
Abstract
Acetaminophen (APAP)-induced liver injury is an important clinical and toxicological problem. Understanding the mechanisms and modes of cell death are vital for the development of therapeutic interventions. The histological and clinical features of APAP hepatotoxicity including cell and organelle swelling, karyolysis, and extensive cell contents release lead to the characterization of the cell death as oncotic necrosis. However, the more recent identification of detailed signaling mechanisms of mitochondrial dysfunction, the amplification mechanisms of mitochondrial oxidant stress and peroxynitrite formation by a mitogen-activated protein kinase cascade, mechanisms of the mitochondrial permeability transition pore opening and nuclear DNA fragmentation as well as the characterization of the sterile inflammatory response suggested that the mode of cell death is better termed programmed necrosis. Additional features like mitochondrial Bax translocation and cytochrome c release, mobilization of lysosomal iron and the activation of receptor-interacting protein kinases and the inflammasome raised the question whether other emerging modes of cell death such as apoptosis, necroptosis, ferroptosis and pyroptosis could also play a role. The current review summarizes the key mechanisms of APAP-induced liver injury and compares these with key features of the newly described modes of cell death. Based on the preponderance of experimental and clinical evidence, the mode of APAP-induced cell death should be termed programmed necrosis; despite some overlap with other modes of cell death, APAP hepatotoxicity does not fulfill the characteristics of either apoptosis, necroptosis, ferroptosis, pyroptosis or autophagic cell death.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Drug hepatotoxicity is a serious clinical problem as well as a concern during drug development. While idiosyncratic drug-induced liver injury is very rare and mechanistically poorly understood, the clinically most relevant drug hepatotoxicity and acute liver failure in the US and many western countries is caused by acetaminophen (APAP) overdose (Larson et al. 2005; Lee 2013). Due to the availability of a mouse model that is highly relevant for the human pathophysiology (Jaeschke 2015; McGill and Jaeschke 2019), APAP is the most extensively studied hepatotoxic drug worldwide and hence the modes of cell death have been well studied in this model. Thus, more is known about the mechanisms of APAP-induced cell death compared to any other drug or chemical (Ramachandran and Jaeschke 2019).
It is important to investigate the mode of cell death caused by APAP or other drugs because the mode of cell death is critically linked to the molecular mechanisms activated. Understanding these mechanisms of cell injury allows the identification of therapeutic targets and consequently development of antidotes. Inhibiting drug-induced cell death is vital for limiting serious liver injury, preventing acute liver failure and promoting regeneration of the damaged liver in patients. To better understand the potential therapeutic targets, the current review analyzes the different established and newly emerging modes of cell death in APAP hepatotoxicity.
Oncotic necrosis and programmed necrosis
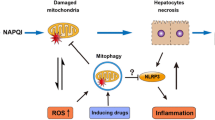
APAP toxicity starts with the formation of a reactive metabolite, N-acetyl-p-benzoquinone imine (NAPQI), by cytochrome P450 enzymes, especially Cyp2E1. Excessive NAPQI generation during an overdose leads to depletion of hepatic GSH levels and protein adduct formation (McGill and Jaeschke 2013). Protein adducts in mitochondria induce an initial oxidant stress, which must be amplified by a MAP kinase cascade (Han et al. 2013). The first step is the activation of redox-sensitive kinases such as ASK-1, MLK3 and GSK3b (Nakagawa et al. 2008; Xie et al. 2015; Sharma et al. 2012; Shinohara et al. 2010). These kinases in turn activate MKK4 (Zhang et al. 2017), which induces the activation (phosphorylation) of c-Jun N-terminal kinase (JNK) (Han et al. 2013; Du et al. 2015). The translocation of phospho-JNK to mitochondria and binding to the anchor protein Sab (Win et al. 2011) enhances the oxidant stress and peroxynitrite formation (Du et al. 2016). The amplified oxidant stress together with iron translocation from the lysosomes triggers the mitochondrial membrane permeability transition pore (MPTP) opening, which causes the collapse of the mitochondrial membrane potential and cessation of ATP synthesis (Ramachandran and Jaeschke 2019). Early Bax translocation to the mitochondria and later mitochondrial matrix swelling with rupture of the outer membrane releases intermembrane proteins such as endonuclease G and apoptosis-inducing factor (AIF) (Bajt et al. 2006, 2008). Both proteins translocate to the nucleus and induce DNA fragmentation (Bajt et al. 2006). Extensive mitochondrial dysfunction and karyolysis leads to cell necrosis, which is accompanied by cell and organelle swelling and cell contents release (Gujral et al. 2002) (Fig. 1). These signaling events causing cellular necrosis can be counteracted in part by adaptive mechanisms of autophagy to remove protein adducts (Ni et al. 2016), mitophagy to remove damaged mitochondria (Ni et al. 2012) and mitochondrial biogenesis to replace damaged mitochondria (Du et al. 2017). Due to the extensive cell swelling and contents release, this type of cell death in APAP hepatotoxicity was previously termed oncotic necrosis (Gujral et al. 2002). The more recently identified intricate signaling pathways involved in the initiation, amplification and propagation of the stress within the cell ultimately leading to cell death also led to the use of the term programmed necrosis.

Forms of cell death discussed in the context of acetaminophen hepatotoxicity. The classical pathways for apoptosis are mediated by the Fas or TNF receptor, whose activation results in their trimerization and activation of caspase 8. While caspase 8 directly activates effector caspases such as caspase 3 in type I cells such as thymocytes, it cleaves the protein Bid in the cytosol in type II cells such as hepatocytes. Truncated Bid (tBid) then translocates to the mitochondria, where along with other Bcl2 family member proteins such as Bax, Bad and Bak it induces mitochondrial permeability transition and release of cytochrome c into the cytosol. This results in assembly of the apoptosome consisting of APAF1, cytochrome c, ATP and procaspase 9 and subsequent activation of caspase 9, which then activates caspase 3. Activation of caspase 3 then results in the characteristic caspase-activated DNase (CAD)-mediated nuclear DNA fragmentation and nuclear condensation, ultimately resulting in apoptotic cell death. In a scenario where TNF activation occurs when caspase 8 is inhibited, however, the signaling shifts to formation of a necrosome complex consisting of the receptor-interacting kinases 1 and 3 and the mixed lineage kinase domain like pseudokinase (MLKL). The phosphorylation and activation of MLKL on this complex result in its translocation to the plasma membrane, and disruption of the membrane resulting in cell death by necroptosis. Acetaminophen (APAP)-mediated hepatocyte cell death has some features of both these modes of cell death, but lacks certain defining features of each, which puts it in a class apart. Cytochrome P450-mediated metabolism of APAP results in formation of the reactive metabolite NAPQI, which induces mitochondrial oxidant stress and activation of the MAP kinase JNK. Translocation of activated JNK as well as Bax to the mitochondria induces an oxidant stress, which amplifies mitochondrial dysfunction and also upregulates RIP3 kinase levels. The subsequent induction of the mitochondrial permeability transition results in release of cytochrome c, endonuclease G and apoptosis-inducing factor into the cytosol. However, cytochrome c release does not result in assembly of the apoptosome and endonuclease G and AIF translocation to the nucleus causes nuclear DNA fragmentation and programmed necrosis
Necroptosis
A more specific form of programmed necrosis is necroptosis (Jouan-Lanhouet et al. 2014; Tang et al. 2019). It was recognized that when caspases were inhibited during TNF-α-induced apoptosis, a necrotic form of cell death emerged (Vercammen et al. 1998). Investigations into the mechanism of this new form of programmed cell death, termed necroptosis, revealed the involvement of the receptor-interacting serine/threonine-protein kinase 1 (RIPK1), RIPK3 and the pseudokinase mixed lineage kinase domain-like protein (MLKL) (Jouan-Lanhouet et al. 2014; Tang et al. 2019). When RIPK1 is activated and caspase-8 is inhibited, RIPK3 and MLKL are recruited to the complex (necrosome), RIPK3 phosphorylates MLKL, which then oligomerizes, and multimers of phospho-MLKL translocate to the cell membrane where it can cause membrane permeabilization (Tang et al. 2019) (Fig. 1). Since the necrosome can also be formed independent of RIPK1, RIPK3 and MLKL are the critical mediators involved in necroptosis (Tang et al. 2019).
Studies with APAP initially showed RIPK1 expression and RIPK3 induction in hepatocytes after APAP overdose (Sharma et al. 2012; Ramachandran et al. 2013). In addition, the RIPK1 inhibitor necrostatin-1 was protective in vitro and in vivo (Sharma et al. 2012; Ramachandran et al. 2013; Zhang et al. 2014). These observations led to the characterization of APAP-induced cell death as necroptotic (Zhang et al. 2014). However, necrostatin-1 has off-target effects and is not specific for RIPK1 (Degterev et al. 2013). Nevertheless, gene deletion experiments with RIPK1 have confirmed the potential role of RIPK1 in APAP toxicity (Dara et al. 2015) but the role of RIPK1 in apoptosis and RIPK1-independent mechanisms of necrosome formation makes RIPK1 not specific for necroptosis. APAP overdose induces RIPK3 in the liver and in hepatocytes and RIPK3 knockout mice as well as morpholino-based gene knockdown has been shown to be protective during the early phase of APAP-induced liver injury (Ramachandran et al. 2013). These in vivo findings with RIPK3 knockout mice were confirmed by some investigators (Deutsch et al. 2015) but not by others (Dara et al. 2015). In addition, an inhibitor of RIPK3 has been shown to protect against APAP-induced cell death in human hepatocytes (Li et al. 2014). We have recently demonstrated that scavenging mitochondrial superoxide inhibited APAP-induced RIPK3 expression and caspase activation and apoptotic morphology was evident in hepatocytes in RIPK3-deficient mice after APAP (Du et al. 2019), suggesting that RIPK3 influenced the mode of cell death after APAP. Nevertheless, the reason for the controversial findings remains unclear (Yang et al. 2016) and a better understanding of the function of RIPK3 in the pathophysiology of APAP is needed. The second critical mediator of necroptosis, MLKL, is expressed in the liver and in hepatocytes but deficiency of this gene did not reduce APAP toxicity (Dara et al. 2015). Thus, despite the expression and induction of RIPKs and MLKL during APAP toxicity, there is no consistent evidence for the involvement of these genes in APAP-induced cell death and, therefore, necroptosis is unlikely to play a critical role. This is also supported by the fact that TNF-α is not a critical mediator of APAP-induced cell death (Boess et al. 1998).
Apoptosis
Apoptosis was the first described form of programmed cell death, and extensive investigations over the last decades revealed many details of the cell death pathway. Based on the initiation, extrinsic and intrinsic pathways are distinguished. During extrinsic apoptosis, ligands, e.g., Fas-ligand or TNF-α, react with their receptors, e.g., Fas receptor or TNF receptor 1, on the cell surface and trigger the trimerization of the receptor. Through the death domain of the receptor, procaspase-8 is bound and activated. Active caspase-8 can either directly cleave and activate effector caspases such as caspase-3 (type I cell) or cleave Bid (type II cell), which then translocates to the mitochondria and together with other Bcl-2 family members (Bax, Bak, Bad) permeabilizes the outer mitochondrial membrane and releases cytochrome c and Smac/Diablo. Cytochrome c together with ATP and procaspase-9 binds to apoptotic protease-activating factor-1 (APAF-1) inducing the activation of caspase-9, which then activates caspase-3 (Fig. 1). Smac/Diablo inhibits the cytosolic inhibitors of apoptosis proteins (IAPs), which prevent inactivation of caspases by IAPs. The intrinsic pathway of apoptosis starts with inducing Bax translation to the mitochondria or other signals that trigger the release of pro-apoptotic mediators from mitochondria, which induces the caspase cascade by activation of caspase-9. In either case, the effector caspases degrade a variety of intracellular macromolecules triggering shrinkage and fragmentation of the cell into apoptotic bodies. In addition, there is chromatin condensation and DNA fragmentation induced by the caspase-activated DNase (CAD). This enzyme is kept inactive in the cytosol by an inhibitor (ICAD), which is cleaved by caspase-3 during apoptosis. This leads to the liberation and translocation of CAD to the nucleus where it cleaves DNA into nucleosomal units. Overall, the most prominent features of apoptosis are the morphological changes including cell shrinkage, nuclear condensation and chromatin margination in the absence of cell or organelle swelling and cell contents release. The biochemical hallmark of apoptosis is the activation of effector caspases, especially caspase-3, and prevention of cell death by caspase inhibitors. Virtually all other signaling events that occur during apoptosis, e.g., mitochondrial cytochrome c release, Bax translocation to mitochondria, etc., can be observed in other forms of cell death and are not specific for apoptosis. Importantly, when many cells undergo apoptosis, the apoptotic signaling pathway cannot be completed due to the decline of cellular ATP levels (Jaeschke and Lemasters 2003). The removal of apoptotic bodies by phagocytosis is then impaired, and cells switch to secondary necrosis. Under these conditions, the cells are swelling and release their contents similar to primary necrotic cell death with the exception that active caspases can still be measured. The complicated signaling events involved, the potential switch from apoptosis to secondary necrosis and the fact that other forms of cell death may use in part similar pathways, and features led to considerable confusion and controversies regarding the role of apoptosis in many disease processes including acute and chronic liver diseases (Jaeschke and Lemasters 2003; Malhi et al. 2006).
In APAP hepatotoxicity, necrosis was the generally accepted cell death mechanisms for decades. In the early 1990s, George Corcoran’s group first reported evidence of nuclear DNA fragmentation using the DNA ladder assay, which shows the cleavage of nuclear DNA into internucleosomal fragments of about 180 base pairs and multiples thereof (Ray et al. 1990, 1993; Shen et al. 1991, 1992). In a follow-up study, Ray et al. (1996) concluded based on the DNA ladder assay and nuclear condensation in histological sections that approximately 40% of the dead cells died by apoptosis. Although the dying cells were also TUNEL-positive (Lawson et al. 1999; Gujral et al. 2002), the morphology did not show the characteristics of apoptosis when compared to positive controls such as Fas- or TNF-induced apoptosis (Gujral et al. 2002; Jaeschke et al. 2011). Both the TUNEL assay and the DNA ladder assay show DNA fragmentation but are not necessarily specific for apoptosis (Grasl-Kraupp et al. 1995; Cover et al. 2005). In addition, there was no evidence of caspase activation during APAP toxicity (Lawson et al. 1999; Adams et al. 2001; Gujral et al. 2002), which suggests that the DNA fragmentation could not have been caused by the caspase-activated DNase. In contrast, subsequent studies have identified the mitochondrial endonucleases, endonuclease G and apoptosis-inducing factor, as the cause of the DNA fragmentation (Bajt et al. 2006, 2011). Furthermore, mitochondrial translocation of Bax and Bid and mitochondrial release of cytochrome c and Smac/Diablo have been observed during APAP hepatotoxicity (Adams et al. 2001; Bajt et al. 2008; El-Hassan et al. 2003; Jaeschke et al. 2006); Bax is responsible for early release of some intermembrane proteins but has no lasting impact on cell death (Bajt et al. 2008). Despite the release of cytochrome c, no relevant caspase activation has been reported (Adams et al. 2001; Knight and Jaeschke 2002; Lawson et al. 1999; El-Hassan et al. 2003) and highly effective caspase inhibitors do not protect (Lawson et al. 1999; Gujral et al. 2002; Williams et al. 2010). In the two studies that claim inhibition of APAP hepatotoxicity with pan-caspase inhibitors (El-Hassan et al. 2003; Hu and Colletti 2010), the solvent DMSO, which is a potent cytochrome P450 inhibitor (Park et al. 1988), was the cause of the protection (Jaeschke et al. 2006, 2011). Thus, despite the significant overlap of signaling pathways between necrosis and apoptosis, APAP-induced cell death in mouse hepatocytes or in vivo is caused by programmed necrosis and there is no evidence of any relevant contribution of apoptosis. This conclusion also applies to APAP overdose in humans and human hepatocytes (McGill et al. 2011, 2012; Xie et al. 2014).
In recent years, a substantial number of manuscripts are being published in the natural product field that claim apoptotic cell death in APAP hepatotoxicity. The conclusions are mainly based on elevated mRNA or protein expression of Bax and caspases after APAP and declining levels of Bcl-2 (reviewed in Jaeschke et al. 2018). Occasionally, a TUNEL assay is performed. The conclusions are questionable as hepatocytes do not need to induce pro-apoptotic proteins to execute apoptosis. Only Bax translocation to mitochondria and activation of caspases are relevant for apoptosis. In addition, the role of Bcl-2 in APAP toxicity is unclear (Adams et al. 2001) and the TUNEL assay indicates DNA strand breaks, which are caused by mitochondria-derived endonucleases (Bajt et al. 2006). Despite some similarities, there are substantial differences in the molecular weight of the DNA fragments between apoptosis and APAP-induced necrosis (Jahr et al. 2001). Thus, most of the recent claims of apoptotic cell death in APAP-induced liver injury are not justified by the data.
Autophagy and autophagy-associated cell death
Macroautophagy (hereafter referred to as autophagy) is a highly conserved intracellular degradation pathway, which degrades cellular components and damaged/excess organelles in response to adverse environmental conditions and stresses for cell survival purpose (Klionsky and Emr 2000; Nakatogawa et al. 2009; Parzych and Klionsky, 2014). Emerging evidence indicates that autophagy protects against APAP-induced liver injury by at least two mechanisms: selective removal of damaged mitochondria and of APAP protein adducts in hepatocytes (Ni et al. 2012, 2013; Chao et al. 2018). Timely removal of damaged mitochondria may help to attenuate APAP-induced mitochondrial damage to maintain mitochondrial homeostasis and sufficient ATP, which protects against APAP-induced liver injury. We demonstrated increased number of GFP-LC3-positive autophagosomes in GFP-LC3 transgenic mouse livers after APAP administration, suggesting the induction of autophagy (Ni et al. 2012, 2013). Autophagic removal of damaged mitochondria is mediated by a selective autophagy process termed as mitophagy. Phosphatase and tensin homolog (PTEN)-induced kinase 1 (PINK1)/Parkin pathway is so far the best characterized signaling pathway regulating mitophagy in mammals. In healthy mitochondria, PINK1 is cleaved by the inner mitochondrial membrane protease PARL to release the truncated PINK1 into the cytosol, which is then degraded by the proteasome via the N-end rule pathway (Jin et al. 2010; Deas et al. 2011; Meissner et al. 2011; Yamano and Youle 2013). In damaged and depolarized mitochondria, PINK1 is stabilized on the outer mitochondrial membrane (Matsuda et al. 2010; Narendra et al. 2010; Vives-Bauza et al. 2010). PINK1 then phosphorylates ubiquitin and Parkin, an E3 ligase, that not only leads to the recruitment of Parkin to mitochondria but also enhances Parkin E3 ligase activity resulting in outer mitochondrial membrane protein ubiquitination (Koyano et al. 2014). Conversely, a recent study showed that PTEN-L, an isoform of PTEN, dephosphorylates ubiquitin via its protein phosphatase activity to negatively regulate mitophagy (Wang et al. 2018). Ubiquitinated mitochondria then bind with autophagy receptor proteins such as optineurin, SQSTM1/p62 (hereafter referred to as p62) and NDP52 to trigger mitophagy (Ni et al. 2015; Rojansky et al. 2016; Pickles et al. 2018; Williams and Ding 2018). We recently demonstrated that APAP administration increases Parkin translocation to mitochondria with concurrent increased ubiquitination of mitochondrial proteins and mitophagy induction in mouse livers (Williams et al. 2015). This mitochondrial translocation and ubiquitination of mitochondrial proteins were blunted in PINK1 knockout (KO) mice (Wang et al. 2019). Using a novel tandem fluorescent-tagged inner mitochondrial membrane protein Cox8 (Cox8-GFP-mCherry) that can be used to monitor mitophagy based on different pH stability of GFP and mCherry fluorescent proteins, we found that single deletion of either Parkin or PINK1 in mice is not enough to blunt APAP-induced mitophagy. However, mitophagy markedly decreased in PINK1/Parkin double KO mouse livers after APAP treatment (Wang et al. 2019). More importantly, PINK1/Parkin double KO mice were more susceptible to APAP-induced liver injury (Wang et al. 2019). These data suggest that PINK1/Parkin-mediated mitophagy protects against APAP-induced liver injury by removing APAP-induced damaged mitochondria. However, it remains unknown whether any of the known mitophagy receptor proteins would be involved APAP-induced liver injury.
Besides the removal of damaged mitochondria, we also demonstrated that autophagy can remove APAP-adducts. We found that APAP-adducts co-localize with GFP-LC3-positive autophagosomes in primary cultured hepatocytes. Moreover, purified hepatic autophagosomes and autolysosomes from APAP-treated mouse livers contain APAP-adducts (Ni et al. 2016). Furthermore, pharmacological inhibition of autophagy increases whereas activation of autophagy decreases APAP-adducts in APAP-treated mouse livers. Furthermore, knockdown of autophagy receptor protein p62 impairs the clearance of APAP-adducts in hepatocytes and increases APAP-induced hepatotoxicity (Ni et al. 2016). Taken together, these findings suggest that activation of autophagy may be beneficial against APAP-induced hepatotoxicity by removing APAP-adducts and damaged mitochondria.
It should be noted that there is a complicated mutual regulatory network among autophagy and cell death. Apoptosis suppresses autophagy by inducing caspase-mediated cleavage of essential autophagy proteins such as Beclin 1 (Li et al. 2011). In addition, necroptosis protein RIPK1 represses basal autophagy by inhibiting TFEB-mediated expression of autophagy-related and lysosomal genes (Yonekawa et al. 2015). In contrast, despite its protective role against cell death, under certain conditions, autophagy also promotes ferroptosis, a form of iron-mediated non-apoptotic cell death (as discussed below). Autophagy can selectively degrade ferritin through NCOA4 (nuclear receptor coactivator 4) (namely ferritinophagy) that increases intracellular iron levels to promote ferroptosis (Mancias et al. 2014; Hou et al. 2016). Currently, it is unclear how cells regulate the balance from cell survival autophagy towards cell injury (via promoting other forms of cell death) that will eventually lead to liver injury after APAP exposure. Moreover, it is also unclear whether and how autophagy may also promote liver repair/regeneration, which is more relevant for treating APAP-induced liver injury. Future work is needed to further elucidate the complicated role of autophagy in APAP-induced liver injury.
Pyroptosis
Pyroptosis is an inflammatory form of programmed necrosis (Bergsbaken et al. 2009; Jorgensen et al. 2017) (Fig. 2). The key mechanism of this mode of cell death is the activation of pyroptotic caspases including caspase-1, -4 and -5 in humans and caspase-11 in mice (Yuan et al. 2016). Caspase-1 is activated by stimulation of inflammasomes such as Nalp3 or Aim2 (Bergsbaken et al. 2009) and the other caspases can be activated through direct binding of bacterial endotoxin (Shi et al. 2014). This links the activation of the pyroptotic caspases not only to pathogen associated molecular patterns (PAMPs) but also to damage-associated molecular patterns (DAMPs) released during a sterile inflammatory response including APAP-induced cell death. The pyroptotic caspases proteolytically cleave gasdermin D and multiple amino-terminal fragments insert into the plasma membrane forming a pore, which triggers the breakdown of ion gradients across the membrane, cell swelling and necrotic cell death (Jorgensen et al. 2017). Caspase-1 can cleave the pro-forms of IL-1β and IL-18 to the active cytokines, which also can be released through the gasdermin pore (Jorgensen et al. 2017). During APAP-induced necrosis, there is release of DAMPs from hepatocytes, which includes mtDNA, nuclear DNA fragments but also ATP (Woolbright and Jaeschke 2017). This causes both the transcriptional activation of cytokines through toll-like receptors, e.g., TLR9, and the activation of the Nlp3 inflammasome leading to increased expression of IL-1β mRNA and formation of pro-IL-1β and the active cytokine (Imaeda et al. 2009; Williams et al. 2010). Caspase-1-deficiency in mice and treatment with a pan-caspase inhibitor prevents the formation of the active IL-1β but not the formation of the mRNA (Imaeda et al. 2009; Williams et al. 2010) indicating the activation of the Nlp3 inflammasome and caspase-1 during APAP hepatotoxicity. Although it was reported that caspase-1-deficient mice have reduced APAP-induced liver injury (Imaeda et al. 2009), this observation could not be reproduced (Williams et al. 2011) and many studies have shown the lack of a protective effect of various caspase inhibitors in this model (Lawson et al. 1999; Gujral et al. 2002; Jaeschke et al. 2006; Williams et al. 2010). In addition, the overall formation of IL-1β after APAP overdose is very limited both in mice (Williams et al. 2010; Zhang et al. 2018) and in humans (Woolbright and Jaeschke 2017). Together these findings do not support that the cell death after APAP is caused by pyroptosis.

Pyroptosis. Pyroptosis is cell death induced by inflammatory mediators such as bacteria, viruses or toxic microbial material mediated by caspase 1, 4, 5 and 11 in humans. Identification of these inflammatory materials by cells results in activation of inflammation sensing complexes such as the NALP3 inflammasome, which results in activation of caspase 1. This is complemented by direct activation of caspases 4, 5 and 11 by bacterial lipopolysaccharides (LPS), where caspase 11 can then activate the NALP3 inflammasome. The activated caspases then cleave gasdermin D, and the insertion of the resulting protein fragments into the plasma membrane form channels which cause cell swelling and necrosis
Ferroptosis
Ferroptosis is considered a new form of cell death that can occur in various cells including hepatocytes. Ferroptosis requires iron mobilization and the inactivation of glutathione peroxidase 4 (GPx4), which specifically reduces phospholipid hydroperoxides and prevents lipid peroxidation (LPO) (Lei et al. 2019). Hence, the fundamental characteristics of ferroptosis involves iron-dependent LPO, which can be inhibited by the key ferroptosis regulator glutathione peroxidase 4 (GPx4), iron chelation, antioxidants and inhibitors of ferroptosis including ferrostatins and liproxstatins (Doll and Conrad 2017; Skouta et al. 2014) (Fig. 3). A recent study suggests that ferroptosis may be involved in APAP-induced cell death in primary mouse hepatocytes based on the partial protection by ferrostatin-1 (Lőrincz et al. 2015). However, ferrostatin-1 is only soluble in DMSO and the authors did not investigate if the compound or the solvent caused some delay in metabolic activation, which may be the cause of the modest protection observed in these cells.

Ferroptosis. Ferroptosis is a mode of cell death mediated by cellular free iron and lipid peroxidation in the context of decreased glutathione peroxidase activity. Intracellular reactive oxygen species such as hydrogen peroxide react with cellular free iron through the Fenton reaction to generate the reactive hydroxyl radical, which attacks lipid membranes to induce lipid peroxidation and membrane instability, which can ultimately result in leakage of cellular material and cell death. This process can be inhibited by compounds such as ferrostatin or chelation of free cellular iron to prevent lipid peroxidation
Interestingly, the role of iron and LPO has been investigated as mechanism of cell death of APAP-induced liver injury in the 1980s, i.e., long before the term ferroptosis was introduced (Jaeschke et al. 2003). Wendel and coworkers provided solid evidence for severe LPO in APAP-treated mice, an effect that could be attenuated by iron chelation and vitamin E pretreatment (Wendel and Feuerstein 1981; Jaeschke et al. 2003). However, what is widely ignored is the fact that these animals were fed a diet deficient in vitamin E and selenium and high in soybean oil resulting in elevated hepatic levels of polyunsaturated fatty acids like arachidonic acid (20:4) and docosahexaenoic acid (22:6), which made these animals extremely susceptible to LPO by GSH-depleting drugs and chemicals such as APAP and allyl alcohol (Jaeschke et al. 1987; Wendel and Feuerstein 1981). In contrast, when mice are fed a normal rodent diet, LPO after APAP is minimal (< twofold above baseline) and quantitatively insufficient to cause cell death (Knight et al. 2003). In addition, lipid-soluble antioxidants like vitamin E do not protect (Knight et al. 2003). Studies comparing cell death and LPO parameters such as hydroxy-eicosatetraenoic acids (HETES), the reaction product of GPx4, and F2-isoprostanes clearly demonstrated that such LPO parameters increase by 20–50-fold above baseline when LPO is the cause of cell death (Mathews et al. 1994), which is consistent with APAP hepatotoxicity in vitamin E-deficient mice (Wendel and Feuerstein 1981; Wendel et al. 1982). Thus, under normal circumstances when animals are not made particularly sensitive to oxidant stress, APAP-induced LPO parameters are at most two-to-threefold above baseline and, therefore, LPO is quantitatively insufficient to cause cell death.
The chelation of intracellular iron by deferoxamine attenuated APAP-induced liver injury (Sakaida et al. 1995; Schnellmann et al. 1999). However, APAP overdose triggers lysosomal instability (Woolbright et al. 2012), which leads to release of lysosomal iron that is being taken up into mitochondria through the electrogenic Ca2+, Fe2+ uniporter where it promotes cell death through the opening of the membrane permeability transition pore (Kon et al. 2010; Hu et al. 2016). The lysosomal-targeted iron-chelator starch-deferoxamine attenuated APAP-induced cell death (Hu et al. 2016). Thus, lysosomal iron promotes APAP hepatotoxicity independent of LPO. Hence, the APAP-induced cell death cannot be characterized as ferroptosis.
Conclusions and future perspectives
Based on the entire experimental evidence, cell death induced by an APAP overdose in mice or patients has all the histological features of oncotic necrosis, which is caused by complex intracellular signaling events justifying the term programmed necrosis. As outlined, each of the other necrotic cell death modes including necroptosis, ferroptosis, pyroptosis or autophagic cell death have characteristics that are not fully met by APAP. Apoptosis is morphologically different from necrosis and has certain biochemical pathways, e.g., caspase activation, that are not present during APAP-induced cell death. When all histological features and intracellular signaling pathways are considered for these modes of cell death and compared to the same parameters in APAP-induced cell death, it is obvious that this is necrosis and does not match the established characteristics of apoptosis or the other, more specialized, necrosis pathways. However, there is clearly overlap in some of the signaling pathways between these modes of cell death. Thus, to avoid wrong conclusions based on one or two ambiguous parameters, it is critical to consider all the signaling events and histological features. In addition, focusing on elucidating critical mechanisms rather than attempts to re-define modes of cell death have a more realistic chance of discovering new therapeutic targets for the clinical problem of APAP-induced liver injury and acute liver failure.
Abbreviations
- AIF:
-
Apoptosis-inducing factor
- AIM2:
-
Absent in melanoma 2
- APAF-1:
-
Apoptotic protease-activating factor-1
- APAP:
-
Acetaminophen
- ASK1:
-
Apoptosis signal-regulating kinase 1
- CAD:
-
Caspase-activted DNase
- DAMP:
-
Damage-associated molecular pattern
- DMSO:
-
Dimethylsulfoxide
- GFP:
-
Green fluorescence protein
- GPx:
-
Glutathione peroxidase
- GSK-3b:
-
Glycogen synthase kinase 3 beta
- HETEs:
-
Hydroxy-eicosatetraenoic acids
- IAP:
-
Inhibitor of apoptosis
- ICAD:
-
Inhibitor of caspase-activated DNase
- IL-1:
-
Interleukin-1
- JNK:
-
c-Jun N-terminal kinase
- LPO:
-
Lipid peroxidation
- MAPK:
-
Mitogen-activating protein kinase
- MKK4:
-
Mitogen-activated protein kinase kinase 4
- MLK3:
-
Mixed-lineage kinase 3
- MLKL:
-
Mixed lineage kinase domain-like protein
- MPTP:
-
Mitochondrial membrane permeability transition pore
- mtDNA:
-
Mitochondrial DNA
- Nalp3:
-
NACHT, LRR and PYD domain-containing protein
- NAPQI:
-
N-acetyl-p-benzoquinone imine
- NCOA4:
-
Nuclear receptor coactivator 4
- PAMP:
-
Pathogen-associated molecular pattern
- PINK1:
-
PTEN-induced kinase 1
- PTEN:
-
Phosphatase and tensin homolog
- RIPK:
-
Receptor-interacting protein kinase
- Smac/Diablo:
-
Second mitochondria-derived activator of caspase/direct inhibitor of apoptosis-binding protein with low pI
- SQSTM1/p62:
-
Sequestosome 1/p62
- TLR:
-
Toll-like receptor
- TUNEL:
-
Terminal deoxynucleotidyl transferase dUTP nick end labeling
- TFEB:
-
Transcription factor EB
References
Adams ML, Pierce RH, Vail ME, White CC, Tonge RP, Kavanagh TJ, Fausto N, Nelson SD, Bruschi SA (2001) Enhanced acetaminophen hepatotoxicity in transgenic mice overexpressing BCL-2. Mol Pharmacol 60:907–915
Bajt ML, Cover C, Lemasters JJ, Jaeschke H (2006) Nuclear translocation of endonuclease G and apoptosis-inducing factor during acetaminophen-induced liver cell injury. Toxicol Sci 94:217–225
Bajt ML, Farhood A, Lemasters JJ, Jaeschke H (2008) Mitochondrial bax translocation accelerates DNA fragmentation and cell necrosis in a murine model of acetaminophen hepatotoxicity. J Pharmacol Exp Ther 324:8–14
Bajt ML, Ramachandran A, Yan HM, Lebofsky M, Farhood A, Lemasters JJ, Jaeschke H (2011) Apoptosis-inducing factor modulates mitochondrial oxidant stress in acetaminophen hepatotoxicity. Toxicol Sci 122:598–605
Bergsbaken T, Fink SL, Cookson BT (2009) Pyroptosis: host cell death and inflammation. Nat Rev Microbiol 7:99–109
Boess F, Bopst M, Althaus R, Polsky S, Cohen SD, Eugster HP, Boelsterli UA (1998) Acetaminophen hepatotoxicity in tumor necrosis factor/lymphotoxin-alpha gene knockout mice. Hepatology 27:1021–1029
Chao X, Wang H, Jaeschke H, Ding WX (2018) Role and mechanisms of autophagy in acetaminophen-induced liver injury. Liver Int 38:1363–1374
Cover C, Mansouri A, Knight TR, Bajt ML, Lemasters JJ, Pessayre D, Jaeschke H (2005) Peroxynitrite-induced mitochondrial and endonuclease-mediated nuclear DNA damage in acetaminophen hepatotoxicity. J Pharmacol Exp Ther 315:879–887
Dara L, Johnson H, Suda J, Win S, Gaarde W, Han D, Kaplowitz N (2015) Receptor interacting protein kinase 1 mediates murine acetaminophen toxicity independent of the necrosome and not through necroptosis. Hepatology 62:1847–1857
Deas E, Plun-Favreau H, Gandhi S, Desmond H, Kjaer S, Loh SH, Renton AE, Harvey RJ, Whitworth AJ, Martins LM, Abramov AY, Wood NW (2011) PINK1 cleavage at position A103 by the mitochondrial protease PARL. Hum Mol Genet 20:867–879
Degterev A, Maki JL, Yuan J (2013) Activity and specificity of necrostatin-1, small-molecule inhibitor of RIP1 kinase. Cell Death Differ 20:366
Deutsch M, Graffeo CS, Rokosh R, Pansari M, Ochi A, Levie EM, Van Heerden E, Tippens DM, Greco S, Barilla R, Tomkötter L, Zambirinis CP, Avanzi N, Gulati R, Pachter HL, Torres-Hernandez A, Eisenthal A, Daley D, Miller G (2015) Divergent effects of RIP1 or RIP3 blockade in murine models of acute liver injury. Cell Death Dis 6:e1759
Doll S, Conrad M (2017) Iron and ferroptosis: a still ill-defined liaison. IUBMB Life 69:423–434
Du K, Xie Y, McGill MR, Jaeschke H (2015) Pathophysiological significance of c-Jun N-terminal kinase in acetaminophen hepatotoxicity. Expert Opin Drug Metab Toxicol 11:1769–1779
Du K, Ramachandran A, Jaeschke H (2016) Oxidative stress during acetaminophen hepatotoxicity: sources, pathophysiological role and therapeutic potential. Redox Biol 10:148–156
Du K, Ramachandran A, McGill MR, Mansouri A, Asselah T, Farhood A, Woolbright BL, Ding WX, Jaeschke H (2017) Induction of mitochondrial biogenesis protects against acetaminophen hepatotoxicity. Food Chem Toxicol 108:339–350
Du K, Ramachandran A, Weemhoff JL, Woolbright BL, Jaeschke AH, Chao X, Ding WX, Jaeschke H (2019) Mito-tempo protects against acute liver injury but induces limited secondary apoptosis during the late phase of acetaminophen hepatotoxicity. Arch Toxicol 93:163–178
El-Hassan H, Anwar K, Macanas-Pirard P, Crabtree M, Chow SC, Johnson VL, Lee PC, Hinton RH, Price SC, Kass GE (2003) Involvement of mitochondria in acetaminophen-induced apoptosis and hepatic injury: roles of cytochrome c, Bax, Bid, and caspases. Toxicol Appl Pharmacol 191:118–129
Grasl-Kraupp B, Ruttkay-Nedecky B, Koudelka H, Bukowska K, Bursch W, Schulte-Hermann R (1995) In situ detection of fragmented DNA (TUNEL assay) fails to discriminate among apoptosis, necrosis, and autolytic cell death: a cautionary note. Hepatology 21:1465–1468
Gujral JS, Knight TR, Farhood A, Bajt ML, Jaeschke H (2002) Mode of cell death after acetaminophen overdose in mice: apoptosis or oncotic necrosis? Toxicol Sci 67:322–328
Han D, Dara L, Win S, Than TA, Yuan L, Abbasi SQ, Liu ZX, Kaplowitz N (2013) Regulation of drug-induced liver injury by signal transduction pathways: critical role of mitochondria. Trends Pharmacol Sci 34:243–253
Hou W, Xie Y, Song X, Sun X, Lotze MT, Zeh HJ 3rd, Kang R, Tang D (2016) Autophagy promotes ferroptosis by degradation of ferritin. Autophagy 12:1425–1428
Hu B, Colletti LM (2010) CXC receptor-2 knockout genotype increases X-linked inhibitor of apoptosis protein and protects mice from acetaminophen hepatotoxicity. Hepatology 52:691–702
Hu J, Kholmukhamedov A, Lindsey CC, Beeson CC, Jaeschke H, Lemasters JJ (2016) Translocation of iron from lysosomes to mitochondria during acetaminophen-induced hepatocellular injury: protection by starch-desferal and minocycline. Free Radic Biol Med 97:418–426
Imaeda AB, Watanabe A, Sohail MA, Mahmood S, Mohamadnejad M, Sutterwala FS, Flavell RA, Mehal WZ (2009) Acetaminophen-induced hepatotoxicity in mice is dependent on Tlr9 and the Nalp3 inflammasome. J Clin Investig 119:305–314
Jaeschke H (2015) Acetaminophen: dose-dependent drug hepatotoxicity and acute liver failure in patients. Dig Dis 33:464–471
Jaeschke H, Lemasters JJ (2003) Apoptosis versus oncotic necrosis in hepatic ischemia/reperfusion injury. Gastroenterology 125:1246–1257
Jaeschke H, Kleinwaechter C, Wendel A (1987) The role of acrolein in allyl alcohol-induced lipid peroxidation and liver cell damage in mice. Biochem Pharmacol 36:51–57
Jaeschke H, Knight TR, Bajt ML (2003) The role of oxidant stress and reactive nitrogen species in acetaminophen hepatotoxicity. Toxicol Lett 144:279–288
Jaeschke H, Cover C, Bajt ML (2006) Role of caspases in acetaminophen-induced liver injury. Life Sci 78:1670–1676
Jaeschke H, Williams CD, Farhood A (2011) No evidence for caspase-dependent apoptosis in acetaminophen hepatotoxicity. Hepatology 53:718–719
Jaeschke H, Duan L, Akakpo JY, Farhood A, Ramachandran A (2018) The role of apoptosis in acetaminophen hepatotoxicity. Food Chem Toxicol 118:709–718
Jahr S, Hentze H, Englisch S, Hardt D, Fackelmayer FO, Hesch RD, Knippers R (2001) DNA fragments in the blood plasma of cancer patients: quantitations and evidence for their origin from apoptotic and necrotic cells. Cancer Res 61:1659–1665
Jin SM, Lazarou M, Wang C, Kane LA, Narendra DP, Youle RJ (2010) Mitochondrial membrane potential regulates PINK1 import and proteolytic destabilization by PARL. J Cell Biology 191:933–942
Jorgensen I, Rayamajhi M, Miao EA (2017) Programmed cell death as a defense against infection. Nat Rev Immunol 17:151–164
Jouan-Lanhouet S, Riquet F, Duprez L, Vanden Berghe T, Takahashi N, Vandenabeele P (2014) Necroptosis, in vivo detection in experimental disease models. Semin Cell Dev Biol 35:2–13
Klionsky DJ, Emr SD (2000) Autophagy as a regulated pathway of cellular degradation. Science 290:1717–1721
Knight TR, Jaeschke H (2002) Acetaminophen-induced inhibition of Fas receptor-mediated liver cell apoptosis: mitochondrial dysfunction versus glutathione depletion. Toxicol Appl Pharmacol 181:133–141
Knight TR, Fariss MW, Farhood A, Jaeschke H (2003) Role of lipid peroxidation as a mechanism of liver injury after acetaminophen overdose in mice. Toxicol Sci 76:229–236
Kon K, Kim JS, Uchiyama A, Jaeschke H, Lemasters JJ (2010) Lysosomal iron mobilization and induction of the mitochondrial permeability transition in acetaminophen-induced toxicity to mouse hepatocytes. Toxicol Sci 117:101–108
Koyano F, Okatsu K, Kosako H, Tamura Y, Go E, Kimura M, Kimura Y, Tsuchiya H, Yoshihara H, Hirokawa T, Endo T, Fon EA, Trempe JF, Saeki Y, Tanaka K, Matsuda N (2014) Ubiquitin is phosphorylated by PINK1 to activate parkin. Nature 510:162–166
Larson AM, Polson J, Fontana RJ, Davern TJ, Lalani E, Hynan LS, Reisch JS, Schiødt FV, Ostapowicz G, Shakil AO, Lee WM, Acute Liver Failure Study Group (2005) Acetaminophen-induced acute liver failure: results of a United States multicenter, prospective study. Hepatology 42:1364–1372
Lawson JA, Fisher MA, Simmons CA, Farhood A, Jaeschke H (1999) Inhibition of Fas receptor (CD95)-induced hepatic caspase activation and apoptosis by acetaminophen in mice. Toxicol Appl Pharmacol 156:179–186
Lee WM (2013) Drug-induced acute liver failure. Clin Liver Dis 17:575–586
Lei P, Bai T, Sun Y (2019) Mechanisms of ferroptosis and relations with regulated cell death: a review. Front Physiol 10:139
Li H, Wang P, Sun Q, Ding WX, Yin XM, Sobol RW, Stolz DB, Yu J, Zhang L (2011) Following cytochrome c release, autophagy is inhibited during chemotherapy-induced apoptosis by caspase 8-mediated cleavage of Beclin 1. Cancer Res 71:3625–3634
Li JX, Feng JM, Wang Y, Li XH, Chen XX, Su Y, Shen YY, Chen Y, Xiong B, Yang CH, Ding J, Miao ZH (2014) The B-Raf(V600E) inhibitor dabrafenib selectively inhibits RIP3 and alleviates acetaminophen-induced liver injury. Cell Death Dis 5:e1278
Lőrincz T, Jemnitz K, Kardon T, Mandl J, Szarka A (2015) Ferroptosis is involved in acetaminophen induced cell death. Pathol Oncol Res 21:1115–1121
Malhi H, Gores GJ, Lemasters JJ (2006) Apoptosis and necrosis in the liver: a tale of two deaths? Hepatology 43(2 Suppl 1):S31–S44
Mancias JD, Wang X, Gygi SP, Harper JW, Kimmelman AC (2014) Quantitative proteomics identifies NCOA4 as the cargo receptor mediating ferritinophagy. Nature 509:105–109
Mathews WR, Guido DM, Fisher MA, Jaeschke H (1994) Lipid peroxidation as molecular mechanism of liver cell injury during reperfusion after ischemia. Free Radic Biol Med 16:763–770
Matsuda N, Sato S, Shiba K, Okatsu K, Saisho K, Gautier CA, Sou YS, Saiki S, Kawajiri S, Sato F, Kimura M, Komatsu M, Hattori N, Tanaka K (2010) PINK1 stabilized by mitochondrial depolarization recruits Parkin to damaged mitochondria and activates latent Parkin for mitophagy. J Cell Biol 189:211–221
McGill MR, Jaeschke H (2013) Metabolism and disposition of acetaminophen: recent advances in relation to hepatotoxicity and diagnosis. Pharm Res 30:2174–2187
McGill MR, Jaeschke H (2019) Animal models of drug-induced liver injury. Biochim Biophys Acta Mol Basis Dis 1865:1031–1039
McGill MR, Yan HM, Ramachandran A, Murray GJ, Rollins DE, Jaeschke H (2011) HepaRG cells: a human model to study mechanisms of acetaminophen hepatotoxicity. Hepatology 53:974–982
McGill MR, Sharpe MR, Williams CD, Taha M, Curry SC, Jaeschke H (2012) The mechanism underlying acetaminophen-induced hepatotoxicity in humans and mice involves mitochondrial damage and nuclear DNA fragmentation. J Clin Investig 122:1574–1583
Meissner C, Lorenz H, Weihofen A, Selkoe DJ, Lemberg MK (2011) The mitochondrial intramembrane protease PARL cleaves human Pink1 to regulate Pink1 trafficking. J Neurochem 117:856–867
Nakagawa H, Maeda S, Hikiba Y, Ohmae T, Shibata W, Yanai A, Sakamoto K, Ogura K, Noguchi T, Karin M, Ichijo H, Omata M (2008) Deletion of apoptosis signal-regulating kinase 1 attenuates acetaminophen-induced liver injury by inhibiting c-Jun N-terminal kinase activation. Gastroenterology 135:1311–1321
Nakatogawa H, Suzuki K, Kamada Y, Ohsumi Y (2009) Dynamics and diversity in autophagy mechanisms: lessons from yeast. Nature reviews. Mol Cell Biol 10:458–467
Narendra DP, Jin SM, Tanaka A, Suen DF, Gautier CA, Shen J, Cookson MR, Youle RJ (2010) PINK1 is selectively stabilized on impaired mitochondria to activate Parkin. PLoS Biol 8:e1000298
Ni HM, Bockus A, Boggess N, Jaeschke H, Ding WX (2012) Activation of autophagy protects against acetaminophen-induced hepatotoxicity. Hepatology 55:222–232
Ni HM, Williams JA, Jaeschke H, Ding WX (2013) Zonated induction of autophagy and mitochondrial spheroids limits acetaminophen-induced necrosis in the liver. Redox Biol 1:427–432
Ni HM, Williams JA, Ding WX (2015) Mitochondrial dynamics and mitochondrial quality control. Redox Biol 4:6–13
Ni HM, McGill MR, Chao X, Du K, Williams JA, Xie Y, Jaeschke H, Ding WX (2016) Removal of acetaminophen protein adducts by autophagy protects against acetaminophen-induced liver injury in mice. J Hepatol 65:354–362
Park Y, Smith RD, Combs AB, Kehrer JP (1988) Prevention of acetaminophen-induced hepatotoxicity by dimethyl sulfoxide. Toxicology 52:165–175
Parzych KR, Klionsky DJ (2014) An overview of autophagy: morphology, mechanism, and regulation. Antioxid Redox Signal 20:460–473
Pickles S, Vigie P, Youle RJ (2018) Mitophagy and quality control mechanisms in mitochondrial maintenance. Curr Biol 28:R170–R185
Ramachandran A, Jaeschke H (2019) Acetaminophen hepatotoxicity. Semin Liver Dis 39:221–234
Ramachandran A, McGill MR, Xie Y, Ni HM, Ding WX, Jaeschke H (2013) Receptor interacting protein kinase 3 is a critical early mediator of acetaminophen-induced hepatocyte necrosis in mice. Hepatology 58:2099–2108
Ray SD, Sorge CL, Raucy JL, Corcoran GB (1990) Early loss of large genomic DNA in vivo with accumulation of Ca2+ in the nucleus during acetaminophen-induced liver injury. Toxicol Appl Pharmacol 106:346–351
Ray SD, Kamendulis LM, Gurule MW, Yorkin RD, Corcoran GB (1993) Ca2+ antagonists inhibit DNA fragmentation and toxic cell death induced by acetaminophen. FASEB J 7:453–463
Ray SD, Mumaw VR, Raje RR, Fariss MW (1996) Protection of acetaminophen-induced hepatocellular apoptosis and necrosis by cholesteryl hemisuccinate pretreatment. J Pharmacol Exp Ther 279:1470–1483
Rojansky R, Cha MY, Chan DC (2016) Elimination of paternal mitochondria in mouse embryos occurs through autophagic degradation dependent on PARKIN and MUL1. Elife 5:e17896
Sakaida I, Kayano K, Wasaki S, Nagatomi A, Matsumura Y, Okita K (1995) Protection against acetaminophen-induced liver injury in vivo by an iron chelator, deferoxamine. Scand J Gastroenterol 30:61–67
Schnellmann JG, Pumford NR, Kusewitt DF, Bucci TJ, Hinson JA (1999) Deferoxamine delays the development of the hepatotoxicity of acetaminophen in mice. Toxicol Lett 106:79–88
Sharma M, Gadang V, Jaeschke A (2012) Critical role for mixed-lineage kinase 3 in acetaminophen-induced hepatotoxicity. Mol Pharmacol 82:1001–1007
Shen W, Kamendulis LM, Ray SD, Corcoran GB (1991) Acetaminophen-induced cytotoxicity in cultured mouse hepatocytes: correlation of nuclear Ca2+ accumulation and early DNA fragmentation with cell death. Toxicol Appl Pharmacol 111:242–254
Shen W, Kamendulis LM, Ray SD, Corcoran GB (1992) Acetaminophen-induced cytotoxicity in cultured mouse hepatocytes: effects of Ca(2+)-endonuclease, DNA repair, and glutathione depletion inhibitors on DNA fragmentation and cell death. Toxicol Appl Pharmacol 112:32–40
Shi J, Zhao Y, Wang Y, Gao W, Ding J, Li P, Hu L, Shao F (2014) Inflammatory caspases are innate immune receptors for intracellular LPS. Nature 514:187–192
Shinohara M, Ybanez MD, Win S, Than TA, Jain S, Gaarde WA, Han D, Kaplowitz N (2010) Silencing glycogen synthase kinase-3beta inhibits acetaminophen hepatotoxicity and attenuates JNK activation and loss of glutamate cysteine ligase and myeloid cell leukemia sequence 1. J Biol Chem 285:8244–8255
Skouta R, Dixon SJ, Wang J, Dunn DE, Orman M, Shimada K, Rosenberg PA, Lo DC, Weinberg JM, Linkermann A, Stockwell BR (2014) Ferrostatins inhibit oxidative lipid damage and cell death in diverse disease models. J Am Chem Soc 136:4551–4556
Tang D, Kang R, Berghe TV, Vandenabeele P, Kroemer G (2019) The molecular machinery of regulated cell death. Cell Res 29:347–364
Vercammen D, Beyaert R, Denecker G, Goossens V, Van Loo G, Declercq W, Grooten J, Fiers W, Vandenabeele P (1998) Inhibition of caspases increases the sensitivity of L929 cells to necrosis mediated by tumor necrosis factor. J Exp Med 187:1477–1485
Vives-Bauza C, Zhou C, Huang Y, Cui M, de Vries RL, Kim J, May J, Tocilescu MA, Liu W, Ko HS, Magrané J, Moore DJ, Dawson VL, Grailhe R, Dawson TM, Li C, Tieu K, Przedborski S (2010) PINK1-dependent recruitment of Parkin to mitochondria in mitophagy. Proc Natl Acad Sci U S A 107:378–383
Wang LM, Cho YL, Tang YC, Wang JG, Park JE, Wu YJ, Wang CX, Tong Y, Chawla R, Zhang JB, Shi Y, Deng S, Lu G, Wu YH, Tan HWS, Pawijit P, Lim GGY, Chan HY, Zhang JZ, Fang L, Yu H, Liou YC, Karthik M, Bay BH, Lim KL, Sze SK, Yap CT, Shen HM (2018) PTEN-L is a novel protein phosphatase for ubiquitin dephosphorylation to inhibit PINK1-Parkin-mediated mitophagy. Cell Res 28:787–802
Wang H, Ni HM, Chao X, Ma X, Rodriguez YA, Chavan H, Wang S, Krishnamurthy P, Dobrowsky R, Xu DX, Jaeschke H, Ding WX (2019) Double deletion of PINK1 and Parkin impairs hepatic mitophagy and exacerbates acetaminophen-induced liver injury in mice. Redox Biol 22:101148
Wendel A, Feuerstein S (1981) Drug-induced lipid peroxidation in mice–I. Modulation by monooxygenase activity, glutathione and selenium status. Biochem Pharmacol 30:2513–2520
Wendel A, Jaeschke H, Gloger M (1982) Drug-induced lipid peroxidation in mice–II. Protection against paracetamol-induced liver necrosis by intravenous liposomally entrapped glutathione. Biochem Pharmacol 31:3601–3605
Williams JA, Ding WX (2018) Mechanisms, pathophysiological roles and methods for analyzing mitophagy—recent insights. Biol Chem 399:147–178
Williams CD, Farhood A, Jaeschke H (2010) Role of caspase-1 and interleukin-1beta in acetaminophen-induced hepatic inflammation and liver injury. Toxicol Appl Pharmacol 247:169–178
Williams CD, Antoine DJ, Shaw PJ, Benson C, Farhood A, Williams DP, Kanneganti TD, Park BK, Jaeschke H (2011) Role of the Nalp3 inflammasome in acetaminophen-induced sterile inflammation and liver injury. Toxicol Appl Pharmacol 252:289–297
Williams JA, Ni HM, Haynes A, Manley S, Li Y, Jaeschke H, Ding WX (2015) Chronic deletion and acute knockdown of Parkin have differential responses to acetaminophen-induced mitophagy and liver injury in mice. J Biol Chem 290:10934–10946
Win S, Than TA, Han D, Petrovic LM, Kaplowitz N (2011) c-Jun N-terminal kinase (JNK)-dependent acute liver injury from acetaminophen or tumor necrosis factor (TNF) requires mitochondrial Sab protein expression in mice. J Biol Chem 286:35071–35078
Woolbright BL, Jaeschke H (2017) Role of the inflammasome in acetaminophen-induced liver injury and acute liver failure. J Hepatol 66:836–848
Woolbright BL, Ramachandran A, McGill MR, Yan HM, Bajt ML, Sharpe MR, Lemasters JJ, Jaeschke H (2012) Lysosomal instability and cathepsin B release during acetaminophen hepatotoxicity. Basic Clin Pharmacol Toxicol 111:417–425
Xie Y, McGill MR, Dorko K, Kumer SC, Schmitt TM, Forster J, Jaeschke H (2014) Mechanisms of acetaminophen-induced cell death in primary human hepatocytes. Toxicol Appl Pharmacol 279:266–274
Xie Y, Ramachandran A, Breckenridge DG, Liles JT, Lebofsky M, Farhood A, Jaeschke H (2015) Inhibitor of apoptosis signal-regulating kinase 1 protects against acetaminophen-induced liver injury. Toxicol Appl Pharmacol 286:1–9
Yamano K, Youle RJ (2013) PINK1 is degraded through the N-end rule pathway. Autophagy 9:1758–1769
Yang X, Chao X, Wang ZT, Ding WX (2016) The end of RIPK1-RIPK3-MLKL-mediated necroptosis in acetaminophen-induced hepatotoxicity? Hepatology 64:311–312
Yonekawa T, Gamez G, Kim J, Tan AC, Thorburn J, Gump J, Thorburn A, Morgan MJ (2015) RIP1 negatively regulates basal autophagic flux through TFEB to control sensitivity to apoptosis. EMBO Rep 16:700–708
Yuan J, Najafov A, Py BF (2016) Roles of caspases in necrotic cell death. Cell 167:1693–1704
Zhang YF, He W, Zhang C, Liu XJ, Lu Y, Wang H, Zhang ZH, Chen X, Xu DX (2014) Role of receptor interacting protein (RIP)1 on apoptosis-inducing factor-mediated necroptosis during acetaminophen-evoked acute liver failure in mice. Toxicol Lett 225:445–453
Zhang J, Min RWM, Le K, Zhou S, Aghajan M, Than TA, Win S, Kaplowitz N (2017) The role of MAP2 kinases and p38 kinase in acute murine liver injury models. Cell Death Dis 8:e2903
Zhang C, Feng J, Du J, Zhuo Z, Yang S, Zhang W, Wang W, Zhang S, Iwakura Y, Meng G, Fu YX, Hou B, Tang H (2018) Macrophage-derived IL-1α promotes sterile inflammation in a mouse model of acetaminophen hepatotoxicity. Cell Mol Immunol 15:973–982
Acknowledgements
This work was supported in part by National Institutes of Health Grants R01 DK102142, R01 DK070195, P20 GM103549 and P30 GM118247.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
H Jaeschke received grant support from McNeil Consumer Health, Inc. All the other authors declare no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Jaeschke, H., Ramachandran, A., Chao, X. et al. Emerging and established modes of cell death during acetaminophen-induced liver injury. Arch Toxicol 93, 3491–3502 (2019). https://doi.org/10.1007/s00204-019-02597-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00204-019-02597-1