
Abstract
Acetaminophen (APAP) is a widely used analgesic and is safe at therapeutic doses. However, an overdose of APAP is hepatotoxic and accidental overdoses are increasingly common due to the presence of APAP in several combination medications. Formation of protein adducts (APAP-CYS) is central to APAP-induced liver injury and their removal by autophagy is an essential adaptive response after an acute overdose. Since the typical treatment for conditions such as chronic pain involves multiple doses of APAP over time, this study investigated APAP-induced liver injury after multiple subtoxic doses and examined the role of autophagy in responding to this regimen. Fed male C57BL/6J mice were administered repeated doses (75 mg/kg and 150 mg/kg) of APAP, followed by measurement of adducts within the liver, mitochondria, and in plasma, activation of the MAP kinase JNK, and markers of liver injury. The role of autophagy was investigated by treatment of mice with the autophagy inhibitor, leupeptin. Our data show that multiple treatments at the 150 mg/kg dose of APAP resulted in protein adduct formation in the liver and mitochondria, activation of JNK, and hepatocyte cell death, which was significantly exacerbated by inhibition of autophagy. While repeated dosing with the milder 75 mg/kg dose did not cause mitochondrial protein adduct formation, JNK activation, or liver injury, autophagy inhibition resulted in hepatocyte death even at this lower dose. These data illustrate the importance of adaptive responses such as autophagy in removing protein adducts and preventing liver injury, especially in clinically relevant situations involving repeated dosing with APAP.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
At therapeutic doses, acetaminophen (APAP) is a safe and effective analgesic and antipyretic drug; however, an overdose can cause severe liver injury or even acute liver failure (Jaeschke 2015; Lancaster et al. 2015; Yoon et al. 2016). Patients either intentionally ingest a single large overdose in a suicide attempt or overdose unintentionally by taking different medications that contain APAP (Alhelail et al. 2011). In the latter case, patients are not aware that numerous over-the-counter drugs including cold and flu mediations and sleep-aids all contain various doses of APAP (Kelly et al. 2018). Even when adhering to the recommended dosing of individual preparations, taking several of them can lead to a moderate overdose of APAP (Alhelail et al. 2011). Because this may occur over several days and the symptoms are initially milder, the patient may not recognize the problem and will not generally seek medical attention immediately, resulting in a delay in treatment. This is thought to be the main reason why the detrimental clinical outcome (liver injury and liver failure) is often more severe in unintentional overdose patients (Lancaster et al. 2015).
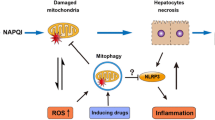
A therapeutic dose of APAP is rapidly metabolized in the liver by phase II reactions (glucuronidation and sulfation), but a small portion of less than 10% of the dose is converted to a reactive metabolite, presumably N-acetyl-p-benzoquinone imine (NAPQI) (McGill and Jaeschke 2013). NAPQI is detoxified by glutathione and only very limited amounts of protein adducts are formed after a therapeutic dose (Curry et al. 2019; Heard et al. 2011; McGill et al. 2013). After an overdose, NAPQI formation is increased, hepatic GSH is depleted and protein adducts are formed in larger quantities (McGill and Jaeschke 2013). Most of the adducts are a reaction of NAPQI with sulfhydryl groups of proteins (Hoffmann et al. 1985). The adducts can be found in both the cytosol and the mitochondria (Tirmenstein and Nelson 1989). However, a comparison between APAP binding and toxicity and its analog N-acetyl-m-aminophenol (AMAP) in mice showed that both compounds caused protein adduct formation in the cytosol, but only APAP caused protein adducts in the mitochondria, which correlated with liver injury (Tirmenstein and Nelson 1989; Xie et al. 2015). Mitochondrial adducts are initially responsible for a mitochondrial oxidant stress (Nguyen et al. 2021), which triggers a complex MAP kinase cascade activation ultimately resulting in activation of c-jun N-terminal kinase (JNK) and the translocation of phosphorylated-JNK to the mitochondria (Hanawa et al. 2008; Win et al. 2018). This amplifies the mitochondrial oxidant stress (Saito et al. 2010), which then triggers the mitochondrial permeability transition pore (MPTP) opening, thereby causing the collapse of the membrane potential and cessation of ATP synthesis (Kon et al. 2004). As a consequence of the MPTP formation, there is matrix swelling and rupture of the outer mitochondrial membrane, with release of intermembrane proteins—some of which (e.g., endonuclease G) translocate to the nucleus and cause DNA fragmentation (Bajt et al. 2006). More recently, adaptive mechanisms to the injury stress came into focus. This includes the removal of damaged mitochondria by mitophagy (Ni et al. 2012; Wang et al. 2019) and replacement by mitochondrial biogenesis (Du et al. 2017). These events are most effective at the periphery of the necrotic area (Ni et al. 2013) where mitophagy and biogenesis limit cell death and promote recovery (Jaeschke et al. 2019).
Although mitochondrial protein adduct formation is considered a critical initiating event in the cell death mechanisms of an acute APAP overdose, the role of cytosolic protein adducts in the pathophysiology remains unclear. We have shown that cytosolic adducts are also removed by autophagy (Ni et al. 2016); however, it is difficult to separate the impact of autophagic removal of mitochondrial adducts from the autophagic degradation of cytosolic proteins after a high overdose that causes severe hepatotoxicity. To address this important issue, we investigated the accumulation of cytosolic adducts after multiple, moderate overdoses and assessed the impact of autophagy inhibition on liver injury.
Materials and methods
Animals
Male C57BL/6 J mice (8–12 weeks old) were purchased from Jackson Laboratories (Bar Harbor, ME) and kept in an environmentally controlled room with 12 h dark/light cycle. The animals had ad libitum access to food and water. Food was removed right before APAP injection. APAP was dissolved in warm saline and injected i.p. with doses of 75 or 150 mg/kg every 2 h. Leupeptin (40 mg/kg) (Sigma, St. Louis, MO) and/or 4-methylpyrazole (50 mg/kg) were dissolved in saline and were co-treated with the first dose of APAP. The animals were supplied with only water during the experiments. Blood was drawn from the caudal vena cava into syringes containing 50 µl of heparin, and plasma was obtained after that by centrifugation at 18,000g for 2 min. A section was taken from the left lobe of the liver and fixed in 10% phosphate-buffered formalin for histology. The caudate and right lobes were used for mitochondrial isolation, and the remaining portions were cut into small pieces and flash frozen in liquid nitrogen for later biochemical analysis. All experimental protocols followed the criteria of the National Research Council for the care and use of laboratory animals and were approved by the Institutional Animal Care and Use Committee of the University of Kansas Medical Center.
Mitochondria isolation
Caudate and right lobes of the liver were homogenized quickly in mitochondria isolation buffer (220 mM Mannitol, 70 mM sucrose, 2.5 mM HEPES, 10 mM EDTA, 1 mM EGTA, and 0.1% bovine serum albumin, pH 7.4) after dissection using a tight-fitting Teflon pestle. Mitochondrial and lysosomal/cytosolic fractions were isolated by differential centrifugation as described in detail (McGill et al. 2012).
Biochemical assays
Plasma ALT activities were measured using an ALT kit (Pointe Scientific, MI). Hepatic levels of glutathione were measured with a modified Tietze assay as described in detail (McGill and Jaeschke 2015).
APAP-protein adduct measurement
Small pieces of liver and isolated mitochondria were homogenized in 10 mM sodium acetate (pH 6.5) and the supernatants were collected after centrifugation at 16,000g for 5 min. To remove low-molecular-weight compounds including APAP-GSH conjugates and its metabolites that might interfere with detection, the liver homogenates were filtered through Bio-Spin 6 columns (Bio-Rad, Hercules, CA), which were pre-washed with 10 mM sodium acetate. The filtered samples were digested with proteases to free APAP-CYS from proteins overnight and then precipitated using 40% TCA for liver tissue or cold acetonitrile for mitochondrial samples. The supernatant of liver tissues was pelleted by centrifugation using filtered tubes. The supernatant of mitochondria samples was evaporated at 55 °C and 16 psi and the protein-derived APAP-CYS containing residues were re-suspended in small volumes of 10 mM sodium acetate buffer with 20% TCA. APAP-CYS was measured using HPLC with electrochemical detection as described (McGill et al. 2012; Muldrew et al. 2002).
Western blots
The primary antibodies used in this study were rabbit anti-JNK (Cat # 9252S), rabbit anti-p-JNK (Cat # 4668S), rabbit anti-LC3-II (Cat #12,741), and rabbit anti-β-actin (Cat # 4970L) from Cell Signaling Technology, Inc. (Danvers, MA).
Histology
Formalin-fixed tissue samples were embedded in paraffin and sections were cut and transferred to glass slides. The sections were stained with hematoxylin and eosin (H&E) for necrosis assessment. Terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) was used to stain for DNA fragmentation with the In Situ Cell Death Detection Kit, AP (Roche Diagnostic, Indianapolis, IN).
Statistics
All data were expressed as mean ± SEM. For normally distributed data, statistical significance was evaluated using the Student’s t test for comparisons between two groups, or one-way analysis of variance (ANOVA) for multiple groups, followed by Student–Newman–Keul’s test. For non-normally distributed data, ANOVA was performed using Kruskal–Wallis Test, followed by Dunn’s multiple comparisons. P < 0.05 was considered significant.
Results
Dose–response of liver injury after multiple doses of APAP
Mice were treated with one to five doses of 75 mg/kg or 150 mg/kg APAP with 2 h between each dose and sacrificed 2 h after the last dose. Multiple doses of 75 mg/kg APAP did not cause any significant increase in plasma ALT activities suggesting no liver injury (Fig. 1a). However, plasma ALT activities started to increase after four doses of 150 mg/kg APAP and then continued to further increase after the fifth dose (Fig. 1a). No necrotic hepatocytes were observed after three doses of 150 mg/kg APAP (Fig. 1b). In contrast, minor necrosis after four doses and extensive centrilobular necrosis was evident after five doses (Fig. 1b). These results showed that repeated administration of subtoxic doses can potentially lead to acute liver injury. Consistent with these findings, JNK activation, a hallmark of APAP hepatotoxicity, was only observed after four or five doses of 150 mg/kg APAP, which correlates with the injury (Fig. 1c). In contrast, no JNK activation was observed after 75 mg/kg dosing (data not shown).

Dose–response of liver injury after multiple doses of APAP. Mice were treated with 1, 2, 3, 4, or 5 doses of APAP (75 mg/kg or 150 mg/kg), 2 h between each dose, and sacrificed 2 h after the last dose. a Plasma alanine aminotransferase (ALT) activities. Data represent mean ± SEM of n = 4 animals per group and time point. *p < 0.05 compared to control (0 dose); b H&E staining and TUNEL staining of representative liver sections after 3, 4, or 5 doses of 150 mg/kg APAP. Asterisks indicate central veins. c Western blots of JNK, P-JNK, and β-actin in controls and after 2–5 doses of APAP
Accumulation of protein adducts with increasing number of doses of APAP
To investigate the relationship between plasma ALT activities and presence of APAP-protein adducts, we measured APAP-protein adducts (APAP-CYS) in the whole liver, mitochondria, and in plasma. Our data showed that doses of 150 mg/kg APAP in fed mice progressively increased protein adducts in the total liver and in mitochondria (Fig. 2a, b). Plasma protein adducts did not increase until after the fourth dose (Fig. 2c). There was only a mild increase of adducts in the liver after the fourth and fifth dose of 75 mg/kg APAP and only barely detectable adduct levels in mitochondria (Fig. 2a, b). Plasma adducts were below the limit of detection (0.005 nmol/ml) after 75 mg/kg (Fig. 2c). Hepatic glutathione levels showed a minor decline after 3–5 doses of 75 mg/kg APAP (Fig. 2d). In contrast, doses of 150 mg/kg triggered a progressive depletion of GSH which reached levels of − 65 to − 75% of baseline values at 3 and 5 doses, respectively (Fig. 2d).

Accumulation of protein adducts after multiple doses of APAP. Protein adducts (APAP-CYS) formation in mice treated with multiple doses of APAP 75 mg/kg or 150 mg/kg. a Total liver. b Mitochondria. c Plasma. d Total glutathione (GSH) content in control livers and after treatment with multiple doses of APAP. Data represent mean ± SEM of n = 4 animals per group and time point. *p < 0.05 compared to control
Effect of autophagy on protein adducts accumulation after multiple doses of APAP
We previously showed that autophagy is essential for adduct removal after a single dose of APAP (Ni et al. 2016). To assess the role of autophagy after multiple subtoxic doses, we treated mice with leupeptin, an inhibitor of multiple proteases, most of which are found in lysosomes. Thus, leupeptin prevents autophagic protein degradation (Ni et al. 2016). Three doses of 150 mg/kg APAP did not cause liver injury (Fig. 3a). In contrast, co-treatment of leupeptin with the first dose of APAP resulted in significant increases of ALT activities (Fig. 3a). These results were confirmed with H&E staining of liver sections showing significant necrosis after three doses of 150 mg/kg APAP and leupeptin treatment (Fig. 3e). In addition, TUNEL-positive cells were found in the centrilobular area. Significant increases in LC3-II in the leupeptin-treated animals support the conclusion that autophagic flux was inhibited (Fig. 3b). Measurement of protein adducts indicated moderate adduct levels after three doses of 150 mg/kg in the whole liver and in mitochondria and very low levels in plasma (Fig. 3d). Leupeptin co-treatment significantly increased adduct levels in the liver, in mitochondria, and in plasma (Fig. 3d). Consistent with the observations of increased mitochondrial adducts formation and injury, leupeptin-treated animals showed JNK activation in the cytosol (Fig. 3c).

Impact of autophagy interference on liver injury after multiple subtoxic doses of APAP. Mice were treated with three doses of APAP 150 mg/kg with or without co-treatment of leupeptin. a Plasma ALT. b Western blot for LC-3II. c Western blot of p-JNK and JNK. d Protein adducts in the liver and mitochondria and in plasma. e Representative liver sections stained with H&E and TUNEL of mice 2 h after the last dose of APAP 150 mg/kg ± leupeptin. Asterisks indicate central veins. Data represent mean ± SEM of n = 4 animals per group. *p < 0.05 compared to APAP alone
When the effect of leupeptin was assessed after three doses of 75 mg/kg APAP, plasma ALT activities increased from baseline levels after APAP alone to about 500 U/L 2 h after the last dose of APAP (Fig. 4a). Although single-cell necrosis is difficult to see in the H&E-stained sections, the TUNEL assay clearly shows that there is no cell death after 3 doses of 75 mg/kg APAP alone, but the additional treatment with leupeptin caused cell death of individual hepatocytes (Fig. 4e). LC3-II levels also increased significantly, indicating that leupeptin indeed inhibited autophagy (Fig. 4b). The limited formation of APAP-protein adducts after APAP alone was again enhanced by 100% in the liver after leupeptin treatment (Fig. 4c). Interestingly, adduct levels in the mitochondria were very low and leupeptin had no effect on mitochondrial adducts (Fig. 4c). There was also no JNK activation after 75 mg/kg APAP alone and only a very mild activation with leupeptin co-treatment (Fig. 4d). The limited JNK activation with the lower dose of APAP + leupeptin is further demonstrated when directly compared to samples after 150 mg/kg APAP + leupeptin (Fig. 4d).

Impact of autophagy interference on liver injury after multiple low doses of APAP. Mice were treated with three doses of APAP 75 mg/kg with or without co-treatment of leupeptin. a Plasma ALT. b Western blot for LC-3II. c Protein adducts in the liver and mitochondria. d Western blot of p-JNK and JNK. e Representative liver sections stained with H&E and TUNEL of mice 2 h after the last dose of APAP 75 mg/kg ± leupeptin treatment. Asterisks indicate central veins. Data represent mean ± SEM of n = 4 animals per group. *p < 0.05 compared to APAP alone
In the previous experiments, liver injury was evaluated 2 h after the last dose of APAP. To investigate whether this injury can progress even when APAP treatments were stopped, plasma ALT activities were measured 15 h after the last dose of 75 mg/kg APAP + leupeptin. The ALT activities more than doubled at that time, indicating that the cell death process continued (Fig. 5a). Nevertheless, co-treatment with the P450 inhibitor 4-methylpyrazole (Akakpo et al. 2018) eliminated the increase in plasma ALT activities in the APAP + leupeptin group (Fig. 5a). The enhanced injury with leupeptin treatment was also confirmed by histology and the TUNEL assay (Fig. 5d). In addition, protein adducts, which were elevated in the liver but barely detectable in the mitochondria or plasma with these doses of APAP, significantly increased in all three compartments by 15 h post-APAP when the animals were co-treated with leupeptin (Fig. 5c). Again, LC3-II levels increased after leupeptin indicating the inhibition of autophagy in these animals (Fig. 5b). However, no JNK activation was detectable after three doses of 75 mg/kg APAP with or without co-treatment of leupeptin by this 15 h time point (data not shown).

Progression of liver injury in mice treated with multiple doses of APAP. Mice were treated with three doses of APAP 75 mg/kg with or without co-treatment of leupeptin. a Plasma ALT. b Western blot for LC-3II. c Protein adducts in the liver, mitochondria, and in plasma. d Representative liver sections stained with H&E and TUNEL of mice 15 h after treatment with three doses of APAP 75 mg/kg ± leupeptin. Asterisks indicate central veins. Data represent mean ± SEM of n = 4 animals per group. *p < 0.05 compared to APAP alone
Discussion
The objective of the current study was to evaluate a potential role of non-mitochondrial protein adducts in the mechanism of toxicity and the impact of autophagy. Using two different subtoxic doses and repeated treatments, we confirmed the role of mitochondrial protein adducts with the higher doses, but could provide evidence for an effect of non-mitochondrial protein adducts when not removed by autophagy.
Mitochondrial APAP-protein adduct accumulation and liver injury after multiple doses of APAP
Treatment of fasted mice with a single dose of 150 mg/kg APAP caused rapid GSH depletion but a full recovery by 4–5 h after APAP administration (McGill et al. 2013). Moderate protein adduct levels were observed in the total liver and in mitochondria together with JNK activation, in part reversible mitochondrial dysfunction, and mild liver injury (McGill et al. 2013; Hu et al. 2016). In the present study, using fed mice with higher baseline GSH levels, a single dose of 150 mg/kg APAP triggered only very limited adduct formation in the liver but not in mitochondria and did not cause JNK activation or ALT increases. However, when these doses were repeated every 2 h, which is not long enough to fully recover hepatic GSH levels (McGill et al. 2013), liver adducts continue to increase, relevant mitochondrial protein adducts were detected after three doses with JNK activation, and ALT increases occurring after four and five doses. This time course of events was accelerated when autophagy was blocked by leupeptin, which is an effective lysosomal protease inhibitor (Ni et al. 2016; Ueno and Komatsu 2019). This is consistent with both cytosolic adducts and damaged mitochondria being removed by autophagy (Ni et al. 2012, 2016), which is an adaptive response to the APAP-induced stress that limits cell necrosis (Chao et al. 2018; Ni et al. 2013). Thus, repeated dosing with 150 mg/kg APAP followed the same sequence of events known to cause liver injury after a single high overdose including substantial protein adducts accumulation in the liver and in mitochondria and JNK activation, which is a prerequisite for the amplification of the mitochondrial dysfunction characteristic of APAP hepatotoxicity (Ramachandran and Jaeschke 2019). In addition, autophagy effectively limited cell necrosis after multiple overdoses; the beneficial effect of autophagy was even more pronounced in this context than after a single high overdose.
Liver injury after cytosolic APAP-protein adduct accumulation after multiple doses of APAP
A dose of 75 mg/kg APAP causes a short-term depletion of GSH and a rapid recovery even in starved mice (McGill et al. 2013). As a result, this dose does only cause mild adduct formation in the liver but not in mitochondria, and no JNK activation or liver injury (Hu et al. 2016; McGill et al. 2013). In the current study using fed mice, the lack of effects on adducts and injury with a single dose of 75 mg/kg APAP could be confirmed. Importantly, even after up to five doses in rapid succession, there was only a very limited increase in total liver adducts, virtually no relevant increase in mitochondrial adducts, and no JNK activation or liver injury. Quantitatively, these data are consistent with the time course of the 150 mg/kg dose. Both the levels of total liver and mitochondrial adducts after five doses of 75 mg/kg APAP were well below the levels observed after three doses of 150 mg/kg where no JNK activation or injury was observed. However, co-treatment with leupeptin increased plasma ALT activities 2 h after the last dose of APAP indicating liver injury. Importantly, a few hours later, ALT activities further increased, which suggests progression of the injury when autophagy is inhibited. Although both total liver and mitochondrial adduct levels increased, there was no JNK activation. Since the mitochondrial adduct levels were almost an order of magnitude below the levels that did not cause JNK activation and liver injury after 150 mg/kg, the results suggest that the injury under these conditions is not caused by the regular mechanism of mitochondrial adducts and JNK activation. Nevertheless, this injury was still eliminated by a potent Cyp inhibitor like 4-methyl-pyrazole, which effectively reduces protein adduct formation after APAP in mice (Akakpo et al. 2018) and humans (Kang et al. 2020). This would indicate that the accumulation of adducts outside mitochondria under conditions of autophagy inhibition can cause liver injury.
Clinical significance of multiple doses of APAP
The multiple subtoxic doses represent the scenario of unintentional overdosing, i.e., where a patient takes various APAP-containing mediations in short order without being aware of the APAP content in each drug. This can lead to severe liver injury after several days. Our data suggest that the cumulative overdosing results in liver injury with mechanism similar to a single large overdose involving mitochondrial protein adducts that trigger a mitochondrial oxidant stress, which, after amplification by the JNK pathway, induce the mitochondrial permeability transition pore opening and necrotic cell death (Ramachandran and Jaeschke 2019). Interestingly, the impact of autophagy inhibition is more profound after multiple subtoxic doses than observed after a single large overdose (Ni et al. 2012, 2016). This is consistent with the concept that autophagy, as an adaptive response to the drug-induced cellular toxicity, is more effective with a more moderate stress (Chao et al. 2018; Ramachandran and Jaeschke 2020).
After multiple, very low doses of APAP, which result in only minor protein adduct formation in the total liver but not in mitochondria, no relevant cellular stress (JNK activation, ALT release) was detectable. However, inhibition of autophagy increased the accumulation of adducts and induces limited cell death but still without the relevant protein adducts in mitochondria or JNK activation. This indicates that the effective elimination of protein adducts by autophagy (Ni et al. 2016) is the main reason why patients can take therapeutic doses of APAP for years and do not develop liver injury despite the continuous generation of very low levels of adducts after each dose (Curry et al. 2019; Heard et al. 2011).
Conclusions
Multiple subtoxic overdoses of APAP can cause accumulation of protein adducts in the liver and in mitochondria, which eventually leads to cell death involving JNK activation and mitochondrial dysfunction. Inhibition of autophagy strongly aggravates these mechanisms leading to more severe injury. Multiple mild overdoses of APAP do not result in relevant mitochondrial protein adduct accumulation and JNK activation, and do not cause liver injury. However, when autophagy is inhibited, the accumulating adducts in hepatocytes eventually cause cell death. Both approaches document the essential role of autophagy in removing protein adducts and damaged mitochondria after any dose of APAP.
Abbreviations
- ALT:
-
Alanine aminotransferase
- AMAP:
-
N-Acetyl-m-aminophenol
- APAP:
-
N-Acetyl-p-aminophenol, acetaminophen
- APAP-CYS:
-
APAP-cysteine derived from protein adducts
- CYP:
-
Cytochrome P450
- GSH:
-
Glutathione
- H&E:
-
Hematoxylin and eosin
- JNK:
-
C-jun N-terminal kinase
- MPTP:
-
Mitochondrial permeability transition pore
- NAC:
-
N-Acetylcysteine
- NAPQI:
-
N-Acetyl-p-benzoquinone imine
- P-JNK:
-
Phospho-JNK
- TUNEL:
-
Terminal deoxynucleotidyl transferase dUTP nick end labeling
References
Akakpo JY, Ramachandran A, Kandel SE, Ni HM, Kumer SC, Rumack BH, Jaeschke H (2018) 4-Methylpyrazole protects against acetaminophen hepatotoxicity in mice and in primary human hepatocytes. Hum Exp Toxicol 37:1310–1322
Alhelail MA, Hoppe JA, Rhyee SH, Heard KJ (2011) Clinical course of repeated supratherapeutic ingestion of acetaminophen. Clin Toxicol (Phila) 49:108–112
Bajt ML, Cover C, Lemasters JJ, Jaeschke H (2006) Nuclear translocation of endonuclease G and apoptosis-inducing factor during acetaminophen-induced liver cell injury. Toxicol Sci 94:217–225
Chao X, Wang H, Jaeschke H, Ding WX (2018) Role and mechanisms of autophagy in acetaminophen-induced liver injury. Liver Int 38:1363–1374
Curry SC, Padilla-Jones A, Ruha AM, O’Connor AD, Kang AM, Wilkins DG, Jaeschke H, Wilhelms K, Gerkin RD (2019) The Acetaminophen Adduct Study Group. The relationship between circulating acetaminophen-protein adduct concentrations and alanine aminotransferase activities in patients with and without acetaminophen overdose and Toxicity. J Med Toxicol 15:143–155
Du K, Ramachandran A, McGill MR, Mansouri A, Asselah T, Farhood A, Woolbright BL, Ding WX, Jaeschke H (2017) Induction of mitochondrial biogenesis protects against acetaminophen hepatotoxicity. Food Chem Toxicol 108(Pt A):339–350
Hanawa N, Shinohara M, Saberi B, Gaarde WA, Han D, Kaplowitz N (2008) Role of JNK translocation to mitochondria leading to inhibition of mitochondria bioenergetics in acetaminophen-induced liver injury. J Biol Chem 283:13565–13577
Heard KJ, Green JL, James LP, Judge BS, Zolot L, Rhyee S, Dart RC (2011) Acetaminophen-cysteine adducts during therapeutic dosing and following overdose. BMC Gastroenterol 11:20
Hoffmann KJ, Streeter AJ, Axworthy DB, Baillie TA (1985) Identification of the major covalent adduct formed in vitro and in vivo between acetaminophen and mouse liver proteins. Mol Pharmacol 27:566–573
Hu J, Ramshesh VK, McGill MR, Jaeschke H, Lemasters JJ (2016) Low dose acetaminophen induces reversible mitochondrial dysfunction associated with transient c-Jun N-terminal kinase activation in mouse liver. Toxicol Sci 150:204–215
Jaeschke H (2015) Acetaminophen: dose-dependent drug hepatotoxicity and acute liver failure in patients. Dig Dis 33:464–471
Jaeschke H, Duan L, Nguyen N, Ramachandran A (2019) Mitochondrial damage and biogenesis in acetaminophen-induced liver injury. Liver Res 3:150–156
Kang AM, Padilla-Jones A, Fisher ES, Akakpo JY, Jaeschke H, Rumack BH, Gerkin RD, Curry SC (2020) The effect of 4-methylpyrazole on oxidative metabolism of acetaminophen in human volunteers. J Med Toxicol 16:169–176
Kelly JP, Battista DR, Shiffman S, Malone MK, Weinstein RB, Kaufman DW (2018) Knowledge of dosing directions among current users of acetaminophen-containing medications. Sci Pract Res 58:492–498
Kon K, Kim JS, Jaeschke H, Lemasters JJ (2004) Mitochondrial permeability transition in acetaminophen-induced necrosis and apoptosis of cultured mouse hepatocytes. Hepatology 40:1170–1179
Lancaster EM, Hiatt JR, Zarrinpar A (2015) Acetaminophen hepatotoxicity: an updated review. Arch Toxicol 89:193–199
Larson AM (2007) Acetaminophen hepatotoxicity. Clin Liver Dis 11:525–548
McGill MR, Jaeschke H (2013) Metabolism and disposition of acetaminophen: recent advances in relation to hepatotoxicity and diagnosis. Pharm Res 30:2174–2187
McGill MR, Jaeschke H (2015) A direct comparison of methods used to measure oxidized glutathione in biological samples: 2-vinylpyridine and N-ethylmaleimide. Toxicol Mech Methods 25:589–595
McGill MR, Williams CD, Xie Y, Ramachandran A, Jaeschke H (2012) Acetaminophen-induced liver injury in rats and mice: comparison of protein adducts, mitochondrial dysfunction, and oxidative stress in the mechanism of toxicity. Toxicol Appl Pharmacol 264:387–394
McGill MR, Lebofsky M, Norris HR, Slawson MH, Bajt ML, Xie Y, Williams CD, Wilkins DG, Rollins DE, Jaeschke H (2013) Plasma and liver acetaminophen-protein adduct levels in mice after acetaminophen treatment: dose-response, mechanisms, and clinical implications. Toxicol Appl Pharmacol 269:240–249
Muldrew KL, James LP, Coop L, McCullough SS, Hendrickson HP, Hinson JA, Mayeux PR (2002) Determination of acetaminophen-protein adducts in mouse liver and serum and human serum after hepatotoxic doses of acetaminophen using high-performance liquid chromatography with electrochemical detection. Drug Metab Dispos 30:446–451
Nguyen NT, Du K, Akakpo JY, Umbaugh DS, Jaeschke H, Ramachandran A (2021) Mitochondrial protein adduct and superoxide generation are prerequisites for early activation of c-jun N-terminal kinase within the cytosol after an acetaminophen overdose in mice. Toxicol Lett 338:21–31
Ni HM, Bockus A, Boggess N, Jaeschke H, Ding WX (2012) Activation of autophagy protects against acetaminophen-induced hepatotoxicity. Hepatology 55:222–232
Ni HM, Williams JA, Jaeschke H, Ding WX (2013) Zonated induction of autophagy and mitochondrial spheroids limits acetaminophen-induced necrosis in the liver. Redox Biol 1:427–432
Ni HM, McGill MR, Chao X, Du K, Williams JA, Xie Y, Jaeschke H, Ding WX (2016) Removal of acetaminophen protein adducts by autophagy protects against acetaminophen-induced liver injury in mice. J Hepatol 65:354–362
Ramachandran A, Jaeschke H (2019) Acetaminophen hepatotoxicity. Semin Liver Dis 39:221–234
Ramachandran A, Jaeschke H (2020) A mitochondrial journey through acetaminophen hepatotoxicity. Food Chem Toxicol 140:111282
Saito C, Lemasters JJ, Jaeschke H (2010) c-Jun N-terminal kinase modulates oxidant stress and peroxynitrite formation independent of inducible nitric oxide synthase in acetaminophen hepatotoxicity. Toxicol Appl Pharmacol 246:8–17
Tirmenstein MA, Nelson SD (1989) Subcellular binding and effects on calcium homeostasis produced by acetaminophen and a nonhepatotoxic regioisomer, 3’-hydroxyacetanilide, in mouse liver. J Biol Chem 264:9814–9819
Ueno T, Komatsu M (2019) Measuring nonselective and selective autophagy in the liver. Methods Mol Biol 1880:535–540
Wang H, Ni HM, Chao X, Ma X, Rodriguez YA, Chavan H, Wang S, Krishnamurthy P, Dobrowsky R, Xu DX, Jaeschke H, Ding WX (2019) Double deletion of PINK1 and Parkin impairs hepatic mitophagy and exacerbates acetaminophen-induced liver injury in mice. Redox Biol 22:101148
Win S, Than TA, Kaplowitz N (2018) The regulation of JNK signaling pathways in cell death through the interplay with mitochondrial SAB and upstream post-translational effects. Int J Mol Sci 19:E3657
Xie Y, McGill MR, Du K, Dorko K, Kumer SC, Schmitt TM, Ding WX, Jaeschke H (2015) Mitochondrial protein adducts formation and mitochondrial dysfunction during N-acetyl-m-aminophenol (AMAP)-induced hepatotoxicity in primary human hepatocytes. Toxicol Appl Pharmacol 289:213–222
Yoon E, Babar A, Choudhary M, Kutner M, Pyrsopoulos N (2016) Acetaminophen-induced hepatotoxicity: a comprehensive update. J Clin Transl Hepatol 4:131–142
Acknowledgements
This work was supported in part by the National Institutes of Health Grants R01 DK102142 (Co-PIs WXD/HJ), P20 GM103549 (HJ), and the Mechanisms of Liver Injury and Diseases COBRE P30 GM118247 (HJ). JYA was supported by a NIH predoctoral fellowship F31 DK1200194.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
The manuscript does not contain clinical studies or patient data.
Conflict of interest
The authors declare that there is no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Nguyen, N.T., Akakpo, J.Y., Weemhoff, J.L. et al. Impaired protein adduct removal following repeat administration of subtoxic doses of acetaminophen enhances liver injury in fed mice. Arch Toxicol 95, 1463–1473 (2021). https://doi.org/10.1007/s00204-021-02985-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00204-021-02985-6