
Abstract
Acetaminophen (APAP) is a widely used analgesic and antipyretic drug. APAP overdose can induce acute liver injury in humans, which is responsible for approximately 50% of total cases of acute liver failure in the United States and some European countries. Currently, the metabolism of APAP in the body has been extensively investigated; however, the exact mechanisms for APAP hepatotoxicity are not well understood. Recent studies have shown that mitochondrial dysfunction, oxidative stress and inflammatory responses play a critical role in the pathogenesis of APAP hepatotoxicity. Autophagy is a catabolic machinery aimed at recycling cellular components and damaged organelles in response to a variety of stimuli, such as nutrient deprivation and toxic stress. Increasing evidence supports that autophagy is involved in the pathophysiological process of APAP-induced liver injury. In this review, we summarized the changes of autophagy in the liver following APAP intoxication and discussed the role and its possible mechanisms of autophagy in APAP hepatotoxicity. Furthermore, this review highlights the crosstalk between mitophagy, oxidative stress and inflammation in APAP-induced liver injury and presents some possible molecular mechanisms by which activated autophagy protects against APAP-induced liver injury.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Acetaminophen (also known as paracetamol, N-acetyl-p-aminophenol; APAP) is a widely used analgesic and antipyretic drug. In many countries, APAP is an over-the-counter drug, and people can choose to buy APAP without a doctor’s prescription. According to the US Food and Drug Administration, approximately 50 million adults in the United States take acetaminophen-containing products each week. The recommended oral dosage for adults is 325–650 mg every 4–6 h, with a maximum recommended daily dose of 4 g (Hinson et al. 2010; Schilling et al. 2010). Although APAP is believed to be safe at therapeutic doses, it produces a centrilobular hepatic necrosis at higher doses (Hinson et al. 2010). In 1966, Davidson and Eastham first reported the cases of acute hepatotoxicity caused by APAP overdose (Davidson and Eastham 1966). In the United States and the United Kingdom, APAP poisoning accounts for approximately 50% of all cases of acute liver failure (Larson et al. 2005; Ostapowicz et al. 2002).
Metabolism and hepatotoxicity of APAP
Acetaminophen toxicity occurs in two phases: a metabolic phase is followed by a toxicity phase (Boobis et al. 1986; Tee et al. 1986). At present, the metabolic phase has been well characterized. In the liver, the major portion of acetaminophen (approximately 85–90%) is metabolized by glucuronidation and sulfonation reactions, only a minor fraction of the drug (up to 10%) undergoes oxidation to form the highly reactive metabolite, N-acetyl-p-benzoquinone imine (NAPQI) (Dahlin et al. 1984; Hodgman and Garrard 2012; Larson 2007; Mitchell et al. 1973a). The metabolizing enzymes responsible for the oxidation of APAP have been identified, liver cytochrome P450 (CYP) enzymes especially CYP2E1 and CYP3A4 are believed to play a key role in the metabolic activation of APAP (Patten et al. 1993; Thummel et al. 1993). When therapeutic doses of APAP are ingested, the small amount of NAPQI is efficiently deactivated by conjugation with reduced glutathione (GSH), forming a mercapturic metabolite that is readily eliminated by the kidneys (Mitchell et al. 1973b). Following exposure to high-dose APAP, however, the endogenous glucuronide and sulfate cofactors, such as UDP-glucuronic acid (UDPGA) and 3′-phosphoadenosine-5′-phosphosulfate (PAPS), become depleted, thus forming increased amounts of NAPQI. Once the endogenous GSH in the liver also becomes depleted, NAPQI can covalently bind to cellular biological macromolecules, such as proteins, nucleic acids and lipids, resulting in mitochondrial damages, endoplasmic reticulum stress, and necrotic cell death (Foufelle and Fromenty 2016; Hinson et al. 2010). By contrast, the toxicity phase is characterized by increased oxygen/nitrogen stress and the mitochondrial permeability transition (MPT); however, the exact mechanisms are poorly understood. A number of molecular events appear to occur in the toxicity phase, including mitochondrial dysfunction, increased oxidative stress, altered ion imbalance, and dysregulated signaling transduction.
Based on the fundamental insight into the mechanism of APAP hepatotoxicity, N-acetylcysteine (NAC) was introduced to scavenge the reactive metabolite in the clinic. The administration of NAC is a highly successful approach for treating APAP overdose; however, this protective effect is only observed in the early stage of APAP hepatotoxicity. In mouse model, NAC no longer plays a protective role at 4 h after APAP overdose. In contrast, therapeutic window of NAC intervention in patients is up to 10 h following APAP overdose. This lag time provides a window of opportunity for optimal treatment with NAC. If given within the first 10 h of overdose, NAC may completely prevent the occurrence of hepatotoxicity (Prescott et al. 1980; Smilkstein et al. 1988). Patients that do not receive NAC in time undergo severe liver injury, which can progress to acute liver failure (ALF). Hence, liver transplantation is the ultimate treatment for patients with ALF.
APAP-induced acute hepatotoxicity demonstrates a necroinflammatory injury pattern. Histologically, liver injury is primarily characterized by a centrilobular hepatic necrosis, which is accompanied by a mild inflammatory infiltrate. Biochemically, the cases of APAP intoxication have marked elevation of serum aminotransferase, such as aspartate aminotransferase (AST) and alanine aminotransferase (ALT). In the rodent model, following the intoxication of APAP, a prominent change in liver was characterized by glycogen loss and vacuolization of centrilobular hepatocytes by 2 h. By 3 h, nuclear changes were observed in centrilobular hepatocytes and single cell necrosis with pycnotic cells. By 6 h, gross necrosis of the entire centrilobular areas were observed (Mitchell et al. 1973a). Furthermore, sterile inflammation and inflammasome activation occurs in both mice and man after APAP overdose (Hinson et al. 2010). In addition, some studies reported that APAP hepatotoxicity was associated with the apoptosis of hepatocytes. However, in contrast to the massive necrotic cells, few apoptotic cells were observed in livers of mice treated with a toxic dose of APAP (Gujral et al. 2002). Collectively, the vast majority of literatures have supported that necrosis rather than apoptosis contributes to the cell death of hepatocytes in APAP-induced acute liver injury.
The mechanisms involved in APAP hepatotoxicity
As mentioned above, the exact mechanisms of APAP hepatotoxicity are not well understood. However, recent studies have suggested that mitochondrial dysfunction can be considered as a critical event in the pathogenesis of APAP hepatotoxicity. Changes in mitochondrial morphology and function have been observed in a multitude of in vivo and in vitro models of acetaminophen hepatotoxicity. In the meantime, toxic doses of APAP result in impaired energy metabolism as well as a lowering of cellular ATP levels and ATP/ADP ratios (McGill et al. 2012a, b; Vendemiale et al. 1996). More importantly, it is recognized that mitochondrial dysfunction is causally linked to the oxidative stress following by APAP overdose. On the one hand, excessive NAPQI generated from APAP can deplete GSH level and affect mitochondrial function in the hepatocytes. On the other hand, damaged mitochondria further lead to overproduction of reactive oxygen species (ROS), forming a vicious cycle. In particular, the formation of protein adducts in mitochondria results in an excessive oxidative stress and activation of mitogen-activated protein kinases including c-jun N-terminal kinase (JNK), which further amplify the oxidant stress (Jaeschke et al. 2012). In the liver, there are three major sources of ROS: cytochrome P450-catalyzed substrate oxidation, macrophage-derived NADPH oxidase, and mitochondria. In terms of APAP hepatotoxicity, however, NADPH oxidase-derived ROS is not the primary cause, because mice with a deficiency in NAPDH oxidase function did not show a decreased oxidative stress or reduced liver injury (James et al. 2003). Similarly, the direct evidence of APAP-induced oxidant stress from cytochrome P450 enzymes in the metabolism phase is also lacking (McGill et al. 2011). By contrast, nowadays, compelling evidence supports that oxidative stress in APAP-induced hepatotoxicity is mainly due to mitochondria-derived ROS and free radicals (Du et al. 2016a; McGill et al. 2012b).
Besides mitochondrial dysfunction and oxidative stress, there is increasing evidence that endoplasmic reticulum (ER) stress can be another important mechanism in APAP-induced liver injury. In this respect, both in vivo and in vitro studies have demonstrated that APAP was able to induce ER stress and the unfolded protein response (UPR), which could play an important role in APAP-induced hepatocyte death (Nagy et al. 2007, 2010; Uzi et al. 2013). For example, CCAAT/enhancer-binding protein homologous protein (CHOP) is a transcription repressor downstream of the PERK and IRE1 pathways of the UPR. Once activated, CHOP inhibits the expression of anti-apoptotic genes and activates pro-apoptotic genes (McCullough et al. 2001). During APAP-induced liver injury, ER stress and UPR activation were coincided with CHOP upregulation. By contrast, deletion of CHOP protects mice from liver damage following APAP intoxication (Uzi et al. 2013). Furthermore, 4-phenylbutyric acid (PBA) treatment dramatically ameliorated the massive hepatocyte death after APAP administration. The underlying protective mechanism of PBA against APAP hepatotoxicity could be attributed to the alleviation of ER stress-induced hepatocytes death, because PBA can significantly prevent the APAP-induced increases in cleaved activating transcription factor 6 (ATF6) and phosphorylation of c-JNK (Kusama et al. 2017).
Although the initial underlying mechanism of APAP-induced hepatotoxicity is the necrosis of hepatocytes, the second step in the liver injury is a sterile inflammation as a response to the necrotic hepatocytes. Intracellular components released from hepatocytes include nuclear DNA, mitochondrial DNA and proteins, they can act as damage-associated molecular patterns (DAMPs) and activate the formation of the inflammasome complex in various cells such as Kupffer cells, thereby causing a release of proinflammatory cytokines including interleukin-1 and tumor necrosis factor-α (TNF-α) (Blazka et al. 1995b; Kubes and Mehal 2012; Laskin and Pilaro 1986). Then, they can result in proinflammatory responses through the activation and hepatic recruitment of neutrophils and monocytes. The sterile inflammation likely amplifies the initial insult and increases overall tissue injury (Woolbright and Jaeschke 2017). By contrast, the Kupffer cell inactivators such as gadolinium chloride and dextran sulfate were reported to decrease acetaminophen toxicity in the mouse (Blazka et al. 1995a; Michael et al. 1999). However, the elimination of Kupffer cells with clodronate liposomes could not protect against the APAP-induced liver injury (Ju et al. 2002). By contrast, it resulted in an increase in liver toxicity, which suggested that KCs may have a beneficial role in the toxicity, such as recruitment of circulating macrophages leading to increased liver repair (Holt et al. 2008). Considering the dual effect of inflammation in APAP hepatotoxicity, we speculate that a moderate inflammatory response may contribute to the tissue repair and hepatocytes regeneration, but excessive inflammatory responses aggravate liver damage.
Autophagy in APAP-induced liver injury
Autophagy is a cellular process responsible for the degradation of excess or aberrant long-lived cytosolic proteins and organelles within lysosomes. Briefly, autophagy initiates as an isolation membrane, then gradually grows into a double-membrane autophagosome, and subsequently matures into an autolysosome after fusion with lysosomes. Finally, the autophagosome-containing cytoplasmic materials is degraded by lysosomal enzymes (Mizushima et al. 2010).
According to the properties of the substrates, autophagy can be further divided into non-selective autophagy and selective autophagy. Upon nutrient deprivation, autophagy catabolizes some non-essential cytoplasmic components non-selectively, including proteins and organelles, into building blocks, such as amino acids (Mizushima and Komatsu 2011). By contrast, selective autophagy mainly targets for some specific substrates such as intracellular protein polymers and the damaged organelles (He and Klionsky 2009; Lamark and Johansen 2012). Mitophagy is a type of autophagy responsible for the selective removal of damaged mitochondria. Nowadays, it is presumed that mitophagy can be completed via ubiquitin-dependent pathway such as PINK1-Parkin-mediated mitophagy, or ubiquitin-independent pathway such as BNIP-NIX, FUNDC1, and Bcl2L13-mediated mitophagy (Hamacher-Brady and Brady 2016). Among them, PINK1-Parkin-mediated mitophagy is the most well-characterized pathway. PINK1 is a mitochondrial serine/threonine kinase, which acts as a molecular sensor to monitor mitochondrial status and protect cells from stress-induced mitochondrial dysfunction. In healthy mitochondria, mitochondrial transmembrane potential drives PINK1 import into the inner mitochondrial membrane (IMM) by the translocase of the outer mitochondrial membrane (OMM). By contrast, mitochondrial damage causes the accumulation of PINK1 on the OMM. Then, PINK1 recruits Parkin to initiate mitophagy (Nguyen et al. 2016). As an E3 ubiquitin ligase, activated Parkin mediates ubiquitination of the outer mitochondrial membrane proteins, which serve as signal to recruit the autophagy adaptors like OPTN, NDP52, and p62. Consequently, the autophagy machinery is recruited to damaged mitochondria for degradation (Kerr et al. 2017; Lazarou et al. 2015; Murata et al. 2013; Nguyen et al. 2016).
There is growing evidence that supports autophagy participates in various metabolic processes of the liver. Under physiological conditions, there is a constitutive, low level of autophagy in hepatocytes, which plays an essential role in maintenance of normal liver function. However, a wide range of conditions such as hunger, oxidative stress, and accumulation of damaged organelles can induce or inhibit the activity of autophagy, thereby affecting liver function and even leading to cell death. It has been found that autophagy was involved in various liver diseases including alcoholic liver disease, non-alcoholic fatty liver disease, liver cancer and viral hepatitis (Hidvegi et al. 2011; Ueno and Komatsu 2017).
The alteration and role of autophagy in APAP-induced liver injury
Recent studies providing compelling evidence that supports autophagy are directly involved in the pathophysiology of APAP-induced liver injury. In 2012, Ni et al. reported that autophagy was associated with APAP-induced liver injury. They found that APAP-induced autophagy in the mouse liver and primary cultured hepatocytes. In the meantime, pharmacological inhibition of autophagy by 3-methyladenine or chloroquine further exacerbated APAP-induced hepatotoxicity. In contrast, induction of autophagy by rapamycin inhibited APAP-induced hepatotoxicity (Ni et al. 2012a). Meanwhile, the study has also found that RAPA does not affect the metabolic activation of APAP, indicating that the protective effect of rapamycin lies in the downstream of APAP metabolism. Treatment with rapamycin 2 h after APAP administration significantly ameliorates APAP-induced liver injury, despite the fact that APAP metabolism and hepatic GSH depletion have already occurred (Ni et al. 2012a). This finding is particularly important, because most patients of APAP poisoning do not receive medical care until they are past the metabolic phase. Therefore, pharmacological induction of autophagy may have a potential therapeutic application in humans with APAP hepatotoxicity. Subsequently, Igusa et al. further demonstrated that hepatocyte-specific ATG7 knockout mice (hepatocyte-specific autophagy deficiency) are more susceptible to APAP-induced liver injury (Igusa et al. 2012). In their study, APAP-induced reactive oxygen species (ROS) production, mitochondrial membrane depolarization, and JNK activation in hepatocytes were accelerated by autophagy deficiency (Igusa et al. 2012). Taken together, autophagy activation in APAP hepatotoxicity is likely to play a protective role, because further elevated autophagy induced by drugs can significantly reduce liver damage.
At present, the mechanism by which APAP induces autophagy activation is not clear, we hypothesize that it is most likely a compensatory response to excessive ROS following APAP intoxication. Autophagy is activated by various stimuli in cells and ROS are one of these autophagy inducers. ROS-induced autophagy is also seen in a variety of oxidative stress conditions, such as NGF deprivation, TNF-induced ROS production, and nutrient starvation. In this respect, the mitochondria represent the principal source of ROS required for autophagy induction signaling (Filomeni et al. 2015; Scherz-Shouval and Elazar 2007; Scherz-Shouval et al. 2007). Mechanistically, the accumulation of ROS can induce autophagy both by direct effect on the core autophagy machinery and by indirect influence on the components of the autophagy-regulatory signaling pathway (Dewaele et al. 2010; Scherz-Shouval and Elazar 2011; Zhang et al. 2016). Indeed, APAP-induced autophagy was suppressed by N-acetylcysteine, suggesting APAP mitochondrial protein binding and the subsequent production of reactive oxygen species may play an important role in APAP-induced autophagy (Ni et al. 2012a).
Surprisingly, however, ATG5 tissue-specific knockout mice have been shown to increase tolerance to APAP-induced acute liver injury (Ni et al. 2012b). Hepatocyte-specific deletion of Atg5 resulted in the loss of autophagic activity and mild liver injury, which is characterized by increased apoptosis and compensatory hepatocyte proliferation; however, they were resistant to APAP-induced liver injury (Ni et al. 2012b). Both ATG5 and ATG7 are the essential genes in the formation of autophagosomes, knocking out either of them can block the occurrence of autophagy. However, ATG5 and ATG7 tissue-specific knockout mice show different effects on APAP hepatotoxicity, and the exact reasons for this seemingly contradictory phenomenon remain unclear. Further investigations revealed an increased basal hepatic GSH content and a faster recovery of GSH after APAP treatment, which can be due to persistent activation of Nrf2, a transcriptional factor regulating drug detoxification and GSH synthesis. In addition, a higher hepatocyte proliferation was observed in the livers of Atg5 liver-specific knockout mice. Therefore, the researchers speculate that the activation of Nrf2-ARE antioxidant pathway and increased hepatocyte proliferation protect against APAP-induced liver injury in Atg5 knockout mice (Ni et al. 2012b). Similarly, Parkin knockout mice were protected against APAP-induced liver injury, which may be due to decreased c-JNK and increased hepatocyte proliferation after APAP treatment (Williams et al. 2015). Furthermore, a novel research has also demonstrated that mice with liver-specific double knockout of Ulk1 and Ulk2, the key component of Atg1/Unc-51-like kinase 1(ULK1) complex in upstream step of autophagy pathway, are more resistant to APAP-induced liver injury. However, mechanistic study has revealed that Ulk1/2 knockout does not affect the autophagic activity in hepatocytes. By contrast, Ulk1/2 deficiency suppresses the activation of JNK via MKK4/7 (Sun et al. 2017). Together, the discrepancy in gene knockout mice experiments above-mentioned is associated with the non-autophagic functions of autophagy-related genes. Hence, a better understanding the function of these autophagy-related proteins could help decipher their distinct role in APAP hepatotoxicity.
The mechanisms for autophagy protects against APAP hepatotoxicity
In liver, once GSH is depleted, NAPQI reacts with many cellular proteins, including mitochondrial proteins, to form protein adducts (Jaeschke and Bajt 2006). Subsequently, APAP protein adducts (APAP-AD) may lead to mitochondrial damage and hepatocyte necrosis. As an essential mechanism of maintaining cell homeostasis, the autophagy can remove the APAP-AD and the damaged mitochondria, which prevents against APAP-induced necrosis (Ni et al. 2012a).
Clearing APAP protein adducts
The formation of APAP-AD in hepatocytes triggers mitochondrial dysfunction and necrosis. Recent studies have found that autophagy selectively eliminates APAP-AD. Following APAP, APAP-AD were detected at 1 h, peaked at approximately 2 h, declined at 6 h and almost full removed at 24 h post treatment with APAP in mouse livers and in primary mouse hepatocytes. In the meantime, the study also found that selective autophagy was responsible for the clearance of protein adducts, because APAP-AD was colocalized with GFP-LC3 positive autophagosomes and Lamp1 positive lysosomes in APAP-treated primary hepatocytes (Ni et al. 2016). More importantly, pharmacological inhibition of autophagy by leupeptin or chloroquine increased, whereas induction of autophagy by Torin 1 decreased serum APAP-AD levels in APAP-treated mice, which correlated with alanine aminotransferase levels and liver necrosis (Ni et al. 2016).
Eliminating the damaged mitochondria
Mitochondria are the power plants inside cells that are responsible for generating ATP for cell survival through oxidative phosphorylation. Furthermore, mitochondria are implicated in critical cellular processes such as programmed cell death and the regulation of inflammatory responses (Green et al. 2011; Nakahira et al. 2011; Weinberg et al. 2015). Dysfunction of mitochondria can lead to a wide range of disorders due to the impact on cellular metabolism and production of ROS (Scheibye-Knudsen et al. 2015).
As mentioned above, mitochondrial dysfunction, oxidative stress and sterile inflammation are critical events in the pathogenesis of APAP-induced liver injury. Recently, the crosstalk between autophagy and them has been found. Under oxidative stress, elevated intracellular ROS can induce autophagy; in turn, activated autophagy can remove damaged mitochondria to reduce the production of ROS and eliminate inflammasomes (such as NLRP3 inflammasome), thus inhibiting the inflammatory response (Chen et al. 2016; Deng et al. 2013). These findings provide new insights into the intrinsic link between APAP-induced liver injury and autophagy. Indeed, emerging evidence suggests that autophagic removal of damaged mitochondria may protect against APAP-induced liver injury. For example, APAP overdose triggers unique pathological zonated changes in the mouse liver, which is characterized by mitochondrial spheroid formation, autophagy and mitochondrial biogenesis. APAP-induced autophagy is believed to limit the expansion of necrosis and promote mitochondrial biogenesis (Ni et al. 2013). Furthermore, Parkin knockdown experiment in mice further supports the above results. Parkin acts an E3 ubiquitin ligase that is directly involved in the PINK1-Parkin-mediated mitophagy pathway. Knockdown of Parkin in mouse livers using adenovirus-shRNA significantly reduced mitophagy but increased JNK activation after APAP administration, which exacerbated APAP-induced liver injury (Williams et al. 2015). However, in contrast to the results of acute knockdown experiment, Parkin KO mice were protected against APAP-induced liver injury. The exact reasons might be associated with the non-autophagic functions of autophagy-related genes, because increased hepatocyte proliferation was observed in Parkin KO mice. In addition, we speculated that chronic KO of Parkin in mice may result in a compensatory activation of other mitophagy pathways. Indeed, regardless of the loss of Parkin function, mitophagy was still observed in hepatocytes, which supports the existence of other mitophagy pathways (Williams et al. 2015).
-
1.
Alleviating oxidative stress via mitophagy
As a defense mechanism against cell death, mitophagy induced by APAP can remove damaged mitochondria. At this aspect, mitochondria are frequently observed within APAP-induced autophagosomes, and the level of mitochondrial proteins is decreased, supporting the role of mitophagy in the removal of damaged mitochondria. Moreover, autophagy inhibition by chloroquine further increased APAP-induced ROS production, whereas induction of autophagy with rapamycin inhibited its production, indicating that mitophagy may attenuate mitochondrial ROS formation and release of pro-death factors (Ni et al. 2012a).
In addition, some drug-intervening experiments also provide the supporting evidence on mitophagy. Metformin, a first-line drug to treat type 2 diabetes mellitus, protected against APAP acute hepatotoxicity in mice (Kim et al. 2015; Saeedi Saravi et al. 2016). In the meantime, the studies found that metformin could attenuate the mitochondrial oxidant stress and mitochondrial dysfunction, which could be attributed to its ability of enhancing mitophagy (Du et al. 2016b). Certainly, a recent study has also found that metformin can suppress inflammation by inhibiting the extracellular activity of HMGB1 in an acetaminophen-induced acute liver injury model (Horiuchi et al. 2017).
-
2.
Inhibiting the activation of inflammasomes via mitophagy
Although the initial stage of APAP hepatotoxicity is mediated by reactive metabolite formation and mitochondrial dysfunction, the later stage of injury is potentially mediated, at least in part, by the recruitment of inflammatory leukocytes such as neutrophils and monocytes (Imaeda et al. 2009; Liu et al. 2006; Marques et al. 2012; Mossanen et al. 2016). For instance, neutrophil depletion with anti-Gr-1 antibody significantly attenuated the hepatoxicity in acetaminophen-treated mice (Liu et al. 2006).
Currently, the most widely accepted viewpoint behind inflammation after APAP overdose is that the process occurs through a sterile inflammatory response. The controversial question is whether the inflammatory response contributes to the injury or whether this inflammation is beneficial for survival. However, more researches support the severity of liver injury may associated with the innate immunity and the activation of the inflammatory response (Connolly et al. 2011; Gardner et al. 2002; Liu et al. 2004).
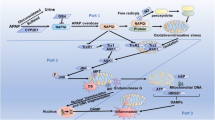
The inflammasomes are a type of multiprotein complexes in the cell, which can sense the external stimulus signal to activate caspase-1 and regulate the processing and secretion of IL-1β, IL-18 and so on. Among them, NLRP3 inflammasome is currently the most studied and well-understood inflammasome. During the pathway of NLRP3 inflammasome activation, mtDNA and mtROS released from damaged mitochondria are the major activators. Recent studies have shown that activated autophagy can relieve inflammatory responses by selectively clearing damaged mitochondria, inhibiting the activation of inflammasomes (Zhong et al. 2016). By contrast, autophagy deficits caused by knocking out LC3 and Beclin 1 activated NLRP3 inflammasome and increased the production of IL-1β and IL-18, thus enhancing the inflammatory response (Lupfer et al. 2013; Nakahira et al. 2011). As for APAP hepatotoxicity, NLRP3 has been identified as a potential mediator in the mouse model of APAP overdose. The formation of NLRP3 inflammasome in particular has directly been attributed to late-stage APAP toxicity (Imaeda et al. 2009; Woolbright and Jaeschke 2017). Given that mitochondrial damage plays a central role in APAP hepatic injury, we hypothesized that mitophagy may selectively remove damaged mitochondria and control the level of oxidative stress in hepatocytes. More importantly, activated mitophagy can inhibit the inflammatory response by inhibiting inflammasome activation and reducing the release of inflammatory mediators, thereby alleviating APAP-induced liver injury (Fig. 1). However, so far, no relevant research has been reported in this regard. Hence, an in-depth study on the crosstalk between mitophagy and NLRP3 inflammasome activation will help clarify the mechanism for autophagy protecting against APAP hepatotoxicity.

Proposed mechanisms for mitophagy in APAP-induced hepatotoxicity. A toxic dose of APAP is metabolized to NAPQI in the liver, which can deplete hepatic GSH and covalently bind to cellular and mitochondrial proteins. Consequently, they lead to increased ROS production and mitochondrial dysfunction. As a result, damaged mitochondria can result in necrotic cell death and further ROS production. Furthermore, DAMP released from necrotic hepatocytes can activate inflammatory response, which further exacerbate liver injury. As a compensatory response under oxidative stress, ROS may trigger autophagy, which helps to remove APAP-AD and damaged mitochondria. Furthermore, autophagy-activating drugs can induce mitophagy, which not only reduce ROS production by removing damaged mitochondria, but also alleviate inflammation by degrading NLRP3 inflammasome, thus attenuating liver injury
The last question need to be mentioned is whether the activation of NLRP3 inflammasome is necessary for APAP hepatotoxicity. For example, mice deficient for NLRP3 inflammasome demonstrated a similar liver injury and sterile inflammation following APAP (Williams et al. 2011). Furthermore, caspase inhibitor could inhibit caspase-1 activity and block the maturation of IL-1β. However, APAP-induced liver injury and neutrophil infiltration were not affected (Williams et al. 2010). Maybe, the discrepancy can be attributed to other pathways. Indeed, in canonical NLRP3 inflammasome pathway, caspase-1 activity is required for the maturation of proinflammatory cytokine; however, the release of IL-1α and IL-1β can be independent of caspase-1 catalytic activity. Calpain, calcium-dependent cysteine protease, is believed to involved in the activation NLRP3 inflammasome and secretion of the proinflammatory cytokine IL-1β (Gross et al. 2012; Valimaki et al. 2016). By contrast, the overexpression of calpastatin, an endogenous inhibitor in mice can inhibit the activation of NLRP3 inflammasome and the production of IL-1α and IL-1β (Hanouna et al. 2017). Therefore, the exact role of NLRP3 inflammasome in APAP hepatotoxicity still needs further investigation. Taken together, there is a complex interaction between mitochondrial damage, oxidative stress and inflammatory response in APAP-induced liver injury. Mitophagy, as a critical mechanism for maintaining mitochondrial homeostasis, can not only reduce ROS production by removing damaged mitochondria, but also alleviate inflammation by degrading NLRP3 inflammasome.
Maintaining the turnover of endoplasmic reticulum
As mentioned above, there is a growing body of evidence that supports ER stress is implicated in APAP hepatotoxicity. ER stress is generally considered an event secondary to NAPQI generation. NAPQI can covalently bind to critical ER proteins such as GSH-S-transferase and calreticulin, thus resulting in an ER stress (Shin et al. 2007; Wang et al. 2006; Zhou et al. 1996). Furthermore, GSH depletion in the ER can result in intraluminal redox imbalance, leading to phosphorylation of eIF2α and activation of ATF6 and CHOP (Nagy et al. 2007, 2010).
Recent studies have found that autophagy play a critical role in the turnover and modulation of ER (Khaminets et al. 2015; Lipatova and Segev 2015; Mochida et al. 2015). Under ER stress condition, autophagy is activated to meet the different cellular requirement. The autophagy induced by ER stress mainly includes the ER stress-mediated autophagy and ER-phagy. Among them, the autophagy that is activated under ER stress condition is usually named as “ER stress-mediated autophagy”, while ER-phagy is a type of selective autophagy that involves the generation of autophagosomes that selectively sequester ER membranes (Smith et al. 2018; Song et al. 2018). Both of them need the UPR and the core autophagy machinery. The major difference is that ER stress-mediated autophagy sequesters and degrades the protein aggregates and damaged organelles, while ER-phagy selectively sequesters ER membranes (Khaminets et al. 2015; Mochida et al. 2015; Song et al. 2018). Moreover, ER-phagy need specific receptors to mediate selective attachment of autophagosomes and ER, FAM134B is a newly identified receptor of ER-phagy in mammalian cells (Khaminets et al. 2015). However, whether ER stress-mediated autophagy and ER-phagy were implicated in APAP hepatotoxicity has not been thoroughly studied so far. Given that the critical role of ER stress, an in-depth study on the causative link between autophagy and ER stress not only helps to elucidate the mechanism of APAP-induced acute liver injury, more importantly, but also promotes the development of treatment strategy for APAP liver injury. Considered the majority of poisoning patients delivered to the hospital for treatment have passed the metabolic phase and progressed to the phase of liver injury, so it is especially important for the treatment of patients with severe acute liver failure. If the progression of liver damage can be controlled by correcting mitochondrial dysfunction and ER stress, it is expected to develop an effective treatment for APAP hepatotoxicity.
References
Blazka ME, Germolec DR, Simeonova P, Bruccoleri A, Pennypacker KR, Luster MI (1995a) Acetaminophen-induced hepatotoxicity is associated with early changes in NF-kB and NF-IL6 DNA binding activity. J Inflamm 47(3):138–150
Blazka ME, Wilmer JL, Holladay SD, Wilson RE, Luster MI (1995b) Role of proinflammatory cytokines in acetaminophen hepatotoxicity. Toxicol Appl Pharmacol 133(1):43–52
Boobis AR, Tee LB, Hampden CE, Davies DS (1986) Freshly isolated hepatocytes as a model for studying the toxicity of paracetamol. Food Chem Toxicol Int J Publ Br Ind Biol Res Assoc 24(6–7):731–736
Chen ZH, Wu YF, Wang PL et al (2016) Autophagy is essential for ultrafine particle-induced inflammation and mucus hyperproduction in airway epithelium. Autophagy 12(2):297–311
Connolly MK, Ayo D, Malhotra A et al (2011) Dendritic cell depletion exacerbates acetaminophen hepatotoxicity. Hepatology 54(3):959–968
Dahlin DC, Miwa GT, Lu AY, Nelson SD (1984) N-acetyl-p-benzoquinone imine: a cytochrome P-450-mediated oxidation product of acetaminophen. Proc Natl Acad Sci USA 81(5):1327–1331
Davidson DG, Eastham WN (1966) Acute liver necrosis following overdose of paracetamol. Br Med J 2(5512):497–499
Deng X, Zhang F, Rui W et al (2013) PM2.5-induced oxidative stress triggers autophagy in human lung epithelial A549 cells. Toxicol In Vitro Int J Publ Assoc BIBRA 27(6):1762–1770
Dewaele M, Maes H, Agostinis P (2010) ROS-mediated mechanisms of autophagy stimulation and their relevance in cancer therapy. Autophagy 6(7):838–854
Du K, Ramachandran A, Jaeschke H (2016a) Oxidative stress during acetaminophen hepatotoxicity: sources, pathophysiological role and therapeutic potential. Redox Biol 10:148–156
Du K, Ramachandran A, Weemhoff JL et al (2016b) Editor’s highlight: metformin protects against acetaminophen hepatotoxicity by attenuation of mitochondrial oxidant stress and dysfunction. Toxicol Sci 154(2):214–226
Filomeni G, De Zio D, Cecconi F (2015) Oxidative stress and autophagy: the clash between damage and metabolic needs. Cell Death Differ 22(3):377–388
Foufelle F, Fromenty B (2016) Role of endoplasmic reticulum stress in drug-induced toxicity. Pharmacol Res Perspect 4(1):e00211. https://doi.org/10.1002/prp2.211
Gardner CR, Laskin JD, Dambach DM et al (2002) Reduced hepatotoxicity of acetaminophen in mice lacking inducible nitric oxide synthase: potential role of tumor necrosis factor-alpha and interleukin-10. Toxicol Appl Pharmacol 184(1):27–36
Green DR, Galluzzi L, Kroemer G (2011) Mitochondria and the autophagy-inflammation-cell death axis in organismal aging. Science 333(6046):1109–1112
Gross O, Yazdi AS, Thomas CJ et al (2012) Inflammasome activators induce interleukin-1alpha secretion via distinct pathways with differential requirement for the protease function of caspase-1. Immunity 36(3):388–400
Gujral JS, Knight TR, Farhood A, Bajt ML, Jaeschke H (2002) Mode of cell death after acetaminophen overdose in mice: apoptosis or oncotic necrosis? Toxicol Sci 67(2):322–328
Hamacher-Brady A, Brady NR (2016) Mitophagy programs: mechanisms and physiological implications of mitochondrial targeting by autophagy. Cell Mol Life Sci 73(4):775–795
Hanouna G, Mesnard L, Vandermeersch S et al (2017) Specific calpain inhibition protects kidney against inflammaging. Sci Rep 7(1):8016
He C, Klionsky DJ (2009) Regulation mechanisms and signaling pathways of autophagy. Annu Rev Genet 43:67–93
Hidvegi T, Mukherjee A, Ewing M, Kemp C, Perlmutter DH (2011) The role of autophagy in alpha-1-antitrypsin deficiency. Method Enzymol 499:33–54
Hinson JA, Roberts DW, James LP (2010) Mechanisms of acetaminophen-induced liver necrosis. Handb Exp Pharmacol 196:369–405
Hodgman MJ, Garrard AR (2012) A review of acetaminophen poisoning. Crit Care Clin 28(4):499–516
Holt MP, Cheng L, Ju C (2008) Identification and characterization of infiltrating macrophages in acetaminophen-induced liver injury. J Leukoc Biol 84(6):1410–1421
Horiuchi T, Sakata N, Narumi Y et al (2017) Metformin directly binds the alarmin HMGB1 and inhibits its proinflammatory activity. J Biol Chem 292(20):8436–8446
Igusa Y, Yamashina S, Izumi K et al (2012) Loss of autophagy promotes murine acetaminophen hepatotoxicity. J Gastroenterol 47(4):433–443
Imaeda AB, Watanabe A, Sohail MA et al (2009) Acetaminophen-induced hepatotoxicity in mice is dependent on Tlr9 and the Nalp3 inflammasome. J Clin Investig 119(2):305–314
Jaeschke H, Bajt ML (2006) Intracellular signaling mechanisms of acetaminophen-induced liver cell death. Toxicol Sci 89(1):31–41
Jaeschke H, McGill MR, Ramachandran A (2012) Oxidant stress, mitochondria, and cell death mechanisms in drug-induced liver injury: lessons learned from acetaminophen hepatotoxicity. Drug Metab Rev 44(1):88–106
James LP, McCullough SS, Knighy TR, Jaeschke H, Hinson JA (2003) Acetaminophen toxicity in mice lacking NADPH oxidase activity: role of peroxynitrite formation and mitochondrial oxidant stress. Free Radical Res 37(12):1289–1297
Ju C, Reilly TP, Bourdi M et al (2002) Protective role of Kupffer cells in acetaminophen-induced hepatic injury in mice. Chem Res Toxicol 15(12):1504–1513
Kerr JS, Adriaanse BA, Greig NH et al (2017) Mitophagy and Alzheimer’s Disease: cellular and molecular mechanisms. Trends Neurosci 40(3):151–166
Khaminets A, Heinrich T, Mari M et al (2015) Regulation of endoplasmic reticulum turnover by selective autophagy. Nature 522(7556):354–358
Kim YH, Hwang JH, Kim KS et al (2015) Metformin ameliorates acetaminophen hepatotoxicity via Gadd45beta-dependent regulation of JNK signaling in mice. J Hepatol 63(1):75–82
Kubes P, Mehal WZ (2012) Sterile inflammation in the liver. Gastroenterology 143(5):1158–1172
Kusama H, Kon K, Ikejima K et al (2017) Sodium 4-phenylbutyric acid prevents murine acetaminophen hepatotoxicity by minimizing endoplasmic reticulum stress. J Gastroenterol 52(5):611–622
Lamark T, Johansen T (2012) Aggrephagy: selective disposal of protein aggregates by macroautophagy. Int J Cell Biol 2012:736905
Larson AM (2007) Acetaminophen hepatotoxicity. Clin Liver Dis 11(3):525–548 (vi)
Larson AM, Polson J, Fontana RJ et al (2005) Acetaminophen-induced acute liver failure: results of a United States multicenter, prospective study. Hepatology 42(6):1364–1372
Laskin DL, Pilaro AM (1986) Potential role of activated macrophages in acetaminophen hepatotoxicity. I. Isolation and characterization of activated macrophages from rat liver. Toxicol Appl Pharmacol 86(2):204–215
Lazarou M, Sliter DA, Kane LA et al (2015) The ubiquitin kinase PINK1 recruits autophagy receptors to induce mitophagy. Nature 524(7565):309–314
Lipatova Z, Segev N (2015) A Role for Macro-ER-Phagy in ER quality control. PLoS Genet 11(7):e1005390
Liu ZX, Govindarajan S, Kaplowitz N (2004) Innate immune system plays a critical role in determining the progression and severity of acetaminophen hepatotoxicity. Gastroenterology 127(6):1760–1774
Liu ZX, Han D, Gunawan B, Kaplowitz N (2006) Neutrophil depletion protects against murine acetaminophen hepatotoxicity. Hepatology 43(6):1220–1230
Lupfer C, Thomas PG, Anand PK et al (2013) Receptor interacting protein kinase 2-mediated mitophagy regulates inflammasome activation during virus infection. Nat Immunol 14(5):480–488
Marques PE, Amaral SS, Pires DA et al (2012) Chemokines and mitochondrial products activate neutrophils to amplify organ injury during mouse acute liver failure. Hepatology 56(5):1971–1982
McCullough KD, Martindale JL, Klotz LO, Aw TY, Holbrook NJ (2001) Gadd153 sensitizes cells to endoplasmic reticulum stress by down-regulating Bcl2 and perturbing the cellular redox state. Mol Cell Biol 21(4):1249–1259
McGill MR, Yan HM, Ramachandran A, Murray GJ, Rollins DE, Jaeschke H (2011) HepaRG cells: a human model to study mechanisms of acetaminophen hepatotoxicity. Hepatology 53(3):974–982
McGill MR, Sharpe MR, Williams CD, Taha M, Curry SC, Jaeschke H (2012a) The mechanism underlying acetaminophen-induced hepatotoxicity in humans and mice involves mitochondrial damage and nuclear DNA fragmentation. J Clin Investig 122(4):1574–1583
McGill MR, Williams CD, Xie Y, Ramachandran A, Jaeschke H (2012b) Acetaminophen-induced liver injury in rats and mice: comparison of protein adducts, mitochondrial dysfunction, and oxidative stress in the mechanism of toxicity. Toxicol Appl Pharmacol 264(3):387–394
Michael SL, Pumford NR, Mayeux PR, Niesman MR, Hinson JA (1999) Pretreatment of mice with macrophage inactivators decreases acetaminophen hepatotoxicity and the formation of reactive oxygen and nitrogen species. Hepatology 30(1):186–195
Mitchell JR, Jollow DJ, Potter WZ, Davis DC, Gillette JR, Brodie BB (1973a) Acetaminophen-induced hepatic necrosis. I. Role of drug metabolism. J Pharmacol Exp Ther 187(1):185–194
Mitchell JR, Jollow DJ, Potter WZ, Gillette JR, Brodie BB (1973b) Acetaminophen-induced hepatic necrosis. IV. Protective role of glutathione. J Pharmacol Exp Ther 187(1):211–217
Mizushima N, Komatsu M (2011) Autophagy: renovation of cells and tissues. Cell 147(4):728–741
Mizushima N, Yoshimori T, Levine B (2010) Methods in mammalian autophagy research. Cell 140(3):313–326
Mochida K, Oikawa Y, Kimura Y et al (2015) Receptor-mediated selective autophagy degrades the endoplasmic reticulum and the nucleus. Nature 522(7556):359–362
Mossanen JC, Krenkel O, Ergen C et al (2016) Chemokine (C–C motif) receptor 2-positive monocytes aggravate the early phase of acetaminophen-induced acute liver injury. Hepatology 64(5):1667–1682
Murata H, Sakaguchi M, Kataoka K, Huh NH (2013) SARM1 and TRAF6 bind to and stabilize PINK1 on depolarized mitochondria. Mol Biol Cell 24(18):2772–2784
Nagy G, Kardon T, Wunderlich L et al (2007) Acetaminophen induces ER dependent signaling in mouse liver. Arch Biochem Biophys 459(2):273–279
Nagy G, Szarka A, Lotz G et al (2010) BGP-15 inhibits caspase-independent programmed cell death in acetaminophen-induced liver injury. Toxicol Appl Pharmacol 243(1):96–103
Nakahira K, Haspel JA, Rathinam VA et al (2011) Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome. Nat Immunol 12(3):222–230
Nguyen TN, Padman BS, Lazarou M (2016) Deciphering the molecular signals of PINK1/Parkin Mitophagy. Trends Cell Biol 26(10):733–744
Ni HM, Bockus A, Boggess N, Jaeschke H, Ding WX (2012a) Activation of autophagy protects against acetaminophen-induced hepatotoxicity. Hepatology 55(1):222–231
Ni HM, Boggess N, McGill MR et al (2012b) Liver-specific loss of Atg5 causes persistent activation of Nrf2 and protects against acetaminophen-induced liver injury. Toxicol Sci 127(2):438–450
Ni HM, Williams JA, Jaeschke H, Ding WX (2013) Zonated induction of autophagy and mitochondrial spheroids limits acetaminophen-induced necrosis in the liver. Redox Biol 1:427–432
Ni HM, McGill MR, Chao X et al (2016) Removal of acetaminophen protein adducts by autophagy protects against acetaminophen-induced liver injury in mice. J Hepatol. https://doi.org/10.1016/j.jhep.2016.04.025
Ostapowicz G, Fontana RJ, Schiodt FV et al (2002) Results of a prospective study of acute liver failure at 17 tertiary care centers in the United States. Ann Intern Med 137(12):947–954
Patten CJ, Thomas PE, Guy RL et al (1993) Cytochrome P450 enzymes involved in acetaminophen activation by rat and human liver microsomes and their kinetics. Chem Res Toxicol 6(4):511–518
Prescott LF, Illingworth RN, Critchley JA, Proudfoot AT (1980) Intravenous N-acetylcysteine: still the treatment of choice for paracetamol poisoning. Br Med J 280(6206):46–47
Saeedi Saravi SS, Hasanvand A, Shahkarami K, Dehpour AR (2016) The protective potential of metformin against acetaminophen-induced hepatotoxicity in BALB/C mice. Pharm Biol 54(12):2830–2837
Scheibye-Knudsen M, Fang EF, Croteau DL, Wilson DM 3rd, Bohr VA (2015) Protecting the mitochondrial powerhouse. Trends Cell Biol 25(3):158–170
Scherz-Shouval R, Elazar Z (2007) ROS, mitochondria and the regulation of autophagy. Trends Cell Biol 17(9):422–427
Scherz-Shouval R, Elazar Z (2011) Regulation of autophagy by ROS: physiology and pathology. Trends Biochem Sci 36(1):30–38
Scherz-Shouval R, Shvets E, Fass E, Shorer H, Gil L, Elazar Z (2007) Reactive oxygen species are essential for autophagy and specifically regulate the activity of Atg4. EMBO J 26(7):1749–1760
Schilling A, Corey R, Leonard M, Eghtesad B (2010) Acetaminophen: old drug, new warnings. Clevel Clin J Med 77(1):19–27
Shin NY, Liu Q, Stamer SL, Liebler DC (2007) Protein targets of reactive electrophiles in human liver microsomes. Chem Res Toxicol 20(6):859–867
Smilkstein MJ, Knapp GL, Kulig KW, Rumack BH (1988) Efficacy of oral N-acetylcysteine in the treatment of acetaminophen overdose. Analysis of the national multicenter study (1976 to 1985). N Engl J Med 319(24):1557–1562
Smith MD, Harley ME, Kemp AJ et al (2018) CCPG1 is a non-canonical autophagy cargo receptor essential for ER-phagy and pancreatic ER proteostasis. Dev Cell 44(2):217–232 e11
Song S, Tan J, Miao Y, Zhang Q (2018) Crosstalk of ER stress-mediated autophagy and ER-phagy: involvement of UPR and the core autophagy machinery. J Cell Physiol 233(5):3867–3874
Sun Y, Li TY, Song L et al (2017) Liver-specific deficiency of unc-51 like kinase 1 and 2 protects mice from acetaminophen-induced liver injury. Hepatology. https://doi.org/10.1002/hep.29759
Tee LB, Boobis AR, Huggett AC, Davies DS (1986) Reversal of acetaminophen toxicity in isolated hamster hepatocytes by dithiothreitol. Toxicol Appl Pharmacol 83(2):294–314
Thummel KE, Lee CA, Kunze KL, Nelson SD, Slattery JT (1993) Oxidation of acetaminophen to N-acetyl-p-aminobenzoquinone imine by human CYP3A4. Biochem Pharmacol 45(8):1563–1569
Ueno T, Komatsu M (2017) Autophagy in the liver: functions in health and disease. Nat Rev Gastroenterol Hepatol 14(3):170–184
Uzi D, Barda L, Scaiewicz V et al (2013) CHOP is a critical regulator of acetaminophen-induced hepatotoxicity. J Hepatol 59(3):495–503
Valimaki E, Cypryk W, Virkanen J et al (2016) Calpain activity is essential for ATP-driven unconventional vesicle-mediated protein secretion and inflammasome activation in human macrophages. J Immunol 197(8):3315–3325
Vendemiale G, Grattagliano I, Altomare E, Turturro N, Guerrieri F (1996) Effect of acetaminophen administration on hepatic glutathione compartmentation and mitochondrial energy metabolism in the rat. Biochem Pharmacol 52(8):1147–1154
Wang X, Thomas B, Sachdeva R et al (2006) Mechanism of arylating quinone toxicity involving Michael adduct formation and induction of endoplasmic reticulum stress. Proc Natl Acad Sci USA 103(10):3604–3609
Weinberg SE, Sena LA, Chandel NS (2015) Mitochondria in the regulation of innate and adaptive immunity. Immunity 42(3):406–417
Williams CD, Farhood A, Jaeschke H (2010) Role of caspase-1 and interleukin-1beta in acetaminophen-induced hepatic inflammation and liver injury. Toxicol Appl Pharmacol 247(3):169–178
Williams CD, Antoine DJ, Shaw PJ et al (2011) Role of the Nalp3 inflammasome in acetaminophen-induced sterile inflammation and liver injury. Toxicol Appl Pharmacol 252(3):289–297
Williams JA, Ni HM, Haynes A et al (2015) Chronic deletion and acute knockdown of parkin have differential responses to acetaminophen-induced mitophagy and liver injury in mice. J Biol Chem 290(17):10934–10946
Woolbright BL, Jaeschke H (2017) Role of the inflammasome in acetaminophen-induced liver injury and acute liver failure. J Hepatol 66(4):836–848
Zhang XL, Cheng XP, Yu L et al. (2016) MCOLN1 is a ROS sensor in lysosomes that regulates autophagy. Nat Commun. https://doi.org/10.1038/Ncomms12109
Zhong Z, Sanchez-Lopez E, Karin M (2016) Autophagy, inflammation, and immunity: a troika governing cancer and its treatment. Cell 166(2):288–298
Zhou L, McKenzie BA, Eccleston ED Jr et al (1996) The covalent binding of [14C]acetaminophen to mouse hepatic microsomal proteins: the specific binding to calreticulin and the two forms of the thiol:protein disulfide oxidoreductases. Chem Res Toxicol 9(7):1176–1182
Acknowledgements
This work was supported by Key research and development plan of Shandong Province (2018GSF118013) and National Natural Science Foundation of China (No. 81673209).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no conflict of interest.
Rights and permissions
About this article

Cite this article
Shan, S., Shen, Z. & Song, F. Autophagy and acetaminophen-induced hepatotoxicity. Arch Toxicol 92, 2153–2161 (2018). https://doi.org/10.1007/s00204-018-2237-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00204-018-2237-5