant effects of metformin, glibenclamide, and repaglinide in alloxan-induced diabetic rats. J Diabetes Res 2016. doi: https://doi.org/10.1155/2016/1635361, 1, 5
Czaja AJ (2014) Hepatic inflammation and progressive liver fibrosis in chronic liver disease. World J Gastroenterol: WJG 20:2515–2532. https://doi.org/10.3748/wjg.v20.i10.2515
Diwan V, Gobe G, Brown L (2014) Glibenclamide improves kidney and heart structure and function in the adenine-diet model of chronic kidney disease. Pharmacol Res 79:104–110. https://doi.org/10.1016/j.phrs.2013.11.007
Dixon LJ, Barnes M, Tang H, Pritchard MT, Nagy LE (2013) Kupffer cells in the liver. Compr Physiol 3:785–797. https://doi.org/10.1002/cphy.c120026
El-Mihi KA, Kenawy HI, El-Karef A, Elsherbiny NM, Eissa LA (2017) Naringin attenuates thioacetamide-induced liver fibrosis in rats through modulation of the PI3K/Akt pathway. Life Sci 187:50–57. https://doi.org/10.1016/j.lfs.2017.08.019
Friedman SL (2004) Mechanisms of disease: mechanisms of hepatic fibrosis and therapeutic implications. Nat Clin Pract Gastroenterol Hepatol 1:98–105. https://doi.org/10.1038/ncpgasthep0055
Gad SC, Cassidy CD, Aubert N, Spainhour B, Robbe H (2006) Nonclinical vehicle use in studies by multiple routes in multiple species. Int J Toxicol 25:499–521. https://doi.org/10.1080/10915810600961531
Hughes FM Jr, Vivar NP, Kennis JG, Pratt-Thomas JD, Lowe DW, Shaner BE, Nietert PJ, Spruill LS, Purves JT (2014) Inflammasomes are important mediators of cyclophosphamide-induced bladder inflammation. American journal of physiology Renal physiology 306:F299–F308. https://doi.org/10.1152/ajprenal.00297.2013
Kewcharoenwong C, Rinchai D, Utispan K, Suwannasaen D, Bancroft GJ, Ato M, Lertmemongkolchai G (2013) Glibenclamide reduces pro-inflammatory cytokine production by neutrophils of diabetes patients in response to bacterial infection. Sci Rep 3:3363. https://doi.org/10.1038/srep03363
Khan S, Jena G (2014a) Sodium butyrate, a HDAC inhibitor ameliorates eNOS, iNOS and TGF-beta1-induced fibrogenesis, apoptosis and DNA damage in the kidney of juvenile diabetic rats. Food Chem Toxicol 73:127–139. https://doi.org/10.1016/j.fct.2014.08.010
Khan S, Jena G (2014b) Sodium valproate, a histone deacetylase inhibitor ameliorates cyclophosphamide-induced genotoxicity and cytotoxicity in the colon of mice. J Basic Clin Physiol Pharmacol: 1-11 https://doi.org/10.1515/jbcpp-2013-0134
Kim K-A, Park J-Y (2003) Inhibitory effect of glyburide on human cytochrome p450 isoforms in human liver microsomes. Drug Metab Dispos 31:1090–1092. https://doi.org/10.1124/dmd.31.9.1090
Kim NH, Lee S, Kang MJ, Jeong HG, Kang W, Jeong TC (2014) Protective effects of diallyl sulfide against thioacetamide-induced toxicity: a possible role of cytochrome P450 2E1. Biomol Ther 22:149–154. https://doi.org/10.4062/biomolther.2014.016
Kimberly WT, Battey TW, Pham L, Wu O, Yoo AJ, Furie KL, Singhal AB, Elm JJ, Stern BJ, Sheth KN (2014) Glyburide is associated with attenuated vasogenic edema in stroke patients. Neurocrit Care 20:193–201. https://doi.org/10.1007/s12028-013-9917-z
Kmiec Z (2001) Cooperation of liver cells in health and disease. Advances in anatomy, embryology, and cell biology 161: iii-xiii, 1-151
Koblihova E, Mrazova I, Vernerova Z, Ryska M (2014) Acute liver failure induced by thioacetamide: selection of optimal dosage in Wistar and Lewis rats. Physiol Res 63:491–503
Koh GC, Weehuizen TA, Breitbach K, Krause K, de Jong HK, Kager LM, Hoogendijk AJ, Bast A, Peacock SJ, van der Poll T, Steinmetz I, Wiersinga WJ (2013) Glyburide reduces bacterial dissemination in a mouse model of melioidosis. PLoS Negl Trop Dis 7:e2500. https://doi.org/10.1371/journal.pntd.0002500
Kwon MJ, Chung HS, Yoon CS, Lee EJ, Kim TK, Lee SH, Ko KS, Rhee BD, Kim MK, Park JH (2013) Low glibenclamide concentrations affect endoplasmic reticulum stress in INS-1 cells under glucotoxic or glucolipotoxic conditions. The Korean journal of internal medicine 28:339–346. https://doi.org/10.3904/kjim.2013.28.3.339
Lamkanfi M, Mueller JL, Vitari AC, Misaghi S, Fedorova A, Deshayes K, Lee WP, Hoffman HM, Dixit VM (2009) Glyburide inhibits the cryopyrin/Nalp3 inflammasome. J Cell Biol 187:61–70. https://doi.org/10.1083/jcb.200903124
Ledda-Columbano GM, Coni P, Curto M, Giacomini L, Faa G, Oliverio S, Piacentini M, Columbano A (1991) Induction of two different modes of cell death, apoptosis and necrosis, in rat liver after a single dose of thioacetamide. Am J Pathol 139:1099–1109
Lee SW, Kim SH, Min SO, Kim KS (2011) Ideal experimental rat models for liver diseases. Korean J Hepatobiliary Pancreat Surg 15:67–77. https://doi.org/10.14701/kjhbps.2011.15.2.67
Lee TF, Mak KM, Rackovsky O, Lin YL, Kwong AJ, Loke JC, Friedman SL (2010) Downregulation of hepatic stellate cell activation by retinol and palmitate mediated by adipose differentiation-related protein (ADRP). J Cell Physiol 223:648–657. https://doi.org/10.1002/jcp.22063
Li S, Tan HY, Wang N, Zhang ZJ, Lao L, Wong CW, Feng Y (2015) The role of oxidative stress and antioxidants in liver diseases. Int J Mol Sci 16:26087–26124. https://doi.org/10.3390/ijms161125942
Liu Y, Meyer C, Xu C, Weng H, Hellerbrand C, ten Dijke P, Dooley S (2013) Animal models of chronic liver diseases. Am J Physiol Gastrointest Liver Physiol 304:G449–G468. https://doi.org/10.1152/ajpgi.00199.2012
Ludwig-Portugall I, Bartok E, Dhana E, Evers BD, Primiano MJ, Hall JP, Franklin BS, Knolle PA, Hornung V, Hartmann G, Boor P, Latz E, Kurts C (2016) An NLRP3-specific inflammasome inhibitor attenuates crystal-induced kidney fibrosis in mice. Kidney Int 90:525–539. https://doi.org/10.1016/j.kint.2016.03.035
Mann DA, Smart DE (2002) Transcriptional regulation of hepatic stellate cell activation. Gut 50:891–896
Maremanda KP, Khan S, Jena G (2014) Zinc protects cyclophosphamide-induced testicular damage in rat: involvement of metallothionein, tesmin and Nrf2. Biochem Biophys Res Commun 445:591–596. https://doi.org/10.1016/j.bbrc.2014.02.055
Maremanda KP, Khan S, Jena GB (2016) Role of zinc supplementation in testicular and epididymal damages in diabetic rat: involvement of Nrf2, SOD1, and GPX5. Biol Trace Elem Res 173:452–464. https://doi.org/10.1007/s12011-016-0674-7
Marumo T, Hishikawa K, Yoshikawa M, Hirahashi J, Kawachi S, Fujita T (2010) Histone deacetylase modulates the proinflammatory and -fibrotic changes in tubulointerstitial injury. Am J Physiol Renal Physiol 298:F133–F141. https://doi.org/10.1152/ajprenal.00400.2009
Masarone M, Rosato V, Aglitti A, Bucci T, Caruso R, Salvatore T, Sasso FC, Tripodi MF, Persico M (2017) Liver biopsy in type 2 diabetes mellitus: steatohepatitis represents the sole feature of liver damage. PLoS One 12:e0178473. https://doi.org/10.1371/journal.pone.0178473
Meng N, Xia M, Lu YQ, Wang M, Boini KM, Li PL, Tang WX (2016) Activation of NLRP3 inflammasomes in mouse hepatic stellate cells during Schistosoma. J Infection Oncotarget 7:39316–39331. https://doi.org/10.18632/oncotarget.10044
Mirzamoradi M, Heidar Z, Faalpoor Z, Naeiji Z, Jamali R (2015) Comparison of glyburide and insulin in women with gestational diabetes mellitus and associated perinatal outcome: a randomized clinical trial. Acta Med Iran 53:97–103
Mokdad AA, Lopez AD, Shahraz S, Lozano R, Mokdad AH, Stanaway J, Murray CJL, Naghavi M (2014) Liver cirrhosis mortality in 187 countries between 1980 and 2010: a systematic analysis. BMC Med 12:145. https://doi.org/10.1186/s12916-014-0145-y
Moreira RK (2007) Hepatic stellate cells and liver fibrosis. Arch Pathol Lab Med 131:1728–1734. https://doi.org/10.1043/1543-2165(2007)131[1728:hscalf]2.0.co;2
Mridha AR, Wree A, Robertson AAB, Yeh MM, Johnson CD, Van Rooyen DM, Haczeyni F, Teoh NC, Savard C, Ioannou GN, Masters SL, Schroder K, Cooper MA, Feldstein AE, Farrell GC (2017) NLRP3 inflammasome blockade reduces liver inflammation and fibrosis in experimental NASH in mice. J Hepatol 66:1037–1046. https://doi.org/10.1016/j.jhep.2017.01.022
Mulder H, Schopman W Sr, van der Lely AJ (1991) Extrapancreatic insulin effect of glibenclamide. Eur J Clin Pharmacol 40:379–381
Ouyang X, Ghani A, Mehal WZ (2013) Inflammasome biology in fibrogenesis. Biochim Biophys Acta 1832:979–988. https://doi.org/10.1016/j.bbadis.2013.03.020
Puche JE, Saiman Y, Friedman SL (2013) Hepatic stellate cells and liver fibrosis. Comprehensive Physiology 3:1473–1492. https://doi.org/10.1002/cphy.c120035
Rowe IA (2017) Lessons from epidemiology: the burden of liver disease. Dig Dis 35:304–309. https://doi.org/10.1159/000456580
Sawada F, Inoguchi T, Tsubouchi H, Sasaki S, Fujii M, Maeda Y, Morinaga H, Nomura M, Kobayashi K, Takayanagi R (2008) Differential effect of sulfonylureas on production of reactive oxygen species and apoptosis in cultured pancreatic beta-cell line, MIN6. Metab Clin Exp 57:1038–1045. https://doi.org/10.1016/j.metabol.2008.01.038
Schmid D, Svoboda M, Sorgner A, Moravcevic I, Thalhammer T, Chiba P, Moslinger T (2011) Glibenclamide reduces proinflammatory cytokines in an ex vivo model of human endotoxinaemia under hypoxaemic conditions. Life Sci 89:725–734. https://doi.org/10.1016/j.lfs.2011.08.017
Sheth KN, Elm JJ, Molyneaux BJ, Hinson H, Beslow LA, Sze GK, Ostwaldt AC, Del Zoppo GJ, Simard JM, Jacobson S, Kimberly WT (2016) Safety and efficacy of intravenous glyburide on brain swelling after large hemispheric infarction (GAMES-RP): a randomised, double-blind, placebo-controlled phase 2 trial. Lancet Neurol 15:1160–1169. https://doi.org/10.1016/s1474-4422(16)30196-x
Sheth KN, Kimberly WT, Elm JJ, Kent TA, Yoo AJ, Thomalla G, Campbell B, Donnan GA, Davis SM, Albers GW, Jacobson S, del Zoppo G, Simard JM, Stern BJ, Mandava P (2014) Exploratory analysis of glyburide as a novel therapy for preventing brain swelling. Neurocrit Care 21:43–51. https://doi.org/10.1007/s12028-014-9970-2
Simard JM, Geng Z, Woo SK, Ivanova S, Tosun C, Melnichenko L, Gerzanich V (2009a) Glibenclamide reduces inflammation, vasogenic edema, and caspase-3 activation after subarachnoid hemorrhage. J Cereb Blood Flow Metab 29:317–330. https://doi.org/10.1038/jcbfm.2008.120
Simard JM, Yurovsky V, Tsymbalyuk N, Melnichenko L, Ivanova S, Gerzanich V (2009b) Protective effect of delayed treatment with low-dose glibenclamide in three models of ischemic stroke. Stroke 40:604–609. https://doi.org/10.1161/strokeaha.108.522409
Socci DJ, Bjugstad KB, Jones HC, Pattisapu JV, Arendash GW (1999) Evidence that oxidative stress is associated with the pathophysiology of inherited hydrocephalus in the H-Tx rat model. Exp Neurol 155:109–117. https://doi.org/10.1006/exnr.1998.6969
Sokolovska J, Isajevs S, Sugoka O, Sharipova J, Paramonova N, Isajeva D, Rostoka E, Sjakste T, Kalvinsh I, Sjakste N (2012) Comparison of the effects of glibenclamide on metabolic parameters, GLUT1 expression, and liver injury in rats with severe and mild streptozotocin-induced diabetes mellitus. Medicina (Kaunas, Lithuania) 48: 532-543
Soliman HM, Selim AO (2012) Role of hepatic stellate cells in fibrogenesis in a model of pomegranate-treated fatty liver induced by junk food in male albino rats immunohistochemical and electron microscopic study. Egyptian Journal of Histology 35:54–66. https://doi.org/10.1097/01.EHX.0000410947.56955.e0
Song R, Chen L, Chen Y, Si X, Liu Y, Liu Y, Irwin DM (2017) Comparison of glyburide and insulin in the management of gestational diabetes: a meta-analysis 12: e0182488 https://doi.org/10.1371/journal.pone.0182488
Spirli C, Fiorotto R, Song L, Santos-Sacchi J, Okolicsanyi L, Masier S, Rocchi L, Vairetti MP, De Bernard M, Melero S, Pozzan T, Strazzabosco M (2005) Glibenclamide stimulates fluid secretion in rodent cholangiocytes through a cystic fibrosis transmembrane conductance regulator-independent mechanism. Gastroenterology 129:220–233. https://doi.org/10.1053/j.gastro.2005.03.048
Szabo G, Petrasek J (2015) Inflammasome activation and function in liver disease. Nat Rev Gastroenterol Hepatol 12:387–400. https://doi.org/10.1038/nrgastro.2015.94
Tsai MK, Lin YL, Huang YT (2010) Effects of salvianolic acids on oxidative stress and hepatic fibrosis in rats. Toxicol Appl Pharmacol 242:155–164. https://doi.org/10.1016/j.taap.2009.10.002
Uno Y, Kojima H, Omori T, Corvi R, Honma M, Schechtman LM, Tice RR, Beevers C, De Boeck M, Burlinson B, Hobbs CA, Kitamoto S, Kraynak AR, McNamee J, Nakagawa Y, Pant K, Plappert-Helbig U, Priestley C, Takasawa H, Wada K, Wirnitzer U, Asano N, Escobar PA, Lovell D, Morita T, Nakajima M, Ohno Y, Hayashi M (2015) JaCVAM-organized international validation study of the in vivo rodent alkaline comet assay for detection of genotoxic carcinogens: II. Summary of definitive validation study results. Mutat Res Genet Toxicol Environ Mutagen 786-788:45–76. https://doi.org/10.1016/j.mrgentox.2015.04.010
Van Doren SR (2015) Matrix metalloproteinase interactions with collagen and elastin. Matrix Biol 0: 224–231 https://doi.org/10.1016/j.matbio.2015.01.005, 44-46
Wan X, Xu C, Yu C, Li Y (2016) Role of NLRP3 inflammasome in the progression of NAFLD to NASH. Can J Gastroenterol Hepatol 2016, doi: https://doi.org/10.1155/2016/6489012, 1, 7
Watanabe A, Sohail MA, Gomes DA, Hashmi A, Nagata J, Sutterwala FS, Mahmood S, Jhandier MN, Shi Y, Flavell RA, Mehal WZ (2009) Inflammasome-mediated regulation of hepatic stellate cells. Am J Physiol Gastrointest Liver Physiol 296:G1248–G1257. https://doi.org/10.1152/ajpgi.90223.2008
WHO (2015) The top 10 cause of death in lower-middle-income economies 2015. WHO & world bank available online on http://wwwwhoint/mediacentre/factsheets/fs310/en/index1html last accessed on september 9, 2017
Wree A, Eguchi A, McGeough MD, Pena CA, Johnson CD, Canbay A, Hoffman HM, Feldstein AE (2014) NLRP3 inflammasome activation results in hepatocyte pyroptosis, liver inflammation, and fibrosis in mice. Hepatology (Baltimore, Md) 59:898–910. https://doi.org/10.1002/hep.26592
Wynn TA (2008) Cellular and molecular mechanisms of fibrosis. J Pathol 214:199–210. https://doi.org/10.1002/path.2277
York JM, Castellanos KJ, Cabay RJ, Fantuzzi G (2014) Inhibition of the NLRP3 inflammasome reduces the severity of experimentally-induced acute pancreatitis in obese mice. Transl Res 164:259–269. https://doi.org/10.1016/j.trsl.2014.06.012
Zhang G, Lin X, Zhang S, Xiu H, Pan C, Cui W (2017a) A protective role of glibenclamide in inflammation-associated injury. 2017: 3578702 https://doi.org/10.1155/2017/3578702
Zhang Z, Zhao S, Yao Z, Wang L, Shao J, Chen A, Zhang F, Zheng S (2017b) Autophagy regulates turnover of lipid droplets via ROS-dependent Rab25 activation in hepatic stellate cell. Redox Biol 11:322–334. https://doi.org/10.1016/j.redox.2016.12.021
Zhu L, Cortes P, Hassett C, Taube DW, Yee J (2012) Glibenclamide induces collagen IV catabolism in high glucose-stimulated mesangial cells. Exp Diabetes Res 2012:183535–183539. https://doi.org/10.1155/2012/183535
Acknowledgements
We wish to acknowledge the financial assistance received from National Institute of Pharmaceutical Education and Research (NIPER), S.A.S. Nagar, India, for carrying out the above experimentation. The authors would like to acknowledge Mr. Vinod Kumar for TEM image analysis. We are also grateful to Dr. Sabbir Khan, Dr. Krishna Prahlad Maremanda, and Dr. Venkateswara Rao Amara for their timely advice and technical support.
Author information
Authors and Affiliations
Contributions
DKD conceived, conducted the experiments, analyzed the data, and wrote the manuscript. GBJ conceived the idea, reviewed the manuscript, and administered the project. Finally, both the authors have read and approved the manuscript.
Corresponding author
Ethics declarations
The experimental protocol was approved by the Institutional Animal Ethics Committee (IAEC) against approval number IAEC/16/54.
Conflict of interest
The authors declare that they have no conflict of interest.
Electronic supplementary material
ESM 1
(DOCX 17354 kb)
Rights and permissions
About this article

Cite this article
Dwivedi, D.K., Jena, G.B. Glibenclamide protects against thioacetamide-induced hepatic damage in Wistar rat: investigation on NLRP3, MMP-2, and stellate cell activation. Naunyn-Schmiedeberg's Arch Pharmacol 391, 1257–1274 (2018). https://doi.org/10.1007/s00210-018-1540-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00210-018-1540-2