
Abstract
Cellular stress elicited by the toxic metal Cd2+ does not coerce the cell into committing to die from the onset. Rather, detoxification and adaptive processes are triggered concurrently, allowing survival until normal function is restored. With high Cd2+, death pathways predominate. However, if sublethal stress levels affect cells for prolonged periods, as in chronic low Cd2+ exposure, adaptive and survival mechanisms may deregulate, such that tumorigenesis ensues. Hence, death and malignancy are the two ends of a continuum of cellular responses to Cd2+, determined by magnitude and duration of Cd2+ stress. Signaling cascades are the key factors affecting cellular reactions to Cd2+. This review critically surveys recent literature to outline major features of death and survival signaling pathways as well as their activation, interactions and cross talk in cells exposed to Cd2+. Under physiological conditions, receptor activation generates 2nd messengers, which are short-lived and act specifically on effectors through their spatial and temporal dynamics to transiently alter effector activity. Cd2+ recruits physiological 2nd messenger systems, in particular Ca2+ and reactive oxygen species (ROS), which control key Ca2+- and redox-sensitive molecular switches dictating cell function and fate. Severe ROS/Ca2+ signals activate cell death effectors (ceramides, ASK1-JNK/p38, calpains, caspases) and/or cause irreversible damage to vital organelles, such as mitochondria and endoplasmic reticulum (ER), whereas low localized ROS/Ca2+ levels act as 2nd messengers promoting cellular adaptation and survival through signal transduction (ERK1/2, PI3K/Akt-PKB) and transcriptional regulators (Ref1-Nrf2, NF-κB, Wnt, AP-1, bestrophin-3). Other cellular proteins and processes targeted by ROS/Ca2+ (metallothioneins, Bcl-2 proteins, ubiquitin–proteasome system, ER stress-associated unfolded protein response, autophagy, cell cycle) can evoke death or survival. Hence, temporary or permanent disruptions of ROS/Ca2+ induced by Cd2+ play a crucial role in eliciting, modulating and linking downstream cell death and adaptive and survival signaling cascades.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Cadmium (Cd) is an environmental contaminant of increasing importance because human activities have increased its availability, and once in the environment, Cd cannot be degraded (Thévenod and Lee 2013; Jarup and Akesson 2009). The major toxic form of Cd is the cadmium ion (Cd2+), which has no known physiological role in humans. As a nonessential metal ion, Cd2+ competes with essential metal ions for entry into cells where it disrupts cellular functions and leads to disease. Therefore, exposure to Cd2+ is a serious environmental and health problem of global dimension. Chronic exposure to low Cd2+ concentrations has emerged as a previously underestimated significant health hazard for ~10 % of the general population that increases morbidity and mortality. It results mainly from dietary sources and cigarette smoking and causes nephrotoxicity, osteoporosis, neurotoxicity, genotoxicity and teratogenicity, or has endocrine and reproductive effects (Nawrot et al. 2010). With a biological half-life of ~20 years, Cd2+ accumulates in organs, particularly kidney (Jarup and Akesson 2009), where it causes fibrosis or failure (Ferraro et al. 2010), or—with Cd2+ being a Class 1 human carcinogen—cancer (Hartwig 2013a).
Organ failure and cancer are the two ends of a continuum of responses to toxicity. The fate of the organ in response to a toxic agent, such as Cd2+, is reflected at the cellular level by death or malignancy. Cellular stress elicited by toxic stimuli triggers detoxification as well as adaptive processes, which allow cell survival until normal function is restored. If the cellular response is inadequate (e.g., due to high concentrations of Cd2+), death pathways are initiated and the cell dies. However, if the level of stress is not sufficient to induce death but impacts on cells for a longer period of time (e.g., in chronic exposure to low Cd2+ concentrations), cells may lose control over adaptive mechanisms (e.g., by alterations of signaling pathways induced by mutations or failure to restore physiological balance) and malignant transformation can ensue. Cellular pathology may also occur due to the disruption of death signaling (“evasion of apoptosis”) (Hanahan and Weinberg 2011), which also facilitates malignant transformation, even if cells have been dysfunctional for a longer time period and are therefore committed to die.
In this review, we describe the main features of death and survival pathways as well as their activation and interactions with cells exposed to Cd2+. In this scenario, Cd2+-induced alterations of the cytosolic concentrations of the intracellular signaling molecules Ca2+ ([Ca2+]cyt) and reactive oxygen species (ROS) play a key role in eliciting, modulating and linking death and adaptive and survival signaling cascades. The magnitude and/or duration of the Ca2+ and ROS signals induced by Cd2+ are crucial determinants of cell fate. But also the extent to which Cd2+-induced functional and/or structural damage to key cellular organelles can be reversed by repair mechanisms dictates the “point of no return”. Although changes in the levels of other intracellular signaling molecules have also been described following Cd2+ exposure, data are not as consistent and/or no recent studies have been published that go beyond the knowledge of pertinent reviews on the topic (Thévenod 2009; Thévenod and Lee 2013; Waisberg et al. 2003).
The Janus face of Ca2+ and reactive oxygen species (ROS)
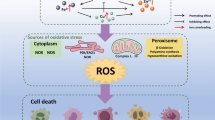
Some of the earliest consistent changes observed after acute Cd2+ exposure are increases in ROS and Ca2+ (see Thévenod 2009 for details), which are commonly involved as 2nd messengers in the physiological regulation of cell function; but at the same time, Cd2+ causes cell damage via ROS and Ca2+, which disrupt cell function and trigger cell death. At first sight, this appears paradoxical, but, in terms of signaling dynamics, Cd2+ induces a temporary or persistent imbalance of Ca2+ and ROS signals, which lead to reversible or permanent perturbations of the cell’s functions. The disequilibrium of the cell’s homeostasis requires adjustments of signaling for normalization or, if the magnitude of the message exceeds the ability of feedback mechanisms to correct the signal, results in damage and/or death of the cell. In other words, low transient levels of ROS or Ca2+ act as signaling molecules that promote cell survival. In contrast, a severe and persistent increase in ROS or Ca2+ can induce cell death. Hence, depending on the magnitude and quality of the increases in [Ca2+]cyt and/or ROS induced by Cd2+ (see Fig. 1), two adaptive responses are possible: (1) A “physiological adaptation” of the cell, which aims to remodel the regulatory imbalance and reestablish homeostasis (Berridge et al. 2003; Droge 2002; Brigelius-Flohe and Flohe 2011); or (2) a “pathological” response, which is triggered by cellular stress and damage and may have various outcomes depending on the strength of the signal and the answer of the cell to it: repair and survival, or disease, cancer and death (Orrenius et al. 2003; Berridge 2012; Trachootham et al. 2008; Roderick and Cook 2008; Fruehauf and Meyskens 2007).

Temporal dynamics and interactions of cell survival and cell death signaling pathways induced by Cd2+. a Note the transient nature of the Ca2+/ROS signal induced by weak Cd2+ stress (e.g., in chronic low Cd2+ exposure). Signal transduction cascades, transcription factors and intracellular organelles all contribute to the survival of the cell to Cd2+ stress and the organelles preserve their function and structure. These adaptive and survival responses of the cell are rapid events that take place as early as 5–15 min after exposure and develop within maximally 3–6 h. b In contrast, severe and acute Cd2+ stress elicits a persistent increase in Ca2+ and ROS. Initial death signaling is a rapid event; however, cellular processing toward a “point of no return” involves additional recruitment of decision and execution processes that result in manifestation of death end points usually after 6 to 24 h. These death signals are also associated with irreversible changes of function and structures of organelles, such as the endoplasmic reticulum (ER) and mitochondria. “?” indicates pathways whose role and/or interactions with other signals is still debated. Please note the different time scales in (a) and (b). The figure is not exhaustive; please refer to the text for further details
What is meant by “physiological adaptation”? Ca2+ and ROS as 2nd messengers are highly plastic homeostatic systems (Droge 2002; Berridge et al. 2003). They show characteristic spatial and temporal signaling dynamics in response to physiological stimuli to induce changes in cell function. If the spatiotemporal properties of the signal are altered by a loss or defect of a key component (e.g., due to the impact of Cd2+ on the cell), compensatory mechanisms are activated to restore normal signaling and function as part of a “remodeling” process, which activates cellular responses, in particular mechanisms of signaling and transcriptional feedback regulation (both positive and negative) that are responsible for maintaining the physiological signaling patterns (Berridge et al. 2003; Brigelius-Flohe and Flohe 2011). Remodeling then results in a novel equilibrium of cell function (“physiological adaptation”) by activating additional signaling as well as nuclear transcriptional processes to restore the physiological homeostasis of signal molecules.
A “pathological response” develops when ROS and Ca2+ induce cell stress, which may lead to the damage of vital cellular compartments (mitochondria, ER, nucleus, cytosol) (Rizzuto et al. 2003; Malhotra and Kaufman 2007; Sedelnikova et al. 2010; Huang et al. 2011; Livnat-Levanon and Glickman 2011; Cereghetti and Scorrano 2006) and may also be characterized by cross talk between both messengers during this process (Hidalgo and Donoso 2008; Feissner et al. 2009; Peng and Jou 2010). Activation of stress signaling (Trachootham et al. 2008; Runchel et al. 2011; Ray et al. 2012; Franklin et al. 2006) as well as reactivation of developmental pathways that are mediated by ROS and Ca2+ (Covarrubias et al. 2008; Slusarski and Pelegri 2007) trigger repair processes, which allow cells to survive and/or adapt. But this may occur at the expense of the development of cellular pathology and disease (Roderick and Cook 2008; Fruehauf and Meyskens 2007; Tsang et al. 2010). Stress and repair signaling pathways may also contribute to counteracting cell death systems (Runchel et al. 2011; Trachootham et al. 2008; Ray et al. 2012). However, if the magnitude of the cell stress inducing Ca2+ or ROS signals overwhelms (i.e., damages) cellular functions and repair mechanisms, cell death ensues due to the activation or predominance of suicidal/apoptosis or necrosis signaling (Sinha et al. 2013; Takeda et al. 2011; Zhivotovsky and Orrenius 2011).
The Ca2+ and ROS toolkits
Ca2+
How do cytosolic Ca2+ signals develop? The homeostasis of resting [Ca2+]cyt is the result of a constant and fine balance between influx into the cytosol via “nonspecific” leak pathways and efflux into stores and extracellular space via specific Ca2+ pumps (“pump-leak system”). Increases in [Ca2+]cyt are evoked in a regulated manner by chemical and/or electrical signals. [Ca2+]cyt increases as the result of controlled opening of channels (e.g., ORAI channel in the plasma membrane; IP3 receptors/channels (ITPR1–3) at the endoplasmic reticulum (ER); ryanodine receptors/channels in the SR (RYR1–3); and NAADP receptors/channels in endosomes/lysosomes (TPC1–3)). Ca2+ binds to various cellular effectors to regulate their function. When the activating signals are turned off, [Ca2+]cyt returns to resting levels, and this is due to the predominance of pumps, transporters and buffers. Hence, signals are quenched by cytosolic buffers, such as calbindin-D-28, and by the activation of Ca2+ pumps in the plasma membrane and ER, which drive Ca2+ into the extracellular space or in the matrix of the ER where Ca2+ is stored. In addition, mitochondria also buffer [Ca2+]cyt by sequestration in their matrix via a Ca2+ uniporter (for further details, see (Berridge et al. 2003; Clapham 2007).
ROS
There is mounting evidence that ROS play a role as physiological signaling molecules (D’Autreaux and Toledano 2007). Similar to other 2nd messengers, ROS can also be induced by physiological stimuli, such as cytokines or mechanical forces (Droge 2002). Nonenzymatic processes contribute to ROS formation, e.g., the oxidative processes at the mitochondrial respiratory chain and these reactions are thought to be not tightly regulated. Although the ROS that are generated may interact or interfere with signaling pathways, they may not constitute a physiologically important system (see, however (Sena and Chandel 2012)). However, ROS can also be generated by the more finely controlled activity of NADPH oxidases (NOX) (Bedard and Krause 2007) that are located at cellular membranes and constitute a physiological signaling pathway (Droge 2002). These enzyme systems have specific subcellular localization, e.g., plasma membranes, ER and nuclear envelope, thereby contributing to a compartmentalization of both ROS production and the signaling response. NOX1 and NOX2 may be activated by phosphatidyinositol 3-kinase (PI3K) and the small GTP-binding protein Rac1, and NOX5 by elevation of [Ca2+]cyt; in contrast, NOX4 is constitutively active and may be expressed in mitochondria (Bedard and Krause 2007; Brown and Griendling 2009).
Classical 2nd messengers, such as cAMP and Ca2+, bind noncovalently to specific effector proteins, and this reversible on–off reaction is merely a function of a reversible macromolecular conformation change, which primarily depends on the 2nd messenger concentration. In contrast, ROS operate in signaling at a submolecular level through chemical reactions with specific atoms of effector molecules, which lead to covalent modifications. For instance, ROS covalently modify redox-sensitive cysteines in a variety of proteins, including phosphatases, kinases, ion channels and transcription factors (Brigelius-Flohe and Flohe 2011; Ray et al. 2012; Winterbourn and Hampton 2008; Janssen-Heininger et al. 2008). These redox-regulated protein switches can only operate in signaling if they are coupled to reducing enzymatic systems, such as thioredoxins or peroxiredoxins (and possibly glutathione (GSH)) (Brigelius-Flohe and Flohe 2011; Ray et al. 2012). Furthermore, in order to operate as physiological 2nd messengers, ROS signals need also to be terminated. Hence, antioxidant enzymes—and possibly endogenous scavengers, such as tocopherol, ascorbic acid, carotenoids, uric acid and polyphenols—are likewise compartmentalized, with specific enzymes for mitochondria (Mn superoxide dismutase (MnSOD), cytosol (CuZnSOD, glutathione peroxidase (GPx)), peroxisomes (catalase) and the extracellular space (extracellular SOD). GSH, the most abundant endogenous antioxidant, is synthesized through two-enzyme reaction catalyzed by glutamate cysteine ligase and glutathione synthetase and also contributes to enzymatic termination of ROS signals (e.g., enzymes such as GPx and peroxiredoxin VI catalyze the reduction in H2O2 by GSH into H2O and glutathione disulfide (GSSG).
Interference of Cd2+ with Ca2+ and ROS toolkits
Cd2+ and Ca2+
Based on theoretical considerations, cellular homeostasis of eukaryotic organisms (and life) is maintained as long as free cellular Ca2+ concentration (~10−7 M) is about 106-fold higher than that of Cd2+ (10−13 M) (Williams 2002). Because the ionic radius of Ca2+ (114 pm) is quite similar to that of Cd2+ (109 pm), any increase in the Cd2+ content of the cell (which reduces the ratio between Ca2+ and Cd2+ concentrations) may result in displacement of Ca2+ from its physiological binding sites and affect their function. Furthermore, Cd2+ binding to functionally relevant SH groups of sarcoplasmic/endoplasmic reticulum calcium ATPase (SERCA) Ca2+ pumps of the ER (Zhang et al. 1990) and interference with the Ca2+-buffering role of mitochondria may contribute to disruption of Ca2+ homeostasis and result in transient or permanent elevations of [Ca2+]cyt. Biagioli et al. showed that Cd2+ treatment of NIH 3T3 cells reduces Ca2+ release induced by bradykinin, which was attributed to the inhibition of the SERCA pumps in the ER and reduction in the ER Ca2+ pools by Cd2+ (Biagioli et al. 2008). This caused ER stress, which induced a so-called unfolded protein response (UPR) and caspase-12 activation (see sections “Endoplasmic reticulum (ER) stress” and “Caspase 12 and caspase 4”). But Cd2+ also caused mitochondrial damage, and all these processes contributed to the activation of apoptosis (Biagioli et al. 2008). At variance with the concept that Cd2+ may change intracellular Ca2+ homeostasis to activate gene transcription and trigger death or survival in a controlled manner, Tvermoes et al. proposed that gene transcription in HEK293 cells exposed to Cd2+ (1 μM) is Ca2+-independent (Tvermoes et al. 2011). A change in the size of ER Ca2+ stores was observed with 30 μM Cd2+ (4 or 24 h) and the authors proposed that the release of Ca2+ is a nonspecific feature of dying cells rather than a primary mechanism by which Cd2+ regulates gene transcription and death signaling. However, these conclusions cannot be justified because cell viability was minimally affected by 30 μM Cd2+ at 4 h though ER Ca2+ stores were reduced by ~40 %, which suggests that Ca2+ release induced by Cd2+ is a cause rather than a consequence of maximal cell death observed at 24 h. Indeed, 15 out of 17 genes affected by thapsigargin (which increases [Ca2+]cyt by blocking SERCA pumps) were upregulated by 30 μM Cd2+ (Tvermoes et al. 2011). It remains to be investigated by more sensitive and accurate tools whether exposure to low micromolar or submicromolar Cd2+ concentrations for longer time periods affects Ca2+ homeostasis as well. In contrast to the work of Tvermoes et al., Cd2+-adapted myelomonocytic lymphoma U937 cells overexpress the Ca2+-binding protein calbindin-D(28 k) and become resistant to the effects of elevation of [Ca2+]cyt and depletion of intracellular Ca2+ stores induced by Cd2+, suggesting that enhanced Ca2+ buffering by upregulated calbindin-D(28 k) contributes to acquiring resistance to Cd2+-induced apoptosis (Jeon et al. 2004). Further support for this Ca2+ dependence of apoptosis signaling comes from other studies in which Cd2+ was not the proapoptotic stimulus. Overexpression of B cell lymphoma 2 (Bcl-2) (see section “Bcl-2 proteins”) or addition of ceramide (see section “Ceramides”) to cells led to structural alterations of ER and mitochondria and caused apoptosis that was mediated by ROS and Ca2+ (Hanson et al. 2008a; Darios et al. 2003), and expression of calbindin (or an enzyme that inhibited IP3-mediated Ca2+ release) reduced cell death caused by these very high levels of Bcl-2 or by ceramide. Similarly, downregulation of ceramide synthase 6/C16-ceramide formation in cancer cell induces activating transcription factor 6 (ATF6)-mediated ER stress (see section “Endoplasmic reticulum (ER) stress”) and apoptosis via perturbation of cellular Ca2+ and the ER/Golgi membrane network; accordingly, ectopic expression of calbindin prevented all the effects of CerS6/C16-ceramide downregulation, including apoptosis (Senkal et al. 2011). At this point, it must be emphasized that all these data also provide strong indications that structural changes to intracellular Ca2+ pools (ER, mitochondria) are also prognostic signs of irreversible cellular damage and apoptosis.
Cd2+ and ROS
Cd2+ affects cellular ROS signals, which are temporarily or permanently increased (reviewed in (Thévenod 2009; Cuypers et al. 2010; Liu et al. 2009). Cd2+ is not a Fenton metal and is therefore not directly redox active (O’Brien and Salacinski 1998). However, it can produce ROS indirectly by displacement of endogenous Fenton metals (e.g., Fe2+) from proteins and thereby increase free redox-active metals (Casalino et al. 2002; Dorta et al. 2003). ROS formation is also elevated by Cd2+ interfering with several of the processes controlling physiological ROS signaling (see section “ROS”). Cd2+ shows high affinity to thiols. It affects the conformation and hence the function of many proteins directly but also alters the cellular redox status by binding to and reacting with exogenous and endogenous antioxidants, in particular GSH (reviewed in (Thévenod 2009; Cuypers et al. 2010; Liu et al. 2009)). Cd2+ damages mitochondria and therefore not only interferes with Ca2+ signaling ((Biagioli et al. 2008); see section “Ca2+ ”), but also increases mitochondrial ROS formation (Pourahmad et al. 2003; Belyaeva et al. 2006). Wang et al. (2004) identified complex III as the sole origin of ROS in isolated mitochondria from three different tissues, which were generated by the formation of unstable semiubiquinones and transfer of one electron to molecular oxygen to form superoxide. Cd2+ may also increase ROS formation by activation of NOX enzymes in liver and blood cells (Souza et al. 2009; Banakou and Dailianis 2010) or increase NOX4 gene expression in mouse kidney (Thijssen et al. 2007). Finally, Cd2+ may increase ROS formation by disrupting cellular ROS depletion and antioxidative defense mechanisms. Cd2+ induces depletion of the reduced GSH pool (Lopez et al. 2006; Ikediobi et al. 2004), possibly also by complex formation (Leverrier et al. 2007). Cd2+ acutely inhibits antioxidative enzyme activity, e.g., CuZnSOD, MnSOD, catalase, GPx, GSH reductase, in vivo and in vitro (Casalino et al. 2002; Ikediobi et al. 2004), probably by direct binding to the enzymes (Huang et al. 2006; Picaud and Desbois 2006). Chronic exposure to Cd2+, on the other hand, results in more complex patterns of antioxidative enzyme activities and alterations of gene expression and therefore may or may not be associated with increased ROS formation (summarized in Thévenod 2009; Waisberg et al. 2003; Cuypers et al. 2010; Liu et al. 2009).
Cell death: a definition
Superfluous, aged and damaged cells are removed by several modes of tightly controlled death routines, which apply to both in vitro and in vivo settings and include apoptosis (extrinsic or intrinsic), necrosis, autophagic cell death and mitotic catastrophe (Edinger and Thompson 2004; Galluzzi et al. 2012). While the focus of this review is predominantly on apoptosis signaling elicited by Cd2+, the roles of other cell death pathways will also be briefly discussed.
Apoptosis, otherwise known as programmed cell death, is a genetically encoded process that is involved in normal development and homeostasis and can be divided into three distinct phases: initiation, integration/decision, and execution/degradation (Kroemer et al. 2007). The initiation phase depends on the nature of the death-inducing signal and may be “extrinsic” (e.g., ligation of a death receptor by ligands, such as FAS ligand, tumor necrosis factor (TNF) or TNF-related apoptosis-inducing ligand (TRAIL) and subsequent caspase-8 activation) or “intrinsic” (due to the action of exogenous or endogenous stressors and leading to damage of any cellular organelle including the nucleus, the ER, lysosomes or mitochondria), which can be caspase-dependent or caspase-independent. The integration/decision phase involves the activation of a specific class of proteases, the caspases (“cysteine protease cleaving after Asp”), which are required for the rapid manifestation of apoptosis: Active caspases acquire the ability to cleave important intracellular substrates, such as structural cytoskeletal proteins (e.g., actin, lamin and fodrin) that result in structural disassembly and morphological changes, as well as to activate DNase and induce DNA condensation/fragmentation associated with apoptosis (Riedl and Shi 2004). Activation of the integration/decision phase represents a “point of no return” and is followed by the execution/degradation phase, which is independent of the initiating stimulus (Kroemer et al. 2007).
Another class of proteases involved in apoptosis (and necrosis) is the calpains. Calpains are a family of Ca2+-dependent intracellular cysteine proteases, including the ubiquitously expressed μ- and m-calpains, as well as a number of distinct tissue-specific calpains. In vitro, m-calpain activity requires Ca2+ at the millimolar range, whereas micromolar concentrations activate μ-calpain. These Ca2+ levels are unlikely to be achieved in vivo; however, highly localized concentrations of Ca2+ may occur transiently in close proximity to ion channels or the ER during stress. Calpains are also negatively regulated by the endogenously expressed peptide inhibitor calpastatin. Similarly to caspases, calpains are synthesized as proenzymes. A small regulatory subunit encoded by the capn4 gene is cleaved during activation by increased Ca2+ levels and active calpains then cleave a wide array of substrates that can lead to apoptosis as well as survival and autophagy (Storr et al. 2011). Apoptotic substrates include caspases, Bcl-2, Bax (see section “Bcl-2 proteins”) and apoptosis-inducing factor (AIF). Interestingly, calpains are also localized in mitochondria, where they cleave AIF for release into the cytosol, and calpains may also be activated by caspase-12 (see section “Caspase 12 and caspase 4”).
Both the extrinsic and the intrinsic routes to apoptosis ultimately lead to cell shrinkage, chromatin condensation, nuclear fragmentation, blebbing and phosphatidylserine exposure on the surface of the plasma membrane (Kerr et al. 1972).
Upstream stress and apoptosis-inducing signaling
Ceramides
Cell stress may initiate the activation of upstream signaling pathways, such as ceramides, which elevate [Ca2+]cyt and/or ROS through their downstream action on mitochondria, ER and other organelles, and also communicate between the different organelles to induce damage and death (Grimm 2012). One such pathway involves the sphingolipid ceramide, the central molecule of sphingolipid metabolism, which is generated by sphingomyelin hydrolysis or by de novo synthesis in response to cell stress and plays a prominent role in apoptosis and tumor suppression, but also in signal transduction of ER stress and autophagy (see sections “Endoplasmic reticulum (ER) stress” and “Autophagy”) (reviewed in (Mullen and Obeid 2012; Gulbins and Li 2006; Ruvolo 2003; Hannun and Obeid 2008)). However, its role as a proapoptotic or survival signal molecule is complex and context-dependent (Grosch et al. 2012). Ceramide formation induces Ca2+/ROS signals but can also occur via increases in ROS and Ca2+ upstream of ceramide. Ceramide targets intracellular Ca2+ pools, such as the ER and mitochondria, that are regulated by the multidomain proapoptotic Bcl-2 members Bax and Bak (see section “Bcl-2 proteins”) and are implicated in apoptosis signaling (Scorrano et al. 2003; Pinton et al. 2001; Darios et al. 2003; Ferrari et al. 2011). But independently from ceramides, ROS and Ca2+ can also increase Ca2+ release from the ER via deregulation of the IP3 receptor, either through direct sensitization, caspase- and calpain-dependent cleavage or through binding of cytochrome c released from mitochondria (reviewed in Roderick and Cook 2008). Ca2+ released from the ER via IP3 receptors is then taken up by mitochondria to induce mitochondrial apoptosis (Sugawara et al. 1997).
Ceramides have also been shown to trigger ROS production in various intracellular compartments (reviewed in Won and Singh 2006). Acute and chronic damaging effects of ceramide on mitochondria have been observed: C16-ceramide induces ROS formation through direct inhibition of mitochondrial complex IV activity, resulting in oxidative stress (Zigdon et al. 2013). Ceramide can also cause apoptosis by inducing the expression of the harikari gene, which is translocated to cause mitochondrial dysfunction (Rizvi et al. 2011). Ceramide may also induce ER stress (see section “Endoplasmic reticulum (ER) stress”) (Chen et al. 2008a) or act downstream of ER stress (Yacoub et al. 2010) to induce the release of Ca2+ and ROS formation and elicit the UPR to trigger apoptosis by activation of caspase-12 (see section “Caspase 12 and caspase 4”) (Tabas and Ron 2011; Ron and Walter 2007; Zhang and Kaufman 2006).
Cd2+ and ceramides
Our laboratory was the first to show that Cd2+ increases ceramide formation, possibly via de novo synthesis, to increase [Ca2+]cyt and ROS formation (Lee et al. 2007; Lee and Thévenod 2008). These studies were performed in rat kidney proximal tubule (PT) cells exposed to 10–50 μM Cd2+, which caused significant formation of ceramide within 3 h (up to 24 h), as measured by a diacylglycerol kinase assay (Lee et al. 2007). Inhibition of ceramide synthase, which participates in de novo ceramide synthesis and in the salvage pathway, with fumonisin B1 (FB1) prevented ceramide formation, but did not reduce ROS, suggesting that ROS formation is upstream of ceramide (Lee and Thévenod 2008). FB1 also abolished Cd2+-induced calpain activation, which was associated with significant attenuation of apoptosis at 3–6 h (Lee et al. 2007). Moreover, addition of exogenous C6-ceramide to PT cells rapidly increased [Ca2+]cyt and activated calpains, suggesting that ceramide may induce Ca2+ release from intracellular pools and/or Ca2+ influx from the extracellular space to activate calpains, similarly as described by others (Poppe et al. 2002; Scorrano et al. 2003; Ferrari et al. 2011). This indicates that Cd2+ enhances de novo ceramide synthesis and that calpains are a downstream target of ceramides in apoptosis execution. Experiments with human D283 medulloblastoma cells showed that ceramide-induced apoptosis is calpain-dependent but independent of mitochondrial damage and caspase activation (Poppe et al. 2002). They are similar to a recent study in rat brain tissue where glutamate-induced apoptosis in neurons involved activation of ceramide synthase-6 and formation of long-chain ceramides, which caused cell death via an increase in mitochondrial Ca2+ as well as calpain activation (Novgorodov et al. 2011). In other cells, ceramide-dependent calpain activation was shown to promote apoptosis by cleavage of the autophagy protein Atg5 (Yousefi et al. 2006). Calpains are not only potent amplifiers and initiators of death signaling, they can also engage apoptotic pathways by processing and activating caspases (Gomez-Vicente et al. 2005; Nakagawa and Yuan 2000). In renal PT cells, apoptosis mediated by C6-ceramide at 24 h was significantly reduced by caspase-3 inhibition (Lee et al. 2007), which indicates cross talk between calpain- and caspase-dependent apoptotic pathways and emphasizes the importance of calpains for Cd2+ apoptosis in renal tissue (Lee and Thévenod 2008). So far, no studies have been performed that investigate the effect of Cd2+ on IP3 receptor function and IP3-induced Ca2+ release. But this mechanism of Cd2+ modulation of IP3 receptor function could contribute to Cd2+-induced ER Ca2+ release, decrease in the size of ER Ca2+ stores and subsequent cell death observed in HEK293 cells or NIH 3T3 cells (Tvermoes et al. 2011; Biagioli et al. 2008).
ASK1–MAPK signaling
The stress-activated mitogen-activated protein kinases (MAPK) are Ser/Thr-specific kinases that regulate activation of various stress responses, such as proliferation, gene expression, differentiation, mitosis, cell survival, and apoptosis and thereby contribute to the cellular answer to stress. MAPK pathways are protein kinase cascades in which signals are relayed through phosphorylation of downstream kinases by activated upstream kinases, leading to the appropriate cellular responses. Apoptosis signal-regulating kinase 1 (ASK1) is a member of the mitogen-activated protein kinase kinase kinase (MAP3K) family that activates via MAP2Ks downstream MAP kinases (MAPKs), c-Jun NH2-terminal protein kinases (JNKs) and p38 MAPKs, but not the extracellular signal-regulated kinase (ERK) pathway, in response to various stresses (Ichijo et al. 1997), including ROS, ER stress and Ca2+ overload (reviewed in (Takeda et al. 2011). Activation of the JNK and p38 pathways by ASK1 then induces, among other stress responses, cell death and in particular mitochondrial apoptosis (Hatai et al. 2000). Thioredoxin (Trx), an oxidoreductase with a dithiol-disulfide active site, is the first ROS-regulated ASK1-interacting molecule to be identified. The reduced but not oxidized form of Trx interacts noncovalently with the N-terminus of ASK1 and suppresses basal ASK1 kinase activity. Interaction between Trx and ASK1 is observed under reducing conditions, and Trx is oxidized and dissociates from ASK1 when cells are exposed to oxidative stress such as ROS (Saitoh et al. 1998). The ROS-dependent dissociation of Trx from ASK1 thus serves as a molecular switch, which converts oxidative stress into a phosphorylation-dependent signal. Phosphorylation of Thr845 in ASK1 is essential for its activation. Following ROS stimulation, Thr845 is autophosphorylated, leading to ASK1 activation. Tumor necrosis factor-α receptor-associated factors (TRAFs) are also important in the regulation of ASK1 activity. For example, ER stress activates ASK1 through the formation of an IRE1-TRAF2-ASK1 complex (see section “Endoplasmic reticulum (ER) stress”) (Nishitoh et al. 2002). Ca2+ signaling also activates the ASK1-p38 pathway through Ca2+/calmodulin-dependent protein kinase II (CaMKII) activation, which increases the phosphorylation of Thr845 in cultured cells, although CaMKII failed to directly phosphorylate Thr845 of ASK1 in vitro (Takeda et al. 2004). ASK1 and ASK2 may cooperate to exert proapoptotic activity in epithelial cells (Iriyama et al. 2009). ASK3 is predominantly expressed in the kidney where it is activated by osmotic stress (Naguro et al. 2012).
JNK is an integral player in the ER stress–apoptosis axis (see sections “Endoplasmic reticulum (ER) stress” and “ER stress and cell death”). Signaled via IRE1-TRAF2- ASK1, JNK is phosphorylated and translocates to the nucleus where it transactivates numerous transcription factors, of note c-Jun (and hence activator protein 1 (AP-1)) (see section “AP-1”), to transcriptionally regulate genes involved in apoptosis or directly phosphorylate target proteins that regulate the life–death balance (Nishitoh et al. 1998, 2002). The Bcl-2 family of proteins (see section “Bcl-2 proteins”) appears to be a particularly important target of JNK. First, Bcl-2 can be directly phosphorylated by JNK such that it can longer sequester proapoptotic Bax and exert its antiapoptotic effects (Park et al. 1997; Bassik et al. 2004); second, proapoptotic members Bax and Bak are essential for executing apoptosis by JNK by initiating cytochrome c release from mitochondria (Lei et al. 2002); third, JNK can release proapoptotic Bim and Bmf from their inhibitory bound states (Lei and Davis 2003). All these Bcl-2 members are linked to ER stress-induced (as well as mitochondrial) apoptosis (Szegezdi et al. 2006).
Cd2+ and ASK1–MAPK signaling
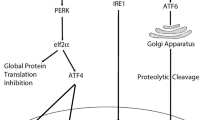
Since oxidative stress is a strong inducer of MAPK signaling, these kinases are therefore modulated by Cd2+, which has been previously reviewed (Thévenod 2009). JNK can also be inhibited by GSH (MacKinnon and Kapron 2010). Generally, Cd2+-activated JNK (and p38) signaling leads to apoptotic cell death, although JNK has been shown to have an antiapoptotic role too (Liu and Lin 2005). Cd2+ is also known to elicit JNK-mediated apoptosis by activating the ER stress-dependent IRE1-XBP1-JNK UPR signaling pathway (see section “Endoplasmic reticulum (ER) stress”) in cultured renal PT cells (Yokouchi et al. 2007; Yokouchi et al. 2008; Komoike et al. 2012) and in rat testicular germ cells in vivo, most likely via mitochondrial apoptosis (Ji et al. 2011). Furthermore, Cd2+ has also been shown to cause cell death via activation of p38 and CAMKII (Liu and Templeton 2008; Chen et al. 2011c) though both kinases were thought to promote cell death independently from each other (possibly via ROS and Ca2+ activation, respectively) (Xiao et al. 2009). Nevertheless, little is known about the role of apoptosis signal-regulating kinases (ASKs) in Cd2+ toxicity. One study demonstrated the ASK1-MKK4-JNK/c-Jun-caspase-3-dependent signaling cascade as being responsible for Cd2+-induced neuronal cell apoptosis (Kim et al. 2005b). The two cysteine residues within the redox-active center of Trx are a particular target for Cd2+. It has been shown in HeLa cells that Cd2+, along with mercury and arsenic, oxidizes Trx-1 and Trx-2 leading to ASK1 autophosphorylation and activation and cell death, whereas copper, iron and nickel had no effect on Trx oxidation (Hansen et al. 2006). A study in C. elegans characterized Cd2+-induced germline apoptosis through the ASK1/2-MKK7-JNK and ASK1/2-MKK3/6-p38 signaling pathways in a caspase-dependent manner (Wang et al. 2008a). Other studies allude to the activation of the IRE1-TRAF2-ASK1-JNK axis by Cd2+ by way of inhibition of eIF2α phosphorylation using salubrinal (Komoike et al. 2012) or by correlating JNK activation with ER stress (Yokouchi et al. 2007). Only the study by Yokouchi et al. has demonstrated that JNK activation is a direct effect of superoxide anion-activated XBP1 by Cd2+, linking ER stress with JNK-mediated apoptosis (Yokouchi et al. 2008). Activated JNK is likely to act via modification of Bcl-2 family members since overexpression of Bcl-2 can attenuate JNK activation and apoptosis by Cd2+ (Qu et al. 2007).
Additional work is necessary to determine a role for apoptosis signal-regulating kinase signaling as an upstream signal of Cd2+-induced mitochondrial apoptosis.
Mitochondrial apoptosis
Although the signaling cascades that trigger intrinsic apoptosis are highly heterogeneous as far as the initiating stimuli are concerned, they are all wired to a mitochondrion-centered control mechanism (Galluzzi et al. 2012). The mitochondrial apoptotic pathway is often utilized by toxic stimuli, such as ROS, UV light, Ca2+, and metals like Cd2+, either indirectly through increases in Ca2+ and ROS (see sections “Ca2+ ” and “ROS”) or by direct damage. These stress stimuli lead to mitochondrial outer membrane permeabilization (MOMP) and the release of proapoptotic factors, such as cytochrome c, from the mitochondrial intermembrane space (IMS) into the cytosol. These factors either activate caspases causing apoptosis or bypass this step and induce caspase-independent apoptosis (e.g., via AIF). Cytosolic cytochrome c binds apoptotic protease-activating factor 1 (APAF1), inducing its conformational change and oligomerization and leading to the formation of a caspase activation platform termed the apoptosome. The apoptosome recruits, dimerizes and activates an “initiator” caspase, caspase-9, which, in turn, cleaves and activates caspase-3 and caspase-7 (Tait and Green 2010). These “executioner” caspases mediate their effects by the cleavage of specific substrates in the cell.
Cd2+ and mitochondrial apoptosis
Cd2+ and mitochondrial apoptosis Cd2+ induces apoptosis in a number of organs in vivo, including the liver and kidneys (Ishido et al. 1998; Habeebu et al. 1998). In cultured cells, Cd2+ elicits a cellular stress response that mainly culminates in the activation of mitochondrial (“intrinsic”) apoptosis pathways (reviewed in Thévenod 2009). In intact cells, Cd2+-induced mitochondrial damage and mitochondrial apoptosis are late events, which occur at least 12–24 h after Cd2+ exposure (Oh et al. 2004; Lee et al. 2006; Li et al. 2000; Biagioli et al. 2008). Therefore, additional, earlier and more indirect cellular mechanisms induced by Cd2+ must contribute to mitochondrial damage. One study used lung fibroblasts to demonstrate Cd2+ (40 μM)-induced caspase-8 activation at 6 h with ROS formation, loss of Δψm, release of cytochrome c and mitochondrial association of proapoptotic Bax (see section “Bcl-2 proteins”) at 12–24-h Cd2+ exposure (Oh et al. 2004). As well as activating the classical mitochondrial pathway involving activation of caspases-9 and -3, Cd2+ can also utilize caspase-independent pathways in a context-dependent manner. Our laboratory has demonstrated early apoptosis in rat kidney proximal tubule cells exposed to 10 μM Cd2+ (3–6 h) that was induced by calpain activation, but with no indication of activation of mitochondrial apoptosis signaling; apoptosis with mitochondrial damage, cytochrome c and AIF release and subsequent caspases-9 and -3 activation were seen only after 24 h, and these effects were partly calpain-dependent because the calpain inhibitor PD-150606 attenuated caspase-3 activity and apoptosis at 24 h (Lee et al. 2006). A similar induction of calpain-dependent apoptosis by Cd2+ has been described by others as well, where calpain was shown to cleave Bax to allow release of cytochrome c (Oh et al. 2004). Other studies have reported similar observations with multiple apoptosis signaling pathways being affected by Cd2+ to induce apoptotic cell death (Li et al. 2000; Mao et al. 2007; Liu and Templeton 2008).
But Cd2+ can also damage mitochondria directly, which has a significant impact on respiration, ATP production and Ca2+ homeostasis. This is mainly the consequence of Cd2+-induced disruption of the electrochemical gradient across the inner membrane, known as the mitochondrial membrane potential (Δψm), which is maintained under normal physiological conditions through extrusion of protons from the matrix. Cd2+-induced loss of Δψm, inhibition of the electron transport chain, mitochondrial swelling and release of proapoptotic factors have been observed in various cell lines and isolated mitochondria (Lemarie et al. 2004; Lee et al. 2005a, b; Wang et al. 2004). How Cd2+ causes loss of Δψm is a matter of debate: Several authors have postulated that Cd2+ induces opening of a pore, the so-called permeability transition pore (PTP) (see for example Belyaeva et al. 2002; Li et al. 2003). The PTP is activated by oxidative stress or high Ca2+ and blocked by cyclosporine A and allows entry of cytosolic molecules <1.5 kDa through the mitochondrial outer (MOM) and inner membrane (MIM) into the matrix (Crompton 1999). Influx of cytosolic solutes through the PTP causes progressive osmotic swelling of the matrix and dissipation of Δψm and ultimately leads to the disruption of the MOM, spilling the intermembrane contents into the cytosol. PTP opening has therefore been associated with apoptosis induction (Crompton 1999). However, the molecular identity of the PTP has remained elusive because most of its postulated protein subunits are not required to induce this phenomenon (reviewed in Siemen and Ziemer 2013). Hence, the studies claiming activation of the PTP by Cd2+ (Belyaeva et al. 2002; Li et al. 2003) need to be taken with caution. Conversely, using isolated rat kidney cortex mitochondria, we have shown that Cd2+ (2–50 μM) induces mitochondrial swelling independently of the presence of PTP blockers (e.g., cyclosporine A or bongkrekic acid); we found that Cd2+ enters the mitochondrial matrix exclusively through the Ru360-blockable Ca2+ uniporter of the MIM (Lee et al. 2005a). In the matrix, Cd2+ activated Ag+-sensitive aquaporin-8 water channels expressed in the MIM to induce osmotic swelling of mitochondria and cytochrome c release (Lee et al. 2005a; Lee and Thévenod 2006). But how Cd2+ crosses the MOM is unknown.
A few studies have described increased activity of the extrinsic pathway, i.e., increased caspase-8 activity and/or FAS ligand levels (Eichler et al. 2006; Nguyen et al. 2013; Oh et al. 2004). But how Cd2+ activates the death receptor-dependent signaling pathway has not been investigated.
Necrotic cell death
Necrotic cell death is also a form of programmed cell death, rather than a passive process of cell swelling and rupture, termed “necroptosis,” and uses many of the tools available for apoptosis induction (Galluzzi et al. 2011; Golstein and Kroemer 2007). Necrosis is often induced by severe and acute injury and/or may develop secondary to apoptosis. Inhibition of cell death signaling pathways can initiate a cell to “re-wire” its cell death program from apoptosis to necrosis through a molecular switch (Nicotera and Melino 2004). Earlier studies put forward ATP as the mediator (Nicotera et al. 1998). Depletion of ATP would favor necrosis, which can be explained by energy demands during apoptosis execution. More recent data report the Ripoptosome as a signaling platform, which governs the form of cell death that will be executed (Darding and Meier 2012; Tenev et al. 2011).
Cd2+ is a well-known necrosis inducer, especially at higher concentrations in vivo and/or in sensitive cell lines, e.g., Templeton and Liu (2010). The scenario for cell death switch in Cd2+ treated cells is complicated, but ATP, GSH status and peroxide accumulation have all been implicated (Sancho et al. 2006; Lopez et al. 2003). Using pharmacological inhibitors, Yang and coworkers (Yang et al. 2007a) provided evidence that Cd2+ (4 μM for 24 h) stimulates Ca2+-dependent necrosis in CHO cells (which are very sensitive to Cd2+ because they do not express metallothioneins) through two separate pathways: Cd2+ reduces Δψm by activating (Ca2+-dependent) calpains and inhibits NF-κB activity (see section “NF-κB”) by increasing ROS levels. The authors suggested that sustained depletion of Ca2+ stores in ER leads to apoptosis while Ca2+ overload might be important for the execution phase of necrotic cell death (Yang et al. 2007a). In a follow-up study by the same group, necrostatin-1, a blocker of receptor-interacting serine/threonine-protein kinase 1 (RIPK1), rescued CHO cells from Cd2+-induced necrosis by attenuating calpain activity and Δψm loss, and by enhancing mitochondrial Ca2+ uptake (Hsu et al. 2009). In line with their previous observations, necrostatin-1 also prevented Cd2+-induced decrease in NF-κB activity, suggesting reduction of this transcription factor is integral to the necrotic response.
Cell survival signaling
Cell survival can be controlled by ROS and Ca2+ at various levels to counteract death signals by regulating signal transduction, transcription and/or execution (Roderick and Cook 2008; Trachootham et al. 2008).
Upstream signaling pathways
ERK1/2
The best studied MAPK cascade is the ERK1/2 pathway, which is usually activated by growth factors and cytokines via stimulation of tyrosine kinase receptors and elicits a signaling cascade involving Ras activation, recruitment of Raf-1 MAP3K to the plasma membrane, and sequential activation/phosphorylation of MEK1/2 and ERK1/2 (Kyriakis and Avruch 2001; McCubrey et al. 2007). Proapoptotic ASKs activate JNK and p38, whereas ERK signaling is not involved in this proapoptotic signaling cascade (Hattori et al. 2009). Indeed, activation of ERK1/2 phosphorylates and activates various transcription factors (e.g., cAMP response element binding (CREB) or ETS domain-containing protein (elk1)) and other protein kinases, e.g., the mammalian target of rapamycin (mTOR) protein kinase (Foster and Fingar 2010), to promote cell survival, anti-apoptosis, differentiation and cell cycle regulation (Matsuzawa and Ichijo 2005). Oxidative stress can activate ERK1/2 signaling at different levels (McCubrey et al. 2007): (1) A number of growth factor receptors, such as epidermal growth factor (EGF) receptor and platelet-derived growth factor (PDGF) receptor, undergo phosphorylation in response to oxidative stress; (2) Ras, a small G protein, is a target of ROS, which transduces a signal from tyrosine kinase receptors to the ERK1/2 pathway; (3) ROS inhibit protein phosphatases, and this results in the activation of ERK1/2; (4) ROS also induce the activation of ERK1/2 in Ras-negative cells via c-Src, which activates phospholipase C (PLC)-γ to generate diacylglycerol (DAG) and increase [Ca2+]cyt, which in turn activate several forms of protein kinase C (PKC). Although PKC can lead to Ras activation, it may also directly activate downstream Raf-1 (reviewed in (McCubrey et al. 2007)). This connection to Ca2+ signaling may be predominant for the stimulation of the ERK1/2 pathway and regulation of gene transcription in excitable cells (Wiegert and Bading 2011; Carrasco et al. 2004).
Cd2+ and ERK1/2
With a few exceptions, the majority of earlier studies have shown that ERK1/2 protects against cell death induced by Cd2+ (reviewed in Thévenod 2009). More recently, a study using rat mesangial cells exposed to Cd2+ (0.5 μM in serum-free medium for ≥30 s up to 6 h) showed increased ROS and CaMKII activity (peak at 0.5 min), followed by activation of epidermal growth factor receptor (EGFR) (peak at 5 min) and ERK1/2 (peak at 10–30 min), which increased cell survival and suggested involvement of Ca2+ and ROS in ERK1/2 activation (Xiao et al. 2009; Liu and Templeton 2008). We have recently shown in renal PT cells that Cd2+ (25 μM in serum-free medium for up to 6 h) elicits an increase in [Ca2+]cyt and downstream ROS formation to induce ER stress and CHOP-dependent apoptosis (see section “CHOP”) (Lee et al. 2012); this is counteracted by Ca2+- and ROS-dependent activation of ERK1/2, which downregulates proapoptotic CHOP and enhances cell survival through increased transcription of the putative Cl− channel bestrophin-3 (see section “Bestrophin-3”). In neuronal cells (SH-SY5Y and PC12), Cd2+ (20 μM for 24 h) induced ROS formation, which resulted in ERK1/2 activation by decreasing the expression and activity of protein phosphatases PP2A and PP5 (Chen et al. 2008b). In primary mouse hepatocytes, Cd2+ (5 μM up to 3 h) induced activation of NOX, and the resulting ROS formation triggered Src and EGFR phosphorylation and downstream phosphorylation of signal transducer and activator of transcription 3 (Stat3) at Tyr705 and Ser727 (Martinez Flores et al. 2013). Furthermore, ROS-dependent Src-EGFR phosphorylation enhanced ERK1/2 phosphorylation, which increased Stat3 phosphorylation at Ser727. The authors concluded that Cd2+-induced ROS formation increases Src-EGFR-ERK1/2 signaling to produce an adaptive survival response mediated by upregulation of metallothionein-2 (MT-2) (see section “Metallothioneins”) (Martinez Flores et al. 2013). It remains to be determined whether this Cd2+ activation of NOX enzymes in mouse hepatocytes is Ca2+-dependent, as shown in biochemical studies for the Ca2+ dependence of NOX5 (Tirone et al. 2010). Early work of Misra et al. (Misra et al. 2002) unmistakably showed rapid PLC- and IP3-dependent Ca2+ mobilization and PKC activation induced by Cd2+ (≤1 μM) in murine peritoneal macrophages (possibly via Cd2+ binding to a G protein-coupled cell surface receptor), which resulted in activation of Ras/MEK/ERK1 signaling and increased the expression of c-Fos and c-Myc and cell proliferation (Misra et al. 2002). Although several recent reports have also proposed ERK-mediated Cd2+ apoptosis via Ca2+ signaling (Chen et al. 2011c; Wang et al. 2008a), these studies used BAPTA-AM and EGTA, respectively, to chelate Ca2+ or measured [Ca2+]cyt with fluorescent derivatives of EGTA without considering that Cd2+ binds to these compounds with higher affinity than Ca2+ and that consequently, the conclusions of these studies must be taken with caution (see (Thévenod 2009)).
PI3K-Akt/PKB
Signal transduction via PI3K-Akt/protein kinase B (PKB) signaling plays an important role in regulating cell growth, proliferation, survival and motility (Manning and Cantley 2007; Hers et al. 2011). After ligand-induced activation of specific receptor tyrosine kinases (RTKs) activated by growth factors, such as epidermal growth factor (EGF), platelet-derived growth factor (PDGF), nerve growth factor (NGF), insulin and vascular endothelial growth factor (VEGF), PI3K can be activated by two mechanisms: A phosphorylated Tyr residue on the receptor serves as a docking site for the p85 regulatory subunit of PI3K. This recruits the catalytic subunit of PI3K, p110, to this complex. Alternatively, upon activation of the cytokine receptor by the appropriate ligand, the Shc protein binds the receptor to enable the Grb-2 and Sos proteins to form a complex, which results in the activation of Ras. Ras is then able to induce the membrane translocation and activation of the p110 subunit of PI3K. Activated PI3K converts phosphatidylinositol 4,5 biphosphate (PIP2) into phosphatidylinositol 3,4,5 phosphate (PIP3) which results in the membrane localization of phosphoinositol-dependent kinase-1 (PDK1) via its pleckstrin homology (PH) domain. Akt is also recruited to the lipid-rich plasma membrane by its PH domain and is phosphorylated at residues Thr308 and Ser473 by PDK1 and another kinase. Akt then regulates the functions of downstream targets through its Ser/Thr kinase activity. This results in inactivation of proapoptotic proteins (e.g., Bad, Bim, procaspase-9), transcription factors (FOXO3A) or/and activation of transcription factors (e.g., NF-κB via phosphorylation of IKKα, β-catenin via inactivation of glycogen synthase kinase-3β (GSK-3β), CREB), which induce the expression of antiapoptotic (Bcl-2, Bcl-xL) or cell cycle proteins (c-Myc, cyclin D1). Akt can also activate Mdm2, which inhibits p53 and thereby prevents apoptosis, as well as activate mTOR signaling, which promotes growth (Zoncu et al. 2011). Furthermore, Akt also exerts its antiapoptotic function by phosphorylation (at Ser83) and inhibition of ASK1 activity (see section “ASK1–MAPK signaling”), which prevents stress-induced apoptosis. The activation of the PI3K/Akt pathway is tightly regulated by phosphatases, especially the reversion of PIP3 back to PIP2 by phosphatase and tensin homolog (PTEN) and the inactivation of receptor tyrosine kinases by protein tyrosine phosphatases (PTPases) (summarized in (Song et al. 2005)). A cross talk exists between PI3K/Akt and ERK1/2 (McCubrey et al. 2007) or nuclear factor erythroid 2 (NF-E2)-related transcription factor (Nrf2) signaling (see section “Ref-1-Nrf2”) (Sakamoto et al. 2009). Redox regulation of the PI3K/Akt pathway can lead to activation or inactivation of signaling (Leslie 2006; Trachootham et al. 2008): (1) Under oxidative stress, this pathway can be activated by oxidative modifications of active Cys in PTPases and PTEN, which results in constitutive activation of tyrosine kinase receptors and PI3K; (2) direct oxidative modification of PI3K and Akt (e.g., formation of a disulfide bridge at Cys297 and Cys311 of Akt, leading to increased binding of PP2A and dephosphorylation of Akt) can result in their inactivation and prevent cell survival; and (3) furthermore, the PI3K/Akt pathway can also promote cellular production of ROS through activation of Rac1 and NOX1. The regulation of the PI3K/Akt signaling pathway by Ca2+ has been studied with less detail but could involve upstream activation of CaMK kinase to overcome stress and promote survival of neurons (Yano et al. 1998) and other cells (Britschgi et al. 2013; Tano et al. 2012).
Cd2+ and PI3K-Akt/PKB
The first relevant study demonstrating Cd2+ activation of PI3K-Akt signaling was performed in the 1LN prostate cell line (1 μM Cd2+ in 0.5 % serum for 24 h) (Misra et al. 2003). Cd2+ caused a rapid increase in [Ca2+]cyt induced by IP3 release that was associated with phosphorylation of MEK1/2, ERK1/2 (as well as p38 MAPK and JNK). Increased phosphorylation of PDK1, the 85-kDa regulatory subunit of PI3K, Akt and p70s6k (a downstream target of mTOR) as well as increased protein levels of Grb-2, Sos, Shc and Raf-1 were also observed, suggesting Ca2+-dependent activation of PI3K/PDK1/Akt/p70s6k signaling and cross talk with the ERK1/2 pathway via Raf-1 signaling. Cd2+ treatment also increased the levels of transcription factors NF-κB and phosphorylated CREB, and c-Fos and c-Myc. These effects were associated with increased survival and cell proliferation (Misra et al. 2003). In rat mesangial cells, Cd2+ (0.5 μM in serum-free medium ≥30 s up to 6 h) increased CaMKII activity (peak at 30 s), followed by EGFR (peak at 5 min), ERK1/2 and Akt activation (at 15 min), which resulted in increased cell survival, suggesting activation of CaMKII (and ROS, see Liu and Templeton 2008) upstream of EGFR activation and simultaneous ERK1/2 and Akt activation as downstream events (Xiao et al. 2009). More recently, Matsuoka and coworkers showed that Cd2+ (10–20 μM in serum-free medium for 0.5–12 h) increased phosphorylation of FOXO3a (at Thr32 and Ser253) and its upstream kinase, Akt (at Thr308 and Ser473) in HK-2 human renal proximal tubular cells (Fujiki et al. 2013). Cd2+-induced phosphorylation of FOXO3a was suppressed by an inhibitor of EGFR, the CaMKII inhibitor, KN-93, and a Src inhibitor; furthermore, the p38 inhibitor, SB203580, suppressed Cd2+-induced phosphorylation of Akt and FOXO3a, suggesting possible cross talk between p38 mitogen-activated protein kinase and Akt. Since FOXO3a siRNA suppressed Cd2+-induced cell damage, the authors concluded that PI3K/Akt-dependent FOXO3a phosphorylation and inactivation promotes HK-2 cell survival (Fujiki et al. 2013). A study in rat pheochromocytoma (PC12) and human neuroblastoma (SH-SY5Y) cells exposed to 20 μM Cd2+ for 24 h showed reduction in PTEN, resulting in increased PI3K/Akt signaling (Chen et al. 2011a). This study also confirmed PI3K/Akt signaling dependence of ROS formation that was induced by upregulation of NOX2, indicating that ROS formation may also occur downstream of PI3K and feedback on Cd2+-induced signaling (Chen et al. 2011a). In another study, immortalized human lung epithelial BEAS-2B or normal human bronchial epithelial cells (NHBE) were exposed to 5–20 μM Cd2+ for ≤6 h, which increased ROS formation and downstream activation of ERK1/2 and Akt signaling pathways and resulted in increased phosphorylation of the mTOR downstream target p70s6k1 as well as increased the expression of the target genes hypoxia-inducible factor 1alpha (HIF-1α) and VEGF (Jing et al. 2012). Most importantly, Cd2+ transformed these epithelial cells in culture: The transformed cells induced tube formation in vitro, angiogenesis on the chicken chorioallantoic membrane, and formed tumors in nude mice, indicating that activation of ERK1/2 and Akt signaling pathways promotes malignant transformation (Jing et al. 2012). Another study also showed that chronic exposure of human lung epithelial BEAS-2B cells to Cd2+ (≤2 μM in 10 % serum for 2 months) induces cell transformation, as evidenced by anchorage-independent growth in soft agar and clonogenic assays, promotes cell invasion and migration, and increases tumor formation in nude mice (Son et al. 2012). Furthermore, chronic Cd2+ exposure led to the activation of signaling cascades involving PI3K, Akt, GSK-3β and β-catenin in cells and tumor tissues. All these effects of Cd2+ were prevented by transfection with catalase, SOD1 or SOD2, suggesting a direct involvement of ROS in Cd2-induced carcinogenesis and implicating a role of PI3K/Akt/GSK-3β/β-catenin signaling in this process (Son et al. 2012).
Transcriptional regulation
In addition to modulating signal transduction, Ca2+ and ROS signals can promote cell survival by regulating the expression and activity of several transcription factors.
Ref-1-Nrf2
Nrf2 is a member of the Cap“n” Collar (CNC) family of basic leucine zipper (bZIP) proteins (Taguchi et al. 2011). It binds to the antioxidant-responsive element (ARE)—also called electrophile-responsive element (EpRE)—within the promoter region of genes that favor cell survival. One major function of Nrf2 is the transactivation of many antioxidant proteins, including heme oxygenase-1 (HO-1), peroxiredoxin 1, catalase, GPx, SOD, Trx and proteins that enhance GSH synthesis and regeneration (Lu 2013). Nrf2 also induces phase 2 detoxification (e.g., via glutathione S-transferase), drug metabolism and elimination (e.g., ATP-binding cassette transporter C1), promotes nucleotide excision repair, inhibits autophagy and also interferes with ER stress-associated apoptosis (see section “Endoplasmic reticulum (ER) stress”) by inducing heat shock proteins and the 26S proteasome (see section “Ubiquitin-proteasome system (UPS)”) to facilitate repair and elimination of unfolded proteins. Furthermore, Nrf2 upregulates anti-apoptotic Bcl-2 proteins (see section "Bcl-2 proteins") (Niture and Jaiswal, 2012, 2013). Hence, Nrf2 is one of the most important transcription factors that protect the organism against exogenous stressors and ROS damage. Under normal conditions, Nrf2 localizes in the cytoplasm, where it interacts with the actin-binding protein, Kelch-like ECH-associating protein 1 (Keap1). Keap1 functions as an adaptor of cullin-3 (Cul3)-based E3 ubiquitin ligase and targets Nrf2 for degradation by the ubiquitin–proteasome (see section “Ubiquitin-proteasome system (UPS)”). Dissociation of Nrf2 from Keap1 is a key step in activating Nrf2, which permits translocation of free Nrf2 to the nucleus where it heterodimerizes with Maf transcription factor proteins in order to bind to ARE/EpRE of responsive genes. Reactive Keap1 cysteines are redox sensors, and upon oxidation by ROS, this results in the dissociation of Nrf2 from Keap1/Cul3, which allows Nrf2 stabilization and translocation into the nucleus (reviewed in (Brigelius-Flohe and Flohe 2011; Ray et al. 2012; Trachootham et al. 2008).
Redox factor-1 (Ref-1) is a multifunctional protein that regulates transcription factor activity and also mediates base excision repair (Tell et al. 2009). The transcriptional regulatory function of Ref-1 occurs through its redox activity on several transcription factors such as AP-1, p53, nuclear factor kappa B (NF-κB) and HIF-1α. Two cysteines on Ref-1 (Cys 65 and 93) are major redox-active sites that are required for the reduction and increased DNA binding of targeted transcription factors. A highly conserved cysteine in various bZIP transcription factors (bZIP-TF), including Jun, Fos, CREB, Maf and Nrf proteins, is subject to redox regulation by Ref-1. Oxidative stress somehow triggers translocation of Ref-1 to the nucleus where Ref-1 reduces and thereby activates the bZIP-TF. This process is redox regulated by nuclear translocated Trx. The BTB and CNC homolog 1 (Bach1), a bZIP transcriptional repressor of ARE/EpRE, also features the Ref-1 redox-regulated conserved cysteine, whose oxidation results in cytoplasmic translocation of Bach1 and hence ARE/EpRE derepression.
Hence, these results suggest that at least two sequential redox events (1) the oxidation of Keap1 in the cytoplasm and subsequent release and nuclear translocation of Nrf2, and (2) the redox regulation of Nrf2 (via Ref-1) and Bach1 in the nucleus, are critical for transcriptional activation of ARE/EpRE-dependent antioxidant genes via the Nrf2 signaling pathway (Brigelius-Flohe and Flohe 2011; Ray et al. 2012; Trachootham et al. 2008). A few studies have shown Ca2+ or CaMKII (as well as ROS) dependence of Nrf2 activation following cellular stress that involved ERK1/2 activation upstream of Nrf2, suggesting interactions between ERK1/2 and Nrf2 signaling to enhance the expression of Nrf2 target genes and promote cell survival (Kim et al. 2010, 2012; Khan et al. 2011).
Cd2+ and Ref-1-Nrf2
So far, only one study has investigated the role of Ref-1 in Cd2+ stress. Following prolonged exposure to Cd2+ (2 month at nontoxic doses) of mouse epidermal JB6P+ cells, a model for tumor promotion and cellular transformation, Ref-1 was increased, but not in control JB6P-cells (Yang et al. 2007b). This was associated with decreased ROS formation and oxidative DNA damage (8-oxo-dG formation). Knockdown of Ref-1 inhibited tumor promoter-induced cell transformation, proliferation and AP-1 transcriptional activity of JB6P+ cells exposed to Cd2+ and increased ROS formation and apoptosis (Yang et al. 2007b). Most studies investigating the effects of Cd2+ on the Ref-1-Nrf2 axis have exclusively focused on Nrf2. Using different cell culture models, Alam et al. described Cd2+-dependent induction of the HO-1 gene that was mediated by Nrf2 (Alam et al. 1999). p38 MAPK activation occurred upstream of Nrf2 activity (Alam et al. 2000), and the activating transcription factor-4 (ATF4), which is involved in the UPR (see section “Endoplasmic reticulum (ER) stress”), was a co-activator of HO-1 (He et al. 2001), thus suggesting interactions between electrophilic and ER stress pathways. Furthermore, Cd2+ delayed the rate of Nrf2 degradation by the ubiquitin–proteasome pathway (see section “Ubiquitin-proteasome system (UPS)”), possibly by inhibition of the degradation pathway, which also resulted in HO-1 gene activation (Stewart et al. 2003). Cd2+ has also been shown to induce nuclear export of Bach1 to relieve the repression of HO-1, which was dependent on ERK1/2 activity (Suzuki et al. 2003). Further studies showed that Cd2+ activation of Nrf2 is ROS-dependent and induces transcriptional activation of cytochrome P450 enzymes, HO-1, Trx reductase-1 and NADPH:quinone oxidoreductase-1 to reduce oxidative stress and apoptosis in hepatic, renal and other cells in vitro (Chen and Shaikh 2009; Abu-Bakar et al. 2004; Sakurai et al. 2005; He et al. 2008). Primary cells and tissues from Snell dwarf mice, which are resistant to cytotoxic stress, have increased protein levels of Nrf2 and increased transcription of antioxidative enzymes and proteins, such as HO-1, NADPH:quinone oxidoreductase-1, glutathione S-transferase A1, GSH and MT-1, and show increased survival to Cd2+ exposure (Leiser and Miller 2010). Acute Cd2+ exposure (3.5 mg Cd/kg, i.p.) in wild-type mice, Nrf2-null mice, Keap1-knockdown mice with enhanced Nrf2, and Keap1-hepatocyte knockout mice with maximal Nrf2 activation showed increased ROS formation in all genotypes of mice, but upregulation of antioxidative genes, such as glutamate cysteine ligase, GPx-2 and sulfiredoxin-1, was only observed in the livers of Nrf2-enhanced mice and not in Nrf2-null mice (Wu et al. 2012). Interestingly, MT upregulation was not dependent on Nrf2 because Cd2+ also markedly induced MT-1 and MT-2 in livers of Nrf2-null mice (Wu et al. 2012) despite the presence of a putative ARE/EpRE in the MT-1 promoter region (Campagne et al. 2000). One report showed that Nrf2-mediated adaptive response to Cd2+-induced toxicity (i.e., increased expression of NADPH:quinone oxidoreductase and HO-1) involves PKCδ in human 1321N1 astrocytoma cells (Lawal and Ellis 2011). Since PKCδ is not sensitive to Ca2+ but activated by ROS, the authors proposed that Cd2+ may either cause oxidative activation of PKCδ, bind to the zinc finger of PKCδ, or operate through a G protein-coupled metal-binding receptor to increase DAG production via the PLC-IP3 pathway, which would cause Nrf2 accumulation and transcription of cytoprotective genes. The authors also proposed that Cd2+ may directly modify Keap1 by reacting with the reactive cysteine residues on Keap1, or increased ROS levels by Cd2+ may oxidize Keap1, resulting in nuclear accumulation of Nrf2 (Lawal and Ellis 2011).
NF-κB
Nuclear factor kappa B (NF-κB) is a transcription factor that regulates immunity, inflammation, development, cell proliferation and survival in response to stimuli such as stress, cytokines, ROS, UV irradiation, oxidized low density lipoprotein (LDL), and bacterial or viral antigens (Ghosh and Hayden 2012). In mammalian cells, the NF-κB family consists of NF-κB1, NF-κB2, RelA (p65), c-Rel and RelB (Ghosh et al. 2012). NF-κB1 and NF-κB2 proteins are synthesized as large precursors, p105 and p100, which undergo processing to generate the mature NF-κB subunits, p50 and p52, respectively. All members are characterized by a Rel homology domain, which mediates DNA binding, dimerization between the family members and the association of NF-κB dimers with a family of inhibitors, called inhibitors of kappa B (IκBs). In unstimulated cells, the NF-κB dimers are sequestered and inactivated in the cytoplasm by IκBs, which contain multiple ankyrin repeats. By virtue of their ankyrin repeat domains, the IκB proteins mask the nuclear localization signals of NF-κB proteins. A wide range of stimuli, including cytokines and ROS, activate NF-κB. This is initiated by the signal-induced degradation of IκB proteins. This occurs primarily via activation of a kinase called the IκB kinase (IKK). Active IKK phosphorylates IκB, leading to dissociation of NF-κB from the inhibitor and degradation by the ubiquitin/proteasome system (UPS) (see section “Ubiquitin-proteasome system (UPS)”). Free NF-κB translocates to the nucleus, binds to DNA at the promoter region and activates the transcription of target genes (Ghosh and Hayden 2012). NF-κB promotes cell survival and proliferation by increasing the expression of antiapoptotic proteins, such as Bcl-xL, the inactive homolog of caspase-8, cFLIPL, and caspase inhibitors, such as cIAPs, which all suppress the execution phase of cell death. NF-κB also induces Gadd45, which inhibits JNK and prevents JNK-induced apoptosis, and it also increases the expression of cyclin B1 and D1, which regulates cell cycle progression. Of note, NF-κB also promotes the expression of antioxidant genes, such as MnSOD, catalase or Trx, which play a major role in scavenging superoxide and in maintaining redox homeostasis (Ghosh and Hayden 2012), as well as proteins that enhance GSH synthesis and regeneration (Lu 2013). Some enzymes that promote the production of ROS, such as NOX2, are also upregulated, especially in cells of the immune system (summarized in (Morgan and Liu 2011).
NF-κB is a redox-sensitive transcription factor (Morgan and Liu 2011). ROS can either activate or inhibit NF-κB activity, depending on the level of ROS, types of stimuli and cell types. Multiple redox-mediated mechanisms can regulate NF-κB activity. Consequently, a difficulty in defining ROS impact on NF-κB signaling is that ROS can often function in multiple places (i.e., upstream or downstream) and sometimes in opposing ways (i.e., inhibitory or stimulatory) (Morgan and Liu 2011). Many of these interactions also occur in a cell type-specific manner. As a rule of thumb, ROS often stimulates the NF-κB pathway in the cytoplasm, but inhibits NF-κB activity in the nucleus. Furthermore, moderate increases in ROS often lead to NF-κB activation, whereas severe increases in ROS may inactivate or decrease NF-κB with corresponding consequences for cell fate. Some of the major effects of ROS on NF-κB signaling can be summarized as follows: (1) ROS may both activate (disulfide bond formation between Cys54 and Cys347, which potentiates dimerization of IKKγ—also called NEMO) or inactivate (oxidation of Cys179 on IKKβ) the IKK complex leading to corresponding effects on downstream targets; (2) ROS have been shown to activate NF-κB through alternative IκBα phosphorylation (phosphorylation of Tyr42 instead of Ser32 and Ser36), which may or may not result in the degradation of IκBα. (3) Lastly, ROS may influence the DNA-binding properties of the NF-κB proteins themselves: Oxidation of p50 on its DNA-binding domain (at Cys62) prevents DNA binding and can be reversed in the nucleus by a Trx1-dependent process involving Ref-1, which enhances NF-κB DNA binding. (4) On the other hand, the phosphorylation of (nuclear) RelA at Ser276 that is influenced by ROS-dependent processes leads to greater NF-κB activation. Additional mechanisms of ROS-mediated NF-κB activation/inactivation include TNFα-induced oxidation of dynein light chain (LC8), which binds to IκBα in the reduced state and prevents its degradation (Jung et al. 2008). Upon oxidation to an intermolecular disulfide, LC8 dissociates and frees the way for IκBα degradation and activation of NF-κB. The oxidation is reversed by the Trx homolog thioredoxin-related protein-14 (TRP14), which is reduced by cytosolic thioredoxin reductase 1 (TrxR1) (Jeong et al. 2004). There is also some evidence for a bidirectional regulation of NF-κB by ER stress (see section “Endoplasmic reticulum (ER) stress”), whereby early ER stress induces activation of NF-κB, whereas in the later phase, UPR signaling inhibits NF-κB (Kitamura 2011), which is likely accounted for by the severity of stress and magnitude of ROS formation. These and many other effects of ROS on NF-κB signaling are discussed in great detail in excellent reviews (Morgan and Liu 2011; Brigelius-Flohe and Flohe 2011). Ca2+-dependent activation of NF-κB has also been demonstrated in certain tissues, in particular brain (Meffert and Baltimore 2005) and the immune system (Rickert et al. 2011). The elevation of [Ca2+]cyt may involve activation of voltage-dependent Ca2+ channels in the plasma membrane or specific immune cell receptor-mediated activation of phospholipase C (PLC)-γ to generate IP3/DAG and the release of Ca2+ from intracellular Ca2+ stores, and activation of a downstream Ca2+-responsive signaling cascade involving CaMKII, calcineurin and/or PKC and leading to the activation of NF-κB.
Cd2+ and NF-κB
Studies investigating Cd2+-induced ROS-dependent regulation of NF-κB signaling have demonstrated its importance for cellular protection and survival, with one notable exception. Cd2+ (≤30 μM for up to 72 h) caused apoptosis of rat lung epithelial cells, which was preceded by the upregulation of oxidant stress genes (glutathione S-transferase-alpha, gamma-glutamylcysteine synthetase and MT-1), activation of redox-sensitive transcription factors AP-1 and NF-κB (which, however, may reflect adaptive and survival responses to Cd2+ stress) and changes in various forms of glutathione (reduced, oxidized and protein-bound), suggesting involvement of ROS in these processes (Hart et al. 1999). Exposure of cultured rat PT cells (WKPT-0293 Cl.2) to Cd2+ (10 μM in 5 % fetal calf serum) caused increased production of ROS and apoptosis, which decreased with prolonged exposure to Cd2+ (up to 72 h), suggesting adaptation to Cd2+ toxicity (Thévenod et al. 2000). Reduction in apoptosis correlated with a time-dependent upregulation of the drug efflux pump multidrug resistance P-glycoprotein (Abcb1) in Cd2+-exposed cells, which was responsible for apoptosis resistance. Both apoptosis and Abcb1 upregulation were dependent on ROS formation because they were abolished by ROS scavengers. Moreover, Cd2+- and ROS-dependent Abcb1 upregulation was caused by activation of NF-κB because ROS scavengers and an IKKα inhibitor prevented both Abcb1 overexpression and proteasomal degradation of IκBα. These data suggested that Abcb1 upregulation, at least in part, provides antiapoptotic protection for PT cells against Cd2+-mediated stress (Thévenod et al. 2000). A subsequent study using cultured rat PT cells overexpressing Abcb1 or Madin-Darby canine kidney cells permanently transfected with human ABCB1 proved that ABCB1 is protective against Cd2+ apoptosis by mediating efflux of proapoptotic ceramides out of cells (see section “Cd2+ and ceramides”) (Lee et al. 2011). These cellular studies demonstrating NF-κB activation by Cd2+ were confirmed in vitro in monocytes (Freitas and Fernandes 2011) and in vivo in kidney of Cd2+-exposed mice (1.2 mg/kg s.c. for 9 weeks) (Fouad and Jresat 2011). A more recent study in murine osteoblast MC3T3-E1 cell lines has confirmed ROS-dependent activation of NF-κB and induction of protective target genes with Cd2+ (≤2.5 μM for 24 h in serum-free medium) (Lizotte et al. 2012). These concentrations of Cd2+ induced progressive depletion of reduced thiol content, stimulated the production of ROS and increased the gene expression of macrophage migration inhibitory factor (MIF), a thiol-containing cytokine with an ability to complex Cd2+; inhibition of NF-κB prevented Cd2+-dependent upregulation of MIF expression and consequently increased Cd2+ cytotoxicity in osteoblasts (Lizotte et al. 2012). In support of these observations, activation of NF-κB (and AP-1 as well as induction of c-Myc and c-Jun) has been associated with malignant transformation of rat liver epithelial cells (TRL 1215) and tumor formation induced by chronic exposure to Cd2+ (1 μM for 28 weeks in 10 % serum) (Qu et al. 2005). Transformed cells showed less ROS formation induced by an acute high dose of Cd2+ (50 μM), suggesting that adaptive survival mechanisms against toxic ROS were mediated by NF-κB. Hence, Cd2+-induced increase in NF-κB activity may promote Cd2+ adaptation and tumorigenesis.
In apparent contrast, Xie and Shaikh (2006a) showed that (1) Cd2+ (20 μM in serum-free medium for 5 h) suppresses basal and TNFα-inducible activity of NF-κB in kidney proximal tubular epithelial cells (NRK-52E) in a concentration-dependent manner; (2) it was associated with downregulation of NF-κB-dependent antiapoptotic gene products cIAP-1 and cIAP-2; (3) antioxidants preserved activity of NF-κB in Cd2+-exposed cells; (4) overexpression of the p65 NF-κB subunit protected cells from Cd2+-induced apoptosis; and (5) suppression of NF-κB rather potentiated apoptosis. The authors concluded that Cd2+-induced apoptosis involves suppression of NF-κB activity which may be mediated by oxidative stress. These data confirmed our study (Thévenod et al. 2000) describing the antiapoptotic effects of NF-κB in Cd2+-exposed renal cells. However, Cd2+-induced ROS formation had opposite effects on NF-κB activity, which may be explained by a difference in the magnitude of ROS formation in both studies (though ROS measurements were not performed in the study by Xie and Shaikh) because of the clearly higher free Cd2+ concentrations in that study (Xie and Shaikh 2006a). In addition, NRK-52E cells are about 5-times more sensitive to Cd2+ than the rat PT cell line (WKPT-0293 Cl.2) used in our studies (F. Thévenod and J.-K. von Sivers, unpublished). NRK-52E cells express intact p53, whereas p53 in WKPT-0293 Cl.2 cells is inactivated (Bork et al. 2010). Interestingly, NF-κB and p53 have adopted opposite strategies to respond to stress and cannot function in the same cell at the same time: On activation of one of these transcription factors, the other is inactivated (Ak and Levine 2010). Hence, activation of p53 to induce Cd2+ apoptosis may also have contributed to the inhibition of NF-κB in NRK-52E cells (Xie and Shaikh 2006a).
A single study has demonstrated Ca2+-dependent NF-κB activation induced by Cd2+ that increased cell proliferation. Misra et al. (2002) showed rapid PLC- and IP3-dependent, Ca2+ mobilization and PKC activation induced by Cd2+ (≤1 μM) in murine peritoneal macrophages (possibly via Cd2+ binding to a G protein-coupled cell surface receptor), which induced activation of NF-κB and resulted in upregulation of genes promoting cell proliferation (Misra et al. 2002).
Wnt
Wnts are evolutionarily conserved secreted Cys-rich proteins, which are encoded by 19 genes in humans (Niehrs 2012). They couple to various receptors and thereby activate different downstream pathways. These pathways have been classified as either canonical (β-catenin-dependent) or noncanonical (β-catenin-independent) signaling pathways. The β-catenin-dependent pathway is the best-characterized pathway. In this pathway, the Wnt proteins signal across the plasma membrane by interacting with seven-span transmembrane serpentine receptors of the Frizzled (Fz) family and with members of the low-density lipoprotein-related protein (LRP) family, such as LRP5/6. The molecular cascade involved in β-catenin-dependent Wnt signaling leads to inhibition of glycogen synthase kinase 3-beta (GSK-3β), which regulates various substrates, including β-catenin. When Wnt proteins bind to the Fz–LRP receptor complex, the cytoplasmic scaffold protein Dishevelled (Dvl) interacts with the heterodimeric receptor complex and co-recruits cytoplasmic transducers. In the canonical pathway, Dvl associates with a β-catenin “destruction” complex, where it inhibits GSK-3β. In unstimulated cells, β-catenin is continuously inactivated by GSK-3β-mediated phosphorylation, ubiquitinated and targeted for proteasomal degradation. In addition to GSK-3β, the destruction complex comprises adenomatosis polyposis coli (APC), axin and casein kinase Iα (CKIα), which also contribute to the inactivation of β-catenin. Hence, upon Wnt activation, cytoplasmic β-catenin is stabilized and enters the nucleus, where it associates with transcription factors, notably T cell factor (TCF) and lymphoid enhancer-binding factor to regulate the transcription of target genes (for an overview of Wnt/β-catenin target genes, see wnt.stanford.edu). This pathway is prominently involved in regulating cell differentiation, proliferation, survival and carcinogenesis (Clevers and Nusse 2012; Thévenod and Chakraborty 2010). Interestingly, by associating with NF-κB subunits, β-catenin can inhibit NF-κB-mediated target gene expression: β-catenin can associate indirectly with p50 and RelA, and this association inhibits NF-κB-mediated DNA binding, trans-activation and target gene expression in human colon and breast cancer cells; moreover, the membrane-bound pool of β-catenin is capable of preventing the transcriptional activity of NF-κB by binding it to the membrane (reviewed in (Thévenod and Chakraborty 2010)). Beside the canonical pathway, there are at least two other noncanonical pathways, the JNK/planar cell polarity (PCP) pathway involved in instrumentation of cell shape changes, movement and asymmetric cell polarities, and a Ca2+ releasing pathway (Niehrs 2012). In the latter, Wnts trigger Frizzled-mediated activation of heterotrimeric G proteins. This activates PLC, which in turn stimulates DAG and IP3 production, Ca2+ release from intracellular stores and activation of effectors such as CaMKII, calcineurin and PKC, which activate the transcriptional regulator nuclear factor associated with T cells (NFAT). This pathway is involved in cancer, inflammation and neurodegeneration (De 2011).
The canonical Wnt signaling is also regulated by ROS (Funato et al. 2006; Funato and Miki 2010). A thioredoxin-related protein, nucleoredoxin (Nrx), governs ROS-stimulated Wnt signaling in a temporal manner. Nrx in the reduced state binds to Dvl. Upon oxidation, Nrx is released and Dvl can associate with the β-catenin degradation complex, where it inhibits GSK-3β and phosphorylation, ubiquitination and degradation of β-catenin. β-catenin moves into the nucleus and activates genes required for proliferation and survival. Recently, upstream signaling of Nrx/Dvl-dependent Wnt/β-catenin signaling has been characterized (Kajla et al. 2012). Wnt treatment of mouse intestinal cells induced production of ROS through NOX1. This NOX1 action was regulated by Rac1 through Wnt-induced activation of the Rac1 guanine nucleotide exchange factor Vav2 by Src-mediated tyrosine phosphorylation. NOX1-generated ROS oxidized and inactivated Nrx, thereby releasing the Nrx-dependent suppression of Wnt-β-catenin signaling through dissociation of Nrx from Dvl. NOX1 siRNA inhibited cell responses to Wnt, such as stabilization of β-catenin, expression of β-catenin/TCF target genes cyclin D1 and c-Myc, and accelerated cell proliferation (Kajla et al. 2012).
Cd2+ and Wnt
With regard to the role of Wnt signaling in Cd2+ toxicity, a recent study showed that chronic exposure of human bronchial epithelial BEAS-2B cells to Cd2+ (≤2 μM in 10 % serum for 2 months) induces carcinogenesis, which was associated with activation of signaling cascades involving upstream ROS formation and downstream signaling mediated by PI3K, Akt, GSK-3β and β-catenin stabilization (Son et al. 2012). ROS formation triggered these effects because they were prevented by transfection with catalase, SOD1 or SOD2. GSK-3β is a downstream effector of the PI3K/Akt pathway, and its activity is inhibited by Akt-mediated phosphorylation at residue Ser9 (Cross et al. 1995). The authors therefore proposed that Akt-dependent phosphorylation of GSK-3β at Ser9 inhibits GSK-3β activity and leads to β-catenin stabilization (Son et al. 2012). Additional roles of ROS in inactivation of GSK-3β, e.g., via oxidation of Nrx and dissociation of Dvl from oxidized Nrx, remain to be demonstrated in Cd2+-exposed cells.
An additional effect of Cd2+ on canonical Wnt signaling involves Cd2+ disruption of the normal association of β-catenin with cadherins and actin at adherens junctions in vitro and in vivo, thus allowing β-catenin to translocate to the nucleus to mimic the canonical Wnt pathway (Thévenod et al. 2007; Chakraborty et al. 2010a, b; Edwards et al. 2013). This results in activation of Wnt target genes c-Jun, c-Myc, cyclin-D1, Abcb1 and markers of epithelial-to-mesenchymal transition (Twist, fibronectin, collagen I), which promote cell proliferation, survival and malignancy. Consequently, in renal PT cells exposed to Cd2+, the multidrug transporter Abcb1 is induced by both NF-κB (see (Thévenod et al. 2000) and section “Cd2+ and NF-κB”) and β-catenin/TCF4 (Chakraborty et al. 2010a, b). Abcb1 mediates evasion of apoptosis by transporting proapoptotic ceramides out of cells (see section “Cd2+ and ceramides”) (Lee et al. 2011). Though the role of ROS (or Ca2+) in eliciting these changes was not directly tested, it is likely that ROS also mediate these alterations in β-catenin distribution and signaling because (1) Cd2+-induced ROS formation increased the expression of proapoptotic ER stress marker CHOP/GADD153/DDIT3 (see section "CHOP"), which binds to TCF4 to inhibit Wnt/β-catenin signaling (Chakraborty et al. 2010a); (2) ROS also disrupt adherens junctional complexes and cellular adhesion in epithelia (DeGennaro et al. 2011; Guntaka et al. 2011; Bailey et al. 2004); and (3) increases in cytosolic ROS and Ca2+ (as well as Cd2+) have been shown to increase E-cadherin cleavage in T47D breast cancer cell lines (Park et al. 2008).
AP-1
AP-1 proteins are transcription factors whose functions involve control of both cell growth and survival or apoptosis (Shaulian and Karin 2002). The AP-1 family consists of several groups of bZIP proteins, including Jun, Fos, Maf and ATF subfamilies. AP-1 proteins form heterodimers and bind to a target DNA sequence. Activation of AP-1 is regulated at both transcript and protein levels. The intracellular levels of c-Jun and c-Fos are controlled mainly by their transcription rates, which are tightly regulated by a variety of stimuli (Shaulian and Karin 2002). MAPKs play a major role in controlling activation of AP-1 proteins through phosphorylation: c-Jun is regulated mainly by JNK and ERK in some cell types; c-Fos is a substrate of ERK, and ATF2 is regulated by JNK and p38 kinases. AP-1 transcription factors are involved in both the induction and prevention of apoptosis, and the exact outcomes are highly tissue and developmental stage specific. For instance, the mechanism by which c-Jun mediates cell survival or death seems to depend on the balance between pro- and antiapoptotic target gene transcription and may be further regulated by p53 and p21 through their cell cycle regulatory activity. Ref-1 modulates gene expression regulated by bZIP proteins, such as Jun, Fos, Maf and ATF. Under oxidative stress, Ref-1 translocates into the nucleus where Ref-1 regulates the activity of bZIP transcription factors by redox mechanisms (see section “Ref-1-Nrf2”) (Ray et al. 2012; Tell et al. 2009). Oxidative stress can also activate AP-1 proteins through phosphorylation by JNK and p38, respectively (Karin and Shaulian 2001). AP-1 proteins then upregulate ARE-mediated expression of antioxidant genes, such as Trx or proteins that enhance GSH synthesis and regeneration, by associating with Nrf2 and Nrf1 and binding with ARE (Venugopal and Jaiswal 1998; Lu 2013). This function may be important in the adaptive response to survive under oxidative stress.
In addition to the regulation of AP-1 by Ca2+-dependent MAPK activation (see sections “ASK1–MAPK signaling” and “ERK1/2”), the four calcium-regulated transcription factors of the NFAT family act synergistically with (MAPK activated) AP-1 (Fos/Jun) proteins on DNA sequences which contain adjacent NFAT and AP-1-binding sites, where they form highly stable ternary complexes to regulate the expression of genes involved in cell survival, apoptosis or differentiation during immune responses (Macian et al. 2001). Ca2+ signaling is critical to NFAT activation because calmodulin (CaM) activates the Ser/Thr phosphatase calcineurin, which dephosphorylates NFAT proteins, thus allowing NFAT nuclear import. NFAT proteins have weak DNA-binding capacity and must cooperate with other transcription factors to induce gene transcription (Macian 2005).
Cd2+ and AP-1
In studies with Cd2+, AP-1 mostly operates as a survival factor. Cd2+ (≤4 μM up to 6 h in serum-free medium) has been shown to activate JNK at 1–6 h (using a phospho-c-Jun antibody at Ser63), rapidly increase c-jun mRNA expression at 1–3 h and promote the binding of AP-1 to DNA at 3–6 h (Hsiao and Stapleton 2004). This process was ROS-dependent because the ROS scavenger carnosol prevented JNK activation. In rat primary mid-brain neuron-glia cultures, Cd2+ induced cytotoxicity (IC50 ~ 2.5 μM after 24 h exposure) and 0.6–1.25 μM Cd2+ for 1 h generated ROS, as determined by 2′,7′-dichlorofluorescein (DCF) signals or electron spin resonance (Yang et al. 2007c). This was associated with AP-1 DNA-binding 6-h posttreatment by gel-shift assay signals, which correlated with increased expression of oxidative stress-related genes 12–24 h after treatment, such as MT, HO-1, glutathione S-transferase-pi and metal transport protein-1 by real-time reverse transcription-PCR, possibly to protect against Cd2+-induced oxidative damage to neurons (Yang et al. 2007c). Prolonged Cd2+ exposure (2 month at nontoxic doses) of mouse epidermal JB6P+ cells, a model for tumor promotion and cellular transformation, increased Ref-1, but not in control JB6P-cells (Yang et al. 2007b). Increased Ref-1 expression was associated with decreased ROS formation. Knockdown of Ref-1 inhibited tumor promoter-induced cell transformation and AP-1 reporter gene activity of JB6P+cells exposed to Cd2+ and increased ROS formation and apoptosis (Yang et al. 2007b). In a subsequent study, gel mobility shift and immunoblot analyses were utilized to determine the distinct AP-1 compositions of the Cd2+ JB6P+ transformants, which showed increased DNA binding of AP-1 complex components c-Jun, JunB, JunD and Fra1 as well as increased JNK and ERK1/2 signaling activities, which may all contribute to a more proliferative phenotype (Yang et al. 2008). When murine embryonic liver BNL CL.2 cells were exposed to Cd2+ (10 μM), ROS formation occurred after 6 h, as determined by DCF fluorescence, and c-Fos and c-Jun expression increased in nuclear extracts after 24 h (Lee and Lim 2011). This was associated with increased expression of matrix metalloproteinase-9 and cell invasion, as determined by a migration assay using Matrigel-coated filters. All these effects were abolished by the presence of an antioxidative 27 kDa glycoprotein from the plant Gardenia jasminoides Ellis (GJE) (Lee and Lim 2011). Malignant transformation of rat liver epithelial cells (TRL 1215) with tumor formation induced by chronic exposure to Cd2+ (1 μM for 28 weeks in 10 % serum) may be triggered by increased activity of AP-1 (and NF-κB) as well as induction of c-Myc and c-Jun, in order to counteract toxic stress induced by ROS (Qu et al. 2005). Finally, several studies have described Cd2+-induced Ca2+ release leading to transcriptional activation of target genes by AP-1 or increased AP-1 DNA binding, but these studies relied on methods for Ca2+ measurements that do not discriminate between Cd2+ and Ca2+, such as BAPTA-AM, Fluo-3, Fluo-4 or Fura-2, and their conclusions need therefore to be taken with caution.
Bestrophin-3
Bestrophins (Best) are members of a family of transmembrane proteins and have been associated with Ca2+-activated Cl− channels in the plasma membrane of epithelial and nonepithelial cells (Hartzell et al. 2008). Bestrophin-1 (Best-1) has also been reported to interact with protein phosphatase 2A and/or to modulate Ca2+ channels. Mutations of Best-1 are linked to Best’s vitelliform macular dystrophy, an autosomal dominant disorder in which lipofuscin accumulates at the retinal pigment epithelium and results in progressive macular degeneration, but the pathology remains unclear (reviewed in Xiao et al. 2010). Recently, human and mouse Best-1 have been found to be expressed in the ER of epithelial cells and were suggested to facilitate Ca2+ release from the ER by acting as a counter-ion pathway, but also to promote dedifferentiation and proliferation of epithelial and fast growing cancer cells (Kunzelmann et al. 2011). However, the exact function of Best proteins remains obscure. Apart from two publications on the role of mammalian Best-3 as a Ca2+-activated Cl− channel for cardiac repolarization in myocytes and contraction of smooth muscle cells (Matchkov et al. 2008; O’Driscoll et al. 2008), there is no further information related to Best-3 function in the current literature. An indication that Best proteins are involved in cell death and cell survival was given by Onuma et al. (2009) who investigated Best proteins in Xenopus embryonic development and noted defects in dorsal axis formation following ectopic expression of xBest-2.
Cd2+ and bestrophin-3
We recently discovered that Best-3 but not Best-1 or Best-2 mRNA was upregulated by Cd2+, which also increases oxidative stress and the UPR (see section “Endoplasmic reticulum (ER) stress”) (Yokouchi et al. 2007; Yokouchi et al. 2008). We reported a novel observation that Best-3 is upregulated by ER stress induced by Cd2+, thapsigargin and tunicamycin (TUN) and contributes to cell survival in kidney proximal tubule cells (Lee et al. 2012). Increase in Best-3 mRNA was signaled through formation of ROS, increase of cytosolic Ca2+ and ERK1/2 phosphorylation. The sources of ROS and Ca2+ were not further investigated. ROS production could be a result of Cd2+ binding to glutathione, NOX activation or inhibition of antioxidative enzymes. A mitochondrial source of ROS is unlikely since mitochondrial dysfunction with Cd2+ was observed only at later time points (>24 h) in these cells (Lee et al. 2006). Inhibition of SERCA pumps by Cd2+ may lead to increased cytosolic Ca2+ (Biagioli et al. 2008; Zhang et al. 1990). Immunofluorescence staining of cultured kidney proximal tubule cells and rat kidney sections located Best-3 to the plasma membrane and intracellular compartments of the proximal tubule, in particular the nucleus. Interestingly, the ER was not labeled with Best-3 indicating that Best-3 does not act from this site. Rather, Best-3 was strongly labeled in the nucleus in the proximal tubule cell line. Knockdown of Best-3 using siRNA increased cell death as indicated by increased sub-G1 phase and poly (ADP-ribose)-polymerase 1 cleavage. These data indicate that Best-3 maintains cell survival even in the absence of stress signals. When ER stress was induced, Best-3 mRNA was upregulated. Furthermore, overexpression of Best-3 attenuated transcription of the proapoptotic transcription factor CHOP induced by ER stress (see section “CHOP”). From these data, we concluded that Best-3 is a survival factor upregulated by ER stress. However, how Best-3 modulates CHOP transcription has yet to be investigated. Preliminary experiments in Best-3 overexpressing cells did not show any change in the level of p-PERK and p-eIF2α (see section “Endoplasmic reticulum (ER) stress”), indicating that Best-3 acts downstream of the UPR (Lee et al. 2012). The protective role of Best-3 in cell death has since been confirmed by others. Intriguingly, Best-3-mediated protection does not seem to be confined to ER stress signaling. Jiang et al. (2013) employed H2O2 as an inducer of apoptosis in smooth muscle cells and found that Best-3 can stabilize the mitochondrial membrane potential to prevent cytochrome c release and caspase activation. However, the target of Best-3 was not clarified. The predominant expression of Best-3 in the nucleus would suggest that Best-3 exerts its cell survival effects from there by influencing gene expression. A very interesting observation was made by Chou and Chan (2011) who investigated potential novel biomarkers for breast cancer. In a secretome analysis comparing nontumorigenic and tumorigenic breast cancer cell lines, they found that larger amounts of Best-3 (>twofold) were secreted from tumorigenic breast cancer cell lines and this was confirmed by immunoblotting. In support of Best-3 as a novel breast cancer biomarker, Best-3 levels were increased by >twofold in plasma samples taken from breast cancer patients (both metastatic and nonmetastatic) compared with healthy donors. It remains to be seen whether Best-3 upregulation, and possibly subsequent secretion, plays a part in the carcinogenic progression caused by chronic exposures to Cd2+ in the kidney and other tissues.
Cellular processes affecting both death and survival decisions
Several cellular proteins, functions and signal cascades, which are targeted by Ca2+ and ROS signals induced by Cd2+, initiate death or survival reactions depending on the recruited molecules, intensity of stress and cellular context.
Metallothioneins
The cysteine-rich Zn2+ and Cd2+ binding proteins, MTs, seem to have an important role in controlling cell death and survival. The mechanisms of transcriptional activation of MT genes are complex: ROS may serve as an upstream signal, which activate ARE/EpRE (see section “Ref-1-Nrf2”) and/or a metal RE (MRE) in the MT promoter region indirectly (reviewed in (Sabolic et al. 2010; Haq et al. 2003). Other described mechanisms for MT induction, such as ERK1/2 or NF-κB signaling (see sections “ERK1/2” and “NF-κB”), are difficult to separate from upstream ROS signaling, or could involve transcriptional regulation mediated by cytokine/stress REs (Haq et al. 2003; Peng et al. 2007). MTs (mainly the most prevalent thionein isoforms MT-1 and MT-2) are known to scavenge Cd2+ (Klaassen et al. 2009; Sabolic et al. 2010) and may also play a role in antioxidant defense (Bell and Vallee 2009; Qu et al. 2013). By these means, MTs contribute to the adaptive and survival response of cells to Cd2+ toxicity (Thévenod 2003; Klaassen and Liu 1998), but they may also promote carcinogenesis by increasing apoptosis resistance of genetically aberrant premalignant cells (Shimoda et al. 2003; Qu et al. 2006) (reviewed in Liu et al. 2009).Ca2+-dependent signaling has also been implicated in the regulation of MT transcription by modulating the activity of the MRE indirectly (Saydam et al. 2002) and has been demonstrated in cells exposed to Cd2+ (Saydam et al. 2002; Shiraishi and Waalkes 1994). Cd2+ causes apoptosis, but MT-3 overexpressing cells show necrosis by Cd2+, which appears to depend on the unique N-terminal sequence of MT-3 (Somji et al. 2006). The mechanism by which MT-3 predisposes cells to necrotic cell death was not investigated further.
Ubiquitin–proteasome system (UPS)
The UPS is the primary cytosolic proteolytic machinery for the selective degradation of various forms of damaged proteins (Ravid and Hochstrasser 2008; Glickman and Ciechanover 2002). In the canonical UPS, both ubiquitin and the 26S proteasome are involved. Substrate proteins of the canonical UPS are first tagged by multiple ubiquitin molecules and then degraded by the 26S proteasome: In the first step, ubiquitin is activated at the C-terminal glycine by the ATP-dependent formation of a thioester at a cysteine of the E1 enzyme. E1 transfers ubiquitin to a cysteine on the ubiquitin-conjugating enzyme E2 and is released from the complex. The ubiquitin-loaded E2 then forms a complex with the E3 enzyme to which the respective substrate is bound. The really interesting new gene (RING) box protein-1 (Rbx1) then targets the substrate for ubiquitination. Ubiquitin can then be passed either directly to a lysine of the RING-bearing E3-bound substrate (Rbx1) or, in case of the homologous to E6AP C terminus-domain bearing E3, to another cysteine on E3, and from there to the substrate lysine. In noncanonical UPS, proteins can be degraded by the 26S or the 20S proteasome without being ubiquitinated (Ravid and Hochstrasser 2008; Glickman and Ciechanover 2002).
Redox modification of proteins is often followed by ubiquitination and selective proteasomal degradation of oxidized and damaged proteins (Shang and Taylor 2011). In mammalian cells, transcription factors are usually present in the cytosol as inactive multi-protein complexes from which they have to be released for gene activation in the nucleus. The activation of the transcription factor requires modification of one or more components of the complex to enable release of the transcription factor and its nuclear import (Muratani and Tansey 2003; Lipford and Deshaies 2003). Accordingly, a redox modification (oxidative modification of Cys or Ser/Thr/Tyr phosphorylation) of a component of a transcription factor is followed by ubiquitination and proteasomal degradation of the same or another component. Hence, in the Nrf2/Keap1 system, oxidant sensing by Keap1 terminates the ubiquitination and degradation of Nrf2 and allows its nuclear translocation and transcriptional activation of target genes (see section “Ref-1-Nrf2”). In the NF-κB system, LC8 is associated with IκB, thereby preventing its degradation and release of NF-κB. Nuclear translocation of NF-κB can take place after oxidative modification and release of LC8 and degradation of the inhibitor IκB (see section “NF-κB”). In the Wnt/β-catenin pathway, GSK-3β phosphorylates β-catenin, thereby facilitating its ubiquitination and degradation and preventing β-catenin-mediated gene expression. Dvl is captured by Nrx. Upon oxidation of Nrx, Dvl is released to inhibit GSK-3β activity. β-catenin is no longer phosphorylated and translocates to the nucleus (see section “Wnt”).
A fully functional UPS is required for cells to cope with oxidative stress, but the activity of the UPS is also modulated by cellular redox status (Shang and Taylor 2011). Mild or transient oxidative stress upregulates the ubiquitination system and proteasome activity in cells and tissues and transiently enhances intracellular proteolysis. For instance, both E1 and E2 activities of the ubiquitin-conjugation system and the levels and activity of the proteasome are increased in response to mild oxidative stress. Thus, increased levels of ubiquitin conjugates in cells seem to be an indicator of mild oxidative stress. Severe or sustained oxidative stress impairs the function of the UPS and decreases intracellular proteolysis. Both the ubiquitin-conjugating enzymes and the proteasome can be inactivated by sustained oxidative stress, especially the 26S proteasome. This can be partly explained by the presence of a cysteine residue in the active sites of E1, E2s, some E3s, and deubiquitinating enzymes. Differential susceptibilities of the ubiquitin-conjugating enzymes and the 26S proteasome to oxidative damage lead to an accumulation of ubiquitin conjugates in cells in response to mild oxidative stress (Shang and Taylor 2011). Consequently, the interaction between the UPS and the cellular antioxidant transcription factors is very complicated and its impact on cell fate therefore varies.
A few studies have investigated the regulation of the UPS by Ca2+ signaling, particularly in neurons. Several reports observed a rapid and dynamic regulation of the proteasome in cultured rat hippocampal neurons by synaptic activity (Djakovic et al. 2009, 2012; Bingol et al. 2010). Blockade of action potentials (APs) inhibited the activity of the proteasome, whereas increasing the frequency of APs increased the activity of the proteasome. The regulation of the proteasome depended upon external Ca2+ entry, in part through N-methyl-D-aspartate (NMDA) receptors and L-type voltage-gated Ca2+ channels, and required the activity of CaMKII. Using in vitro and in vivo assays, CaMKII was shown to stimulate proteasome activity and directly phosphorylate Rpt6, a subunit of the 19 S (PA700) subcomplex of the 26 S proteasome, at Ser120 (Djakovic et al. 2009, 2012; Bingol et al. 2010). In apparent contrast to the previous studies, excitotoxic stimulation with glutamate rapidly decreased ATP levels and the proteasome activity and induced the disassembly of the 26S proteasome in cultured rat hippocampal neurons (Caldeira et al. 2013). Downregulation of the proteasome activity was mediated by Ca2+ entry through NMDA receptors and led to an accumulation of ubiquitinated proteins. On the other hand, proteasomal enzymatic activity in activated human platelets was increased by elevation of [Ca2+]cyt, possibly through activation of Ca2+-dependent downstream effectors like calpain and PKC (Nayak et al. 2011). Accordingly, in human megakaryocytic MEG-01 cells, cyclooxygenase-1 and hematopoietic prostaglandin D synthase were rapidly ubiquitinated and degraded through the UPS in response to elevation of the intracellular Ca2+ level, which could reflect increased UPS activity (Yazaki et al. 2012).
Cd2+ and UPS
In most initial studies, the effect of Cd2+ on the UPS consistently suggested increased UPS activity and protection against Cd2+ toxicity, but controversies have recently arisen though in our opinion the conclusions of these recent studies are not supported by the actual data. The first study describing the effects of Cd2+ on the UPS goes back to 1993, was performed in yeast and demonstrated that the ubiquitin-dependent proteolysis pathway is activated in response to Cd2+ exposure (10–100 μM) to promote survival; indeed, mutants deficient in specific ubiquitin-conjugating enzymes were hypersensitive to Cd2+, possibly because of toxicity of Cd2+-induced formation of abnormal proteins (Jungmann et al. 1993). Subsequent studies in yeast showed that Cd2+ (50 μg/ml for 90 min) increased the degradation of newly synthesized cytosolic proteins ~threefold following oxidative damage of these proteins, and mutations in components of ubiquitin conjugates, proteasome and endoplasmic reticulum-associated protein degradation (ERAD) (see section “ER-associated degradation (ERAD)”) confirmed that Cd2+ increases the activity of the UPS and ERAD system (Medicherla and Goldberg 2008). The conclusions of these studies were subsequently confirmed in mouse neuroblastoma HT4 cells where Cd2+ exposure (5–10 μM for 24 h in serum) caused oxidative stress, which coincided with decreased GSH levels and/or increased protein mixed disulfides, and survival of cells was dependent on normal function of the UPS (Figueiredo-Pereira and Cohen 1999), whereas expression of mutant ubiquitins (Tsirigotis et al. 2001) or mutant proteasome (Li et al. 2004) caused increased Cd2+ sensitivity. In renal PT cells, Cd2+ (5 μM in 5 % FBS for up to 72 h) induced ROS formation and triggered increased protein ubiquitination and proteasomal degradation that correlated with increased cell survival (Thévenod and Friedmann 1999), activation of NF-κB and increased expression of survival genes (Thévenod et al. 2000). In accordance with these observations, Cd2+ (≤50 μM in 10 % FBS for 4–16 h) elicited an increased redox/proteasome-dependent degradation of the HIF-1α protein in hypoxic Hep3B cells (Chun et al. 2000). A study in maize leaf segments indicated that the activity of the proteasome is differentially affected by the Cd2+ concentration: 50 μM Cd2+ increased both trypsin- and peptidylglutamyl-peptide hydrolyzing (PGPH)-like activities of the 20S proteasome, which were probably caused by an increased level of 20S proteasome oxidation (Pena et al. 2007). However, when leaf segments were treated with 100 μM Cd2+, the chymotrypsin- and trypsin-like activities of the 20S proteasome decreased.
In contrast to all these reports, Faustman et al. exposed primary rat Sertoli cell-gonocyte cocultures to Cd2+ concentrations (10–20 μM), which resulted in 50–90 % cell death after 24 h exposure that was apparently caused by disruption of the UPS with accumulation of toxic high-molecular weight polyubiquitinated (HMW-polyUb) proteins (Yu et al. 2008). However, with one notable exception (40 % inhibition of PGPH-like activity with a maximally toxic concentration of 20 μM Cd2+ for 24 h) under all concentrations and exposure times tested, the chymotryptic and PGPH-like activities of the proteasome were either significantly increased or remained unchanged compared to control activities and could be significantly inhibited by specific proteasomal inhibitors. In two subsequent studies, Cd2+ toxicity in NRK-52E rat kidney PT cells or mouse embryonic fibroblasts was proposed to be the consequence of increased p53 accumulation due to disruption of the UPS induced by Cd2+ (Yu et al. 2011; Tokumoto et al. 2011). One study showed accumulation of HMW-polyUb conjugates induced by Cd2+ (5–20 μM in 10 % serum for up to 24 h), suggesting normal ubiquitination and decreased proteasomal degradation (Yu et al. 2011), whereas the other study observed Cd2+ (2–10 μM for 24 h in serum-free medium)-dependent downregulation of the mRNA for the ubiquitin-conjugating enzymes Ube2d1–4 but proteasomal activity was unaffected (Tokumoto et al. 2011). However, in the study by Tokumoto and colleagues, no Ube2d1–4 protein expression was shown, no protein ubiquitination was determined, and the mRNA for other gene products (p53, Mdm2) was downregulated as well, suggesting unspecific effects of Cd2+ on mRNA stability or expression. Overall, the interpretation of these recent studies needs to be taken with caution. Hence, most studies indicate that the activity of the UPS is either not affected or increased by Cd2+, and this may have significant consequences for the regulation of the activity of transcription factors mediating adaptive responses to ROS- or Ca2+-mediated Cd2+ stress.
Bcl-2 proteins
Whether a cell undergoes apoptosis appears to be determined by the balance of Bcl-2 family proapoptotic (e.g., Bax, Bak) and antiapoptotic (e.g., Bcl-2, Bcl-xL) proteins and their effect on the MOM permeability (Youle and Strasser 2008; Bender and Martinou 2013). Following cell stress, direct activator Bcl-2 homology 3 (BH3)-only proteins or p53 induce Bax or Bak to form pores in the MOM to release cytochrome c; Bcl-2, Bcl-xL and Mcl-1 can block this process at the MOM and are, in turn, regulated by derepressor BH3-only proteins (reviewed in Chipuk et al. 2006, 2010). Ca2+ mobilization can directly affect the Bcl-2 protein family, e.g., through dephosphorylation of the BH3-only protein Bad by calcineurin, resulting in its dissociation from 14-3-3, translocation to the mitochondria and cell death (Wang et al. 1999). Ca2+-dependent calpains can mediate proteolysis of Bcl-2, thereby decreasing its ability to protect cells from death and promoting mitochondrial permeabilization and cytochrome c release (Gil-Parrado et al. 2002). On the other hand, Bcl-2 and Bcl-xL may inhibit apoptosis by modulating intracellular Ca2+ signals (reviewed in (Roderick and Cook 2008). Bcl-2 diminishes the magnitude of Ca2+ efflux from the ER by either inhibiting IP3 receptors or by decreasing Ca2+ levels in the ER lumen (Pinton et al. 2001; Chen et al. 2004; Hanson et al. 2008b). Reduced ER Ca2+ levels and Ca2+ signals have also been reported in apoptosis-resistant Bax- and Bak-knockout mouse embryonic fibroblasts (Scorrano et al. 2003). Bcl-2 may reduce ER Ca2+ by inhibiting SERCA pumps (Dremina et al. 2004). Moreover, Bcl-2 appears to decrease the sensitivity of the mitochondrial uptake process as well as increasing the capacity of mitochondria to accumulate more Ca2+ (Roderick and Cook 2008; Hanson et al. 2008b). Bcl-2 may also modulate mitochondrial ROS formation to prevent apoptosis and enhance survival (reviewed in Krishna et al. 2011). Overexpression of Bcl-2 increases mitochondrial oxygen consumption and ROS formation, which blocks death signaling and produces a favorable environment for survival and proliferative signaling: For instance, Bcl-2 increases cytochrome c oxidase (COX) activity and mitochondrial ROS, which exert signaling functions. But under conditions of oxidative stress, Bcl-2 can also reduce COX activity and thereby excessive mitochondrial buildup of ROS and cell death (Krishna et al. 2011).
Cd2+ and Bcl-2 proteins
In Cd2+-exposed cell lines, in general, antiapoptotic Bcl-2 family members are decreased and/or proapoptotic Bax translocate to the mitochondria where Bax can release proapoptotic proteins. These processes involve an increase in [Ca2+]cyt and calpain activation, ROS formation or activation of JNK as upstream activators (Son et al. 2010; Oh et al. 2004; Li and Lim 2007; Papadakis et al. 2006; Lawal and Ellis 2012). The dissociation of the 14-3-3/Bax complex via JNK-dependent phosphorylation of 14-3-3 may constitute a possible mechanism by which JNK promotes the accumulation of active Bax at the mitochondria. However, Cd2+-dependent cytochrome c release that occurs independently of Bcl-2 family proteins has also been reported in fibroblasts deficient in JNK (Papadakis et al. 2006). A reduction in antiapoptotic Bcl-2 proteins (e.g., Bcl-xL) induced by Cd2+ could be the consequence of decreased NF-κB-dependent transcriptional activity caused by dramatic increases in ROS levels, which inactivate or decrease antiapoptotic NF-κB with corresponding consequences for cell fate (Lemarie et al. 2004; Yang et al. 2007a) (see section “NF-κB”). This, however, does not occur when moderate Cd2+-induced ROS levels activate NF-κB and elicit transcription of survival genes, including Bcl-xL. Indeed, Bcl-2 overexpression can protect cells from undergoing Cd2+-induced apoptosis, e.g., by blocking JNK signaling (Ishido et al. 2002; Biagioli et al. 2001; Qu et al. 2007). In support of these observations, Cd2+ exposure (~1 nM for 24–72 h in serum-containing medium) increased proliferation and apoptotic resistance of mouse testicular Leydig cells (TM3), which was associated with increased Bcl-2 and decreased Bax expression (Singh et al. 2009); decreased expression of genes for the maintenance of DNA methylation, DNMT1, and DNA repair, OGG1 and MYH, was also observed and coincided with genomic instability, suggesting that these processes may contribute to malignant transformation. Along those lines, pretreatment of the myelomonocytic lymphoma cell line U937 with 0.1–1 μM Cd2+ for 72 h to stimulate an adaptive survival mechanism prior to acute Cd2+ exposure (50 μM for 12 h in serum) reduced ROS generation and JNK activation and attenuated the reduction in antiapoptotic Mcl-1 and the increase in proapoptotic Bim, Noxa and tBid induced by acute Cd2+ exposure (Cui et al. 2011). In support of these observations, apoptosis-resistant Cd2+ (10 μM for 8 weeks)-transformed RWPE-1 prostate epithelial cells show decreased JNK activity and an increased ratio of Bcl-2/Bax (Qu et al. 2007).
Endoplasmic reticulum (ER) stress
The ER has a highly specialized environment, which is more oxidizing than the cytoplasm and is rich in Ca2+, to provide optimal conditions for the posttranslational modification and folding of proteins that take place there. ER stress develops when ER function is perturbed by accumulation of misfolded proteins, depletion of Ca2+ stores or oxidative stress in the ER lumen (Ron and Walter 2007). A Ca2+ pool with high levels of ER intraluminal Ca2+ is important for the translocation, folding, glycosylation, disulfide bonding and sorting of secretory proteins in the ER through its direct interactions with a variety of ER-resident molecular chaperones and enzymes (Meldolesi and Pozzan 1998). In fact, the ER provides a unique oxidizing folding environment that favors the formation of the disulfide bonds via several protein disulfide isomerases (PDIs) that are required to ensure proper disulfide bond formation and prevent formation of illegitimate disulfide bonds (Malhotra and Kaufman 2007). Thus, protein oxidation in the ER is connected with generation of ROS. Changes in the redox state and the presence of ROS may also affect the Ca2+ homeostasis by modulating the function of ER-based Ca2+ transporters, channels and buffering chaperones and may even disrupt both protein folding reactions and protein chaperone functions (Gorlach et al. 2006; Rao et al. 2004). Moreover, ATP is required for chaperone function to maintain Ca2+ stores and redox homeostasis (Malhotra and Kaufman 2007). It is thus clear to see that perturbations in antioxidative capacity or lumenal ER Ca2+ will most likely result in activation of the unfolded protein response as a result of compromised protein folding ability in the ER from the loss of an optimal environment (Gorlach et al. 2006). The ER has developed an effective defense system against fluctuations in the lumenal environment during periods of cell stress, high protein turnover or high metabolic demands (Santos et al. 2009). In terms of redox status, the ER contains glutathione, thioredoxins and peroxidases to maintain the oxidizing environment required for protein folding but also preventing the generated ROS levels from escalating out of control.
ER stress is sensed by three upstream signaling proteins: protein kinase RNA-like endoplasmic reticulum kinase (PERK), inositol-requiring kinase 1 (IRE1) and activating transcription factor 6 (ATF6), collectively known as the unfolded protein response (UPR). They are found in the lumenal ER membrane where, under ER stress, GRP78, also known as BiP, chaperone binds to unfolded proteins and dissociates from the ER sensors allowing them to homodimerize and activate their downstream signaling cascades (see (Hetz 2012) for a recent review). All three branches of the UPR control the expression of a subset of genes, through either transcriptional repression or activation, through induction or modification of transcription factors, such as an increase in ATF4, cleavage of ATF6 and splicing of Xbox-binding protein 1 (XBP1) mRNA. Initially, the cell tries to prevent accumulation of unfolded proteins by selectively halting transcription through phosphorylation of PERK and the transcription initiation factor eIF2α and to ameliorate the burden of unfolded proteins by initiating ER-associated degradation (ERAD) or autophagy (see sections “ER-associated degradation (ERAD)” and “Autophagy”) to promote cell survival. But chronic or excessive ER stress can lead to the activation of apoptosis mediated by the induction of CHOP (see section “CHOP”), phosphorylation of JNK (see section “ASK1–MAPK signaling”) or activation of caspase-12 (see section “Caspase 12 and caspase 4”) (Tabas and Ron 2011). The importance of ER stress in mediating cellular life and death decisions is demonstrated by its involvement in a number of major modern diseases, including cancer, neurodegenerative and heart diseases. Some cancers have increased expression of GRP78 and GRP94, which could contribute to the ability of a cancer cell to resist proapoptotic challenges (Moenner et al. 2007).
ER stress is particularly susceptible to upstream ROS or Ca2+ signals since the oxidizing and Ca2+-rich ER lumenal environment is critical for optimal protein synthesis and folding. It is not entirely clear where the ROS are coming from as the majority of the literature on oxidative and ER stress is based on pharmacological ROS scavengers, such as N-acetylcysteine or α-tocopherol. Possible sources include mitochondria, NOX enzymes or reduction in antioxidative defense mechanisms, such as glutathione or ROS-metabolizing enzymes. Intriguingly, ER–mitochondria tethering seems to be important in ROS sensing mediated by PERK (see section “BiP/GRP78”). Close contact between mitochondria and the ER suggests that excess ROS signals are passed onto the ER lumen where a change in the oxidizing environment will lead to ER stress that is sensed by PERK (Verfaillie et al. 2012). In the case of Ca2+, accumulating evidence indicates that a partial loss of Ca2+ from ER stores is sufficient to induce the UPR (Mekahli et al. 2011; Paredes et al. 2013), which could be brought about by activation of IP3 receptors or inhibition of SERCA pumps. Disturbances in Ca2+ homeostasis through increased Ca2+ influx from the extracellular surroundings (Chen et al. 2011b) or from mitochondria (Xu et al. 2004) have also been implicated in inducing ER stress. Interestingly, positive signaling loops lead to further increases in ROS and Ca2+ release, and UPR targets, such as CHOP and ERO1α (see section “CHOP”), are involved in amplifying the signals and causing induction of protective mechanisms and/or apoptosis (Higa and Chevet 2012; Li et al. 2009). The sequence of ROS and Ca2+ has not been completely deciphered. In our own work with the ER stressor TUN, we found that ROS are upstream of Ca2+ by using BAPTA-AM and measuring H2O2 levels (Lee et al. 2012). But the signaling loops between Ca2+ and ROS in governing ER stress appear to be far more complicated (Gorlach et al. 2006).
Cd2+ and ER stress
ER stress response genes are highly upregulated in liver and kidney by Cd2+ treatment in vivo (Hiramatsu et al. 2007), and the pattern of genes affected appears to be distinct from other divalent heavy metals (Permenter et al. 2011). While most common toxic heavy metals have overlapping components of activated signaling pathways, e.g., ROS or Ca2+, a very intriguing observation is that the UPR is differentially activated. From a range of heavy metals tested, including mercury and lead, Cd2+ increased GRP78 alongside eIF2α phosphorylation (p-eIF2α) and ATF4, indicating that it is a potent ER stress inducer, whereas GRP78 induction by other metals (mercury, lead, zinc and manganese) did not reach significance (Liu et al. 2006). But notably, mercury significantly increased p-eIF2α and ATF4. Furthermore, DNA microarray analysis demonstrated that Cd2+, nickel and chromium induce distinct subsets of genes. Chaperone genes Dnajb1, Dnajc3, Fkbp5, Hspb8, Cryab, Hspa1a and Hspa1b were induced in a concentration-dependent manner by Cd2+ but to a much lesser extent by nickel or chromium (Permenter et al. 2011). This further demonstrates the specificity of the UPR engagement to Cd2+ and highlights the fact that cellular effects by toxic and carcinogenic heavy metals cannot simply be generalized because of the different stress responses that are elicited.
All three arms of the UPR are activated by Cd2+ (Lee et al. 2012; Kato et al. 2013). Proapoptotic CHOP (see section “CHOP”) is strongly upregulated by Cd2+, and this is mediated by both the PERK and IRE1 arms of the UPR because of increases in their targets, p-eIF2α and XBP1 mRNA splicing, respectively (Yokouchi et al. 2007, 2008; Biagioli et al. 2008). Our laboratory detected phosphorylated PERK (p-PERK) (Lee et al. 2012). Furthermore, Cd2+ increases GRP78 indicating augmented unfolded proteins, but GRP94 has not been as reproducible. Cd2+-induced ER stress can cause phosphorylation of JNK (Yokouchi et al. 2007), caspase activation (Biagioli et al. 2008) and cross talk with mitochondria to release cytochrome c and induce apoptosis (Ji et al. 2011).
What are the consequences of ER stress by Cd2+? Like all other stress responses, this ultimately depends on the concentration and exposure time to Cd2+. Acute exposures to Cd2+ may give rise to a small load of misfolded proteins that are safely degraded or correctly folded within a given time frame and the UPR is minimally engaged. The cells recover and survive the stress by Cd2+ (Hiramatsu et al. 2007) (see section “ER stress and survival”). However, prolonged exposures to Cd2+ augment the unfolded protein load considerably and the UPR is continuously at work to try and ease this load. When the unfolded protein load cannot be reduced with a given time frame, apoptosis pathways are brought into play. Of particular importance are the downstream apoptosis-inducing proteins CHOP, JNK and caspases (see section “ER stress and cell death”).
With Cd2+, several laboratories could attenuate ER stress and apoptosis by applying a pharmacological antioxidant or by overexpressing ROS-metabolizing enzymes (Yokouchi et al. 2008; Kitamura and Hiramatsu 2010; Chakraborty et al. 2010a; Lee et al. 2012; Wang et al. 2012). Yokouchi et al. further defined superoxide anion (O ·−2 ) as the responsible ROS for inducing ER stress by using the superoxide generator menadione and cells overexpressing MnSOD. Interestingly, peroxynitrite anion (ONOO−) and hydrogen peroxide (H2O2) did not increase ER stress though H2O2 induced apoptosis (Yokouchi et al. 2008; Kitamura and Hiramatsu 2010). The involvement of disturbed Ca2+ homeostasis by Cd2+ in eliciting ER stress has been clearly demonstrated by Biagoli et al. (2008) using the aequorin Ca2+ indicator rather than EGTA-based compounds such as BAPTA-AM or Fura2-AM, which also bind Cd2+ ions: ER stress induction could be correlated with inhibition of the SERCA pump in cells exposed to 15 μM Cd2+ for 12 h.
ER stress and cell death
CHOP
At the nexus of ER stress and downstream cell death pathways is C/EBP homologous protein (CHOP), also known as growth arrest- and DNA damage-inducible gene 153 (GADD153), an apoptosis activating protein. CHOP is transcribed from the DNA damage-inducible transcript 3 (DDIT3) gene and has two known functional domains: an N-terminal transcriptional activation domain and a bZIP domain. The bZIP domain contains the DNA-binding region and is required for CHOP’s apoptosis-inducing abilities. The promoter region of CHOP contains at least two ER stress response elements and one amino acid regulatory element. CHOP is ubiquitously expressed and present at low levels in the cytosol under physiological conditions but is one of the highest inducible genes during ER stress (Okada et al. 2002). Exerting its actions through ATF4, ATF6 and XBP1, the UPR induces direct upregulation of CHOP through increased promoter binding during prolonged ER stress. The induction of CHOP is almost absent in PERK null cells indicating that the PERK-eIFα-ATF4 arm of the UPR is dominant in augmenting CHOP levels. Despite the aim of the PERK-eIF2α pathway to reduce global protein translation, selective CHOP transcription is permitted based on a ribosomal bypass mechanism (Palam et al. 2011). Further regulatory mechanisms for CHOP transcription have recently been described in the form of microRNAs. miR-211 is upregulated by PERK and suppresses CHOP transcription by targeting the promoter, where it increases histone methylation and represses CHOP expression (Chitnis et al. 2012). The differential regulation of CHOP by PERK could be related to the extent of the ER stress signal. During low ER stress signals, which do not activate ER stress-induced cell death, CHOP levels are kept low by miR-211, whereas prolonged ER stress initiates cell death by activating CHOP transcription. Posttranslational modification of CHOP by p38 MAPK-mediated phosphorylation is required for CHOP activation, accumulation in the nucleus and transcriptional activity (Wang and Ron 1996).
The proapoptotic effects of CHOP have been well studied and are a result of CHOP acting as a transcription factor to modulate genes involved in cell death and survival. Based on previous observations that CHOP translocates to the nucleus from the cytosol when stress is applied (Chiribau et al. 2010; Lorz et al. 2004), a recent study could demonstrate that CHOP accumulates not only in the nucleus upon stress by TUN but also increases in the cytosol where it exerts its effects in an indirect manner (Jauhiainen et al. 2012). Furthermore, CHOP appears to have distinct functions depending on its localization: for example, in the nucleus, CHOP transcriptionally regulates gene expression, whereas in the cytosol, CHOP predominantly inhibits cell migration. However, in overexpressing cells, both nuclear and cytosolic CHOP modulate genes involved in cell death, cell proliferation and cellular development though they are distinct subsets of genes (Jauhiainen et al. 2012).
Transcriptional targets of CHOP include anti- and proapoptotic members of the Bcl-2 family (Bcl-2, Bim, Bax, Bad), oxidative stress (ERO1α, SODs, glutathione synthesis), metabolic (Ppara, Cebpa, Srebf1) and protein synthesis genes (GADD34, aminoacyl-tRNA synthetases, initiation factors). The majority of genes are upregulated by CHOP, but a few genes, such as Bcl-2 or SODs, are repressed. Forced CHOP expression does not always induce apoptosis indicating that further factors are required. Rather, CHOP sensitizes cells to ER stress signals. In our own experiments, CHOP overexpression in renal PT cells did not alter cell viability when compared to empty vector controls. Furthermore, Cd2+ toxicity was unchanged in CHOP overexpressing cells further supporting the hypothesis that additional factors are required for CHOP activity (Chakraborty et al. 2010a). It is well described that ATF4 is the dominant downstream UPR arm that induces CHOP. The laboratory of Randal Kaufman showed recently that target genes exhibit maximum upregulation when both ATF4 and CHOP are overexpressed and ER stress-induced apoptosis by TUN was absent in Atf4−/− and Chop−/− mice. Moreover, ATF4 and CHOP can interact with each other. Thus, it appears that CHOP works in synergy with ATF4 to transcriptionally modulate target genes (Han et al. 2013). The downstream CHOP response following ER stress varies depending on the cell type and stress signal. For example, together with ATF4, CHOP increased protein synthesis of genes causing protein overload that led to stress and apoptosis, whereas proapoptotic Bcl-2 family members were not augmented in TUN treated mouse embryonic fibroblasts (Han et al. 2013). In contrast, Bcl-2 expression led to replenishment of GSH and a reduction in levels of ROS, and it protected cells from ER stress-induced cell death in A94 cells, a fibroblast cell line with constitutive CHOP and c-Myc expression (McCullough et al. 2001). Furthermore, additional pathways contributing to apoptosis signaled through CHOP are emerging, such as the ERO1α-IP3 receptor-Ca2+ pathway, which activates CaMKII, calpains and subsequently caspase 12 (see section “Caspase 12 and caspase 4”) (Li et al. 2009), suppression of ERK (Liao et al. 2013) or modulation of Mdm2-p53 (Engel et al. 2013) and p21/Waf (Mihailidou et al. 2010).
Cd 2+ and CHOP
Generally, CHOP is induced by Cd2+ in a concentration and time-dependent manner regardless of cell type and has been investigated in vitro in renal PT cells (Chakraborty et al. 2010a; Lee et al. 2012; Komoike et al. 2012), neuroblastoma cells (Kim et al. 2013), lung fibroblasts (Lim et al. 2010), mesangial cells (Wang et al. 2009) and in vivo in the mouse testes (Ji et al. 2011, 2012) and placenta (Wang et al. 2012). High concentrations of Cd2+ were used in the in vivo studies (2–4.5 mg/kg Cd2+, injected intraperitoneally), which are more representative of acute toxicity, and CHOP levels were examined after 24 h. A two–fourfold increase in CHOP was associated with apoptosis occurrence and could be attenuated by the antioxidant vitamin C in the testes. In the cell lines, at low concentrations of 10–15 μM Cd2+, CHOP is increased after 12 h, whereas higher Cd2+ concentrations of 20–30 μM induce CHOP already after 3–6 h and is preceded by ER stress as evidenced by eIF2α phosphorylation or GRP78 increase. One exception described an induction of CHOP without a concomitant increase in GRP78. Rather, DNA damage was responsible for the marginal induction of CHOP induced by Cd2+ (LC50–50 μM) in human liver carcinoma cells (HepG2) cells (Tchounwou et al. 2001), which pertains to its initial discovery as a growth and DNA damage gene. ER stress and CHOP induction by Cd2+ is linked to upstream ROS production and/or disturbances in Ca2+ since antioxidants (ascorbic acid, α-tocopherol, catalase, N-acetylcysteine) and the Ca2+ chelator, BAPTA-AM, could attenuate ER stress and CHOP induction (see for instance (Chakraborty et al. 2010a; Ji et al. 2012; Kim et al. 2013; Wang et al. 2009, 2012; Yokouchi et al. 2008). Of note, data in Cd2+ studies employing N-acetylcysteine, Fura2-AM or BAPTA-AM should be taken with caution since these compounds are also capable of chelating Cd2+ and can produce false-negative data. Downstream targets of CHOP that have been shown to be activated or increased by Cd2+ include Bak (Kim et al. 2013), cytochrome c release (Ji et al. 2011), caspase-12 and caspase-3 (Wang et al. 2009), and JNK phosphorylation (Yokouchi et al. 2007; Komoike et al. 2012; Ji et al. 2011).
How CHOP acts under Cd2+ stress has yet to be investigated. Despite the fact that ATF4 is crucial in many cases for CHOP transcriptional activity, the levels of ATF4 with relationship to CHOP activity in response to Cd2+ has not been determined though the upstream PERK-eIF2α phosphorylation status has been clearly demonstrated (Lee et al. 2012; Yokouchi et al. 2007). However, ATF4 is not only responsible for apoptosis execution in concert with CHOP but also has a considerable role in initiating cell survival programs, such as autophagy, to combat stress signals. Komoike et al. (2012) highlighted this protective role of ATF4 when the percentage of trypan blue positive cells was exacerbated by knocking down ATF4 in renal HK-2 cells exposed to 20 μM Cd2+ for 24 h. CHOP may also interact with other transcription factors to regulate the subset of genes to be activated or repressed. Our laboratory found that CHOP is co-immunoprecipitated with the Wnt/β-catenin target TCF4 under Cd2+, but although this was insufficient to block Wnt signaling, it may have consequent effects on the transcription of other target genes (Chakraborty et al. 2010a).
Caspase 12 and caspase 4
Caspases have long been known to be intimately involved in the apoptosis cell death program where the inactive proenzymes are activated by cleavage in an orchestrated sequence of events. The mammalian caspase family comprises 14 members with caspases-3, -6, -7, -8, -9 and -10 classically seen as the apoptotic members. However, this segregation is not as clear as once thought. These and other caspase family members have been shown to exhibit nonapoptotic functions in the immune system and in cell proliferation (Kuranaga and Miura 2007). While caspases-3, -6, -7 and -9 are found as inactive procaspases in the cytosol and caspases-8 and -10 are associated with death receptor signaling, the laboratory of Junying Yuan discovered the presence of caspase-12 on the ER where it was responsible for executing apoptosis specifically as a result of ER stress by unfolded proteins or Ca2+ disturbance (Nakagawa et al. 2000). In humans, caspase-12 is a pseudogene that has been silenced by mutations during evolution and it is thought that human caspase-4 is the counterpart of murine caspase-12 (Hitomi et al. 2004; Fischer et al. 2002). In the study by Nakagawa and Yuan (2000), cycloheximide, TNF and anti-Fas had no effect on caspase-12 cleavage. But how does caspase-12 sense changes in the ER lumen from its localization on the cytoplasmic side of the ER? Nakagawa and Yuan found that the Ca2+-activated protease calpains were activated following Ca2+ disturbance and cleave caspase-12 at two sites. Interestingly, a calpain cleavage site was also found in the antiapoptotic Bcl-2 family member, Bcl-xL, suggesting that its antiapoptotic effect is counteracted to ensure apoptosis predominates. Conversely, it has been postulated that caspase-7 translocates from the cytosol to activate caspase-12 in the ER (Rao et al. 2001). Further, as a counteracting mechanism, GRP78/BiP forms a complex with caspase-12 preventing its cleavage and release from the ER (Rao et al. 2002). Caspase-12 goes on to activate effector caspases for apoptosis execution. The relevance of caspase-12 in executing ER stress-induced apoptosis has, however, been contested due to a small or lack of reduction in apoptosis in fibroblasts from caspase-12 null mice and B16/B16 melanoma cells (Kalai et al. 2003; Lamkanfi et al. 2004; Nakagawa et al. 2000).
Cd2+ and caspase-12
Only a few studies have investigated caspase-12 in Cd2+-treated cells. Caspase-12 was activated by Cd2+ as determined by cleavage of procaspase-12 (Biagioli et al. 2008). Biagoli and colleagues demonstrated correlation of caspase-12 activation with ER stress in the form of XBP1 mRNA splicing and lowered refilling of ER Ca2+ using ER-targeted aequorin in NIH 3T3 fibroblasts, whereas Wang et al. (2009) used the EGTA-based and Cd2+-chelating compound BAPTA-AM to show that Ca2+ is required for Cd2+-induced CHOP elevation and caspase-12 activation in mesangial cells.
ER stress and survival
BiP/GRP78
The HSP70 homolog glucose-related protein 78 (GRP78), also known as binding immunoglobulin protein (BiP), is encoded by the Hspa5 gene and has a major role in the regulation of ER stress and mediating activation of the UPR. GRP78 normally occupies the lumenal portion of the transmitters of ER stress, PERK, ATF6 and IRE1, maintaining them in an inactive state. As the number of misfolded and unfolded proteins accumulates in the ER, they sequester GRP78 in an ATP-dependent manner in an attempt to properly fold with assistance from GRP78. When GRP78 becomes dissociated from PERK, ATF6 and IRE1, this allows the UPR to be engaged. Furthermore, GRP78 is transcriptionally upregulated through the action of ATF4 and ATF6 as a survival mechanism to reduce the load of misfolded proteins and prevent prolonged UPR. By increasing chaperone assistance, the likelihood of successful protein folding is augmented. Once the number of misfolded proteins decreases, GRP78 is free again to associate with the lumenal domains of the UPR proteins, returning them to their inactive states and preventing the activation of proapoptotic signaling pathways. Unsurprisingly, introduction of GRP78 prevents apoptosis induced by ER stress. Though it is becoming apparent that GRP78 functions extend outside of the ER to the nucleus, mitochondria and even extracellular space (Ni et al. 2011), GRP78’s antiapoptotic actions come presumably from within the ER.
Cd 2+ and GRP78
GRP78 is the quintessential ER stress indicator used in studies with Cd2+. Within 4–6 h, GRP78 mRNA and protein is strongly increased by 10–25 µM Cd2+, which can occur in the absence of apoptosis, and is maintained for up to 24 h (Lee et al. 2012; Liu et al. 2006; Yokouchi et al. 2007). Phosphorylated PERK, p-eIF2α and ATF4 activation were observed at earlier time points (2–3 h) indicating that increased GRP78 is a result of ATF4 transcriptional activity (Lee et al. 2012; Liu et al. 2006). Cd2+-treated mice (<2.0 mg/kg, single dose, intraperitoneally) exhibited increased GRP78 levels (up to threefold) in the testes after 24 h that correlated with p-eiF2α, XBP1 splicing, p-JNK, CHOP induction and apoptosis (Ji et al. 2011). Similarly, in placentas collected 24 h after injection of pregnant mice with 4.5 mg/kg Cd2+ (single dose, intraperitoneally) on gestational day 9, GRP78, p-eIF2α, CHOP and apoptosis were induced by up to threefold (Wang et al. 2012). In line with GRP78’s known protective effect, introduction of GRP78 protects cells against apoptosis induced by Cd2+ (Yokouchi et al. 2007) or GRP78 knockdown by siRNA exacerbates Cd2+ apoptosis (Liu et al. 2006).
ER-associated degradation (ERAD)
Analogous to the cell cycle, every protein synthesized in the ER is subjected to a quality control checkpoint to assess its folding status. If the nascent protein is misfolded or has not yet reached its native structure, it is pulled aside and given a period of time to correctly fold into its native structure. Should the protein fail to do so, it will be retrotranslocated to the cytosol, marked for degradation through ubiquitination by integral E3 ligases and subsequently delivered to the 26S proteasome in the cytosol (see section “Ubiquitin-proteasome system (UPS)”). Through a combination of genetic mutations and mistakes in transcription and translation, a significant number of newly synthesized proteins in the ER never reach their native states (Brodsky 2012). Furthermore, significant proportions of proteins with slow, cumbersome and complicated folding procedures, which under the right circumstances could have been folded correctly, are overzealously destroyed by ERAD, of which a classical example is the cystic fibrosis transmembrane conductance regulator (CFTR) (Ward et al. 1995).
ER chaperones and chaperone-like proteins are essential in overseeing the protein folding pathway. Key members are the HSP70s, which includes BiP/GRP78, and lectins. While BiP/GRP78 directs unglycosylated proteins to the proteasome, glycosylated ERAD substrates are recognized by ER mannosidase and ER-degradation-enhancing α-mannosidase-like protein 1 (EDEM1). Interactions with calnexin and calreticulin alongside reglucosylation prolong folding attempts and facilitate successful protein folding. Retrotranslocation of ERAD substrates, regardless of whether they are soluble substrates or integral proteins, requires the AAA (ATPase Associated with diverse cellular Activities)-ATPase, known as Cdc48 in budding yeast or p97 in mammals (Hampton and Sommer 2012). The driving force, energy requirements and exit channels out of the ER for retrotranslocation remain unknown.
Cd 2+ and ERAD
To date, only two studies have investigated potential effects of Cd2+ on the ERAD machinery. Using Cd2+ as an inducer of oxidative stress, newly synthesized proteins were shown to become more sensitive to degradation by ERAD in yeast (Medicherla and Goldberg 2008). Further, Adle and colleagues demonstrated that Cd2+ prevents the ERAD-mediated degradation of Pca1, a Cd2+ exporter, in yeast as a protective mechanism to prevent Cd2+ toxicity (Adle et al. 2009). Interestingly, the N-terminal degron of Pca1, the peptide sequence that directs the starting point of degradation, contains potential metal sensing residues. Cd2+ was found to bind to the N-terminal domain of Pca1, inducing a conformational change, such that it is no longer recognized by ERAD machinery and evades degradation. It remains to be seen whether this mechanism of direct metal binding to a protein degradation sequence contributes to Cd2+-induced ER stress by preventing degradation of misfolded proteins or contributes to Cd2+ cell death and survival decisions by creating an imbalance of synthesis/degradation of anti- and proapoptotic proteins.
Autophagy
Autophagy is a process of self-eating. By degrading intracellular components, damaged proteins and organelles, the cell can increase its chances of survival during stress or nutrient starvation conditions and works complementary to ERAD (Mizushima and Komatsu 2011). The formation of double-membrane-bound vesicles called autophagosomes is regulated by a number of proteins and complexes including mTOR, Beclin-1, autophagy (Atg) proteins and Class III PI3K (Yang and Klionsky 2010). Autophagosomes “engulf” cytosolic contents, which subsequently fuse with lysosomes to form autophagolysosomes that degrade the engulfed contents. Autophagy can be detected by: (1) the conversion of microtubule-associated protein 1A/1B-light chain 3 (LC3) from LC3-I to its lipidated form LC3-II or the redistribution of LC3 from cytosol to puncta; (2) uptake of monodansylcadaverine or acridine orange into autophagolysosomes; and (3) changes in autophagy-specific proteins, e.g., increase in Beclin-1. Persistence of autophagy can lead to cell death although cell death is not always a direct consequence of autophagy. In fact, the existence of autophagic cell death has been contested (Denton et al. 2012).
Upstream, autophagy can be regulated by ROS (Li et al. 2012; Scherz-Shouval and Elazar 2011) and Ca2+ release (Decuypere et al. 2011a). With a few exceptions, the general consensus is that ROS are inducers of autophagy. The source of ROS appears to be mitochondria (Chen et al. 2007), whereas NOX is involved in antimicrobial autophagy (Huang et al. 2009). However, starvation or rapamycin-induced autophagy in HeLa cell-induced JAK2/STAT3 activation seemed to be linked to NOX activation because pharmacological inhibition of NOX with DPI or knockdown of the NOX subunit p22-phox abolished STAT3 activation suggesting that NOX enzymes could be a source of ROS in autophagy of cancer cells (Yoon et al. 2010). By increasing autophagy, oxidative damaged proteins are degraded, which are thought to be delivered into autophagosomes by the autophagy cargo protein, p62. The regulation of autophagy by ROS occurs at both the transcriptional and nontranscriptional levels. Important redox-reactive transcription factors are HIF1, p53, Nrf2 and Foxo3. By modifying the expression or activity of autophagy-related proteins, autophagy or disinhibition of autophagy is increased. For example, Bcl-2 that is complexed to Beclin-1 is sequestered by increased BNIP3 and Nix expression (Bellot et al. 2009; Burton and Gibson 2009) so that Beclin-1 is released to induce autophagosome formation. Oxidative stress can also act directly on AMPK leading to disinhibition of mTOR and increased autophagy (Scherz-Shouval and Elazar 2011).
In contrast to the straightforward mechanism of ROS-mediated activation of autophagy, [Ca2+]cyt is more promiscuous and can activate as well as inhibit autophagy (Cardenas and Foskett 2012; Decuypere et al. 2011a). Inhibition of autophagy by [Ca2+]cyt has mainly been demonstrated through knockout or blockade of the IP3 receptor (IP3R), which augments autophagy occurrence. It has been suggested that IP3Rs regulate autophagy through binding of Bcl-2 and Beclin-1 in a multimeric complex, which also requires the nutrient-deprivation autophagy factor 1 (NAF-1) to provide the spatial capacity for Bcl-2 to antagonize Beclin-1 without affecting agonist-activation of IP3R and Ca2+ release (Vicencio et al. 2009; Chang et al. 2010). This, however, could not be confirmed in IP3R triple knockout cells expressing mutated IP3Rs with no channel function where autophagy markers were similar to the controls (Cardenas et al. 2010). A further model postulates that because mitochondria require a constant source of Ca2+ provided by IP3R opening, loss of IP3R function results in decreased ATP production, AMPK activation and autophagy induction (Cardenas and Foskett 2012).
Ca2+ also activates autophagy. Some studies have employed BAPTA-AM to chelate Ca2+ released by thapsigargin and could show that autophagy induction is reduced (Decuypere et al. 2011b; Lam et al. 2008; Sakaki et al. 2008). In addition to its antiapoptotic function, Bcl-2 has a key role in regulating ER Ca2+ homeostasis. By lowering ER Ca2+, agonist-induced Ca2+ fluxes are prevented, and therefore, downstream autophagy cannot be executed (Hoyer-Hansen et al. 2007). Similarly, the IP3R has a dual function: It is not only involved in Ca2+ release to inhibit autophagy, but has also been demonstrated to induce autophagy. Taking these opposing roles of IP3Rs in autophagy, it has been hypothesized that IP3R acts as a pivot in governing autophagy induction: mitochondria of normal cells are supplied with Ca2+ provided by IP3R opening to fuel ATP production and prevent autophagy, whereas nutrient-starved cells use IP3Rs for Ca2+-induced autophagic flux (Parys et al. 2012).
In spite of the fact that UPR proteins are not required for autophagy induction (Sakaki et al. 2008), numerous studies evidence autophagy activation as a direct consequence of ER stress. The definitive molecular bridge between the two processes has yet to be elucidated though there have been postulations from various groups. The laboratory of Randalf Kaufman demonstrated the requirement of PKCΘ in ER stress-induced autophagy, which was independent of the UPR and amino acid starvation (Sakaki et al. 2008). In other work, the laboratory of John Reed revealed the ER membrane protein Bax inhibitor-1 (BI-1) as an IP3R-dependent autophagy promoter (Sano et al. 2012). By reducing steady-state levels of ER Ca2+ via IP3Rs, BI-1 influences mitochondrial bioenergetics. The mitochondria receive less Ca2+ that is required for functioning of the tricarboxylic acid cycle; oxygen consumption is reduced, impacting cellular ATP levels and stimulating autophagy. Similarly to Sakaki et al., the UPR was not involved in induction of autophagy by BI-1. In contrast, the data from Castillo et al. (2011) describe IRE1 regulation by BI-1 in the suppression of autophagy. The IP3Rs have been proposed as a fourth arm of the UPR, which, like the canonical UPR branches, is regulated by GRP78. During initial ER stress, GRP78 is replaced by endoplasmic reticulum resident protein 44 (ERp44), also known as Trx domain-containing protein 4 (TXNDC4), which decreases IP3-induced Ca2+ release to induce at first autophagy and promote survival, but as ER stress is prolonged, ERO1α is upregulated in a CHOP-dependent manner, yielding H2O2 as a by-product, leading to hyperoxidation of the ER and IP3Rs and dissociation of ERp44, which causes increased Ca2+ release and excessive Ca2+ transfer to mitochondria, culminating in cell death (Kiviluoto et al. 2013). A very recent study suggests that the ER-located transmembrane 208 (TMEM208) protein is integral in controlling and linking ER stress and autophagy. Using gene expression modification experiments, the authors found that TMEM208 negatively regulates ER stress and autophagy (Zhao et al. 2013). Though changes in proapoptotic CHOP were observed, which indicate that the UPR was engaged, upstream regulation of TMEM208 by UPR components was not tested.
The transduction from the UPR to the autophagic machinery has been postulated to be mediated by IRE1/XBP1 regulation of the SLC33A1/AT-1 transporter, which supplies the ER with acetyl-CoA for protein acetylation, and the acetylation status is sensed by ER-residing Atg9A in the ER that in turn modulates autophagy activation (Pehar et al. 2012). The authors hypothesize that high acetyl-CoA levels inhibit autophagy through acetylation of Atg9A but lead to the formation of abnormal protein aggregates, whereas low acetylation of Atg9A induces autophagy with consequent cell death. However, for fine control of autophagy induction, a middle ground must exist between these two extremes. To further complicate matters, additional molecular bridges between the UPR and autophagy have been reported: The E3 ligase TRIM13 is stabilized during ER stress and governs autophagy through interaction with the ubiquitin-binding scaffold protein p62/SQSTM1 (Tomar et al. 2012); the UPR’s opposite regulation of the survival kinase Akt (depending on the duration of stress) influences the mTORC1 complex, which inhibits autophagy when activated (Appenzeller-Herzog and Hall 2012); direct binding of spliced XBP1 mRNA to the Beclin-1 promoter region (Margariti et al. 2013).
While autophagy may prevent ER stress-associated cell death as part of an early adaptive and survival response (Ogata et al. 2006), there is also evidence that autophagy is invoked as a means of killing cells when ER stress is prolonged and substantial (Ding et al. 2007; Ullman et al. 2008). The study by Ding et al. (2007) shows a differential role of autophagy in promoting survival of cancer cells or death of nontransformed cells, which may be related to the level at which ER stress can be compensated. Similarly, Ullman et al. (2008) demonstrate that autophagy has opposite effects on cell fate in response to ER stress in apoptosis-competent cells in which autophagy serves as a survival mechanism, and in apoptosis-deficient cells that utilize autophagy as a means to promote nonapoptotic cell death. Overall, these observations indicate that autophagy can contribute to ER stress-induced cell death depending on a cell type-specific context. Factors that are able to direct autophagy from a survival to a death pathway have been recently reported. JNK is involved in autophagic cell death, but not in autophagy-induced cell survival (Shimizu et al. 2010). Furthermore, the tumor suppressor DAPk (calmodulin-regulated serine/threonine kinase Death-Associated Protein kinase) has been proposed to convert autophagy from a cell survival mechanism to one for the initiation of cell death (Bialik and Kimchi 2010). Several autophagy-related (Atg) proteins have been implicated in apoptosis, such as Beclin-1, Atg4D and Atg5, which function in autophagy in their unmodified form, but also have a role in apoptosis after cleavage by calpains or caspase 3 (Betin and Lane 2009; Djavaheri-Mergny et al. 2010; Madden et al. 2007; Yousefi et al. 2006). Thus, it is likely that autophagy plays a dual role in determining cell fate, depending on specific cell types and stimuli.
Cd2+ and autophagy
Cd2+-induced autophagy has been shown to be mediated via ROS formation, GSK-3β, AMPK, p38 or ERK activation and decreases in mTOR, PARP, ATP and Ca2+ signaling (though Cd2+ binding BAPTA-AM was employed) as indicated by the presence of LC3-II, formation of double-membrane autophagolysosomes or increase in Atg genes (reviewed in Thévenod and Lee 2013). The consequence of autophagy induction by Cd2+ remains unclear. In some cases, autophagy by Cd2+ appears to suppress apoptosis execution because changes in autophagy markers were observed in the absence of apoptosis in low Cd2+ (<10 μM)-treated rat kidneys in vivo after 3–5 days (Chargui et al. 2011), hematopoietic stem cells after 48 h (Di Gioacchino et al. 2008) and kidney proximal tubule cells after 1 h (W.-K. Lee and F. Thévenod, unpublished data). This has lead to the proposal of autophagy as an early biomarker of Cd2+ toxicity (Chargui et al. 2011). However, the outcome (death versus survival) following autophagy is currently debated. A handful of studies have evidenced apoptotic cell death as a direct consequence of autophagy induction, whereas others showed autophagy as a protective mechanism and inhibition of autophagy exacerbates cell death. Kato and colleagues found that rapamycin, an inhibitor of mTORC1 that is a negative regulator of autophagy, could prevent 10 μM Cd2+-induced cell death in rat kidney NRK-52E cells after 24 h suggesting that autophagy is protective against Cd2+-induced cell death (Kato et al. 2013). These data are apparently supported in two reports by Chen and coworkers in neurons exposed to Cd2+ (Chen et al. 2008c, 2011a). The authors postulated that Cd2+-induced ROS formation activates mTOR, which promotes cell death, whereas rapamycin, an inhibitor of mTOR, is protective. However, downregulation of mTOR by RNA interference was barely protective (Chen et al. 2008c). Furthermore, in the second study, cell viability was not determined; rather, measurements of intracellular Ca2+ with Fluo-3 were used as an indicator of cell death that most likely detected changes in cytosolic Cd2+ concentration (Chen et al. 2011a). In contrast, our laboratory could not reproduce the data of Kato et al. (2013). Using the same concentration of rapamycin (100 nM), Cd2+-induced cell death in NRK-52E cells remained unaffected in the presence of rapamycin after 24–72 h, whereas 3-methyladenine (3-MA), which blocks autophagy by inhibiting autophagosome formation, was protective suggesting that cell death is a consequence of autophagy (J.-K. von Sivers, W.-K. Lee, and F. Thévenod; unpublished data). This is supported by observations from other studies where Cd2+ toxicity was attenuated by 3-MA (Chiarelli et al. 2013). Moreover, Cd2+-induced autophagy in mesangial cells appears to work alongside the classical intrinsic apoptosis mitochondrial signaling pathway to induce cell death, further linking autophagy induction with Cd2+-induced cell death (Wang et al. 2008b). The discrepancy in all these data cannot be explained by concentrations of Cd2+ or exposure times since all studies used Cd2+ in the range of 10–25 μM and observed autophagy within 1–3 h. Thus, the relationship between autophagy and cell death by Cd2+ awaits clarification.
A link between ER stress induced by Cd2+ and downstream autophagy induction may exist but has not been definitely shown. Lim et al. (2010) showed that ER stress and autophagy induction followed similar time frames but did not investigate autophagy in the presence of ER stress inhibitors. In contrast, Komoike et al. employed salubrinal to block the eIF2α-ATF4 pathway. While ER stress-induced apoptosis following Cd2+ exposure was significantly reduced, no effect on LC3-II formation was observed (Komoike et al. 2012). It remains to be seen whether the other branches of the UPR are involved in autophagy induction by Cd2+. As described above, the integration of ER stress and autophagy signaling by identified molecular means is only just becoming apparent and already shows complexity through multiple levels of regulation. Thus, no literature on the effect of Cd2+ on candidates that bridge ER stress to autophagy is available at the time of writing. This could open up an exciting area of research for the Cd2+ field.
Cell cycle regulation and arrest
Healthy cells progress through the G1, S, G2 and M (mitosis) phases to complete a normal cell cycle, which is regulated by multiple signaling pathways and stress surveillance systems to ensure that cell division takes place with fidelity. Physiological oscillations of the cellular redox state are integrated with cell cycle progression and oxidative stress leads to cell cycle arrest (Burhans and Heintz 2009). Redox sensing of cell cycle regulation involves reactive cysteine thiols that function as redox sensors in cell cycle regulators. By modulating cell cycle regulators, these redox-active thiols ensure that cell division is executed at the right redox environment (Chiu and Dawes 2012). In G1, ROS stimulate mitogenic pathways that control the activity of cyclin-dependent kinases (Cdks) and phosphorylation and inactivation of the tumor suppressor protein retinoblastoma protein (pRB), thereby regulating S-phase entry. In response to oxidative stress, Nrf2 and Foxo3a (see sections “PI3K-Akt/PKB” and “Ref-1-Nrf2”) promote cell survival by inducing the expression of antioxidant enzymes and factors involved in cell cycle withdrawal, such as the cyclin-dependent kinase inhibitor (Cki) p27. In the S phase, ROS induce S-phase arrest via PP2A-dependent dephosphorylation of pRB. Replication stress in the S phase activates the DNA damage response and induces cell senescence. Interactions of ROS with the G1 Cdk/Cki network play a fundamental role in senescence, which is considered a barrier to tumorigenesis (Burhans and Heintz 2009). Similarly, changes in [Ca2+]cyt have been detected as a cell passes through G1, G1/S and mitosis (reviewed in (Roderick and Cook 2008)). Ca2+ is required in the early G1 phase for activation and/or expression of transcription factors of AP-1 (e.g., c-Fos, c-Jun, c-Myc), CREB and NFAT families. These factors coordinate the expression of cell cycle regulators, notably the D-type cyclins, which are required for the activation of cyclin D–Cdk4 complexes. Ca2+ is also required later in G1 to ensure phosphorylation and inactivation of Rb and entry into S phase. Ca2+ oscillations at the G1/S and G2/M transitions are thought to be important for the centrosome cycle. Ca2+ acts in concert with CaM, CaMK-II and CP110, a centrosomal protein and Cdk2 substrate, to initiate centrosome duplication at the G1/S transition. CP110 also inhibits centrosome separation allowing temporal coordination of the centrosome cycle by Ca2+. Centrosome duplication starts when cells exit G1 and enter S phase, and cyclin E–Cdk2 has a key role in the process, by activating Rho-associated, coiled-coil containing protein kinase 2 (ROCK2) and monopolar spindle 1 (MPS1), two protein kinases involved in centrosome duplication (Roderick and Cook 2008).
DNA-damaging agents (for example, irradiation or toxic metals) favor the accumulation of DNA double-strand breaks either directly or by interfering with DNA synthesis. Cells normally detect DNA lesions and arrest the cell cycle at either the G1–S or the G2–M transition (known as the G1 and G2 checkpoint, respectively). This allows the DNA repair machinery time to attempt to recover genome integrity (Jackson and Bartek 2009). If the degree of damage is beyond recovery, cells never enter mitosis but undergo apoptotic cell death or senescence, most often through p53-dependent mechanisms. The tumor suppressor p53 regulates DNA damage-induced cell cycle arrest by directly stimulating the expression of p21Waf1/CIP1, an inhibitor of cyclin-dependent kinases (Cdks). Through its negative effects on various Cdks, p21Waf1/CIP1 inhibits both the G1/S and the G2/M transitions (Vogelstein et al. 2000). The protein kinase ataxia-telangiectasia mutated (ATM) and ataxia telangiectasia and Rad3-related kinase (ATR) are master activators of a DNA damage response cascade at double-strand break sites, which transduce the damage signal to downstream effectors. ATM/ATR are PI3K-related proteins that phosphorylate multiple substrates in response to DNA damage or replication blocks; in addition, they recognize DNA strand breaks and contain signaling and repair activity. Two of the best studied ATM/ATR targets are the protein kinases Chk1 and Chk2, which, together with p53 (which is also regulated by ATM/ATR), act to reduce Cdk activity by various mechanisms (Jackson and Bartek 2009). Inhibition of Cdks slows down or arrests cell cycle progression at the G1-S, intra-S and G2-M cell cycle checkpoints, which is thought to increase the time available for DNA repair before replication or mitosis ensues. ATM/ATR activates both cell survival and cell death pathways, and the final outcome depends on a delicate balance between these opposing processes (Shiloh and Ziv 2013; Chen et al. 2012).
Cells with defects in the mitotic apparatus or chromosomes do not proceed through the M phase but become arrested in mitosis due to the activation of mitotic catastrophe. Cells then die without exiting mitosis (‘mitotic death’), reach the G1 phase of the subsequent cell cycle and then undergo cell death, or exit mitosis and undergo senescence (Vitale et al. 2011; Galluzzi et al. 2012). A functioning spindle assembly checkpoint, which controls progression from metaphase into anaphase, appears to be required for mitotic catastrophe to occur (Nitta et al. 2004). Nuclear alterations, such as micro- and multinucleation, have been used as morphological markers of mitotic catastrophe (Vitale et al. 2011).
Cd2+ and cell cycle regulation and arrest
Only a few studies have attempted to thoroughly investigate the role of Cd2+ on cell cycle regulation although it is well established that the toxic and carcinogenic effects of Cd2+ are partly caused by DNA damage. DNA damage occurs by interference with cellular redox regulation and induction of oxidative stress that increases oxidative DNA damage, but also by inhibition of major DNA repair systems. This results in accumulation of critical mutations and development of genomic instability (Beyersmann and Hartwig 2008; Hartwig 2013b; Filipic 2012) when cells evade mitotic arrest/catastrophe, and death or senescence (Hader et al. 1996). In human lung adenocarcinoma cells, Cd2+ (40–160 μM for 2 h)-induced ROS formation was associated with cell cycle arrest in the mitotic phase, micronucleation and apoptosis that were increased by p38 MAPK and reduced by ERK (Chao and Yang 2001). An inhibitor of p38 MAPK significantly decreased the induction of micronuclei, mitotic arrest and apoptosis, whereas an inhibitor of the ERK upstream activators MKK1/2 enhanced micronucleation and apoptosis in Cd2+-treated early-G2 cells (Chao and Yang 2001). Exposure of murine macrophages (J774A.1) expressing functional p53 to Cd2+ (20 μM for 24 h) caused ROS- and ERK-dependent cell cycle arrest at G2/M that was associated with induction of p21Waf1/CIP1 as well as apoptotic and necrotic cell death; inhibition of ERK activation resulted in G0/G1 arrest and partially released Cd2+-mediated G2/M arrest (suggesting mitotic catastrophe) and also strongly attenuated Cd2+-induced necrotic cell death, but did not prevent caspase-3 activation and DNA fragmentation (Kim et al. 2005a). Because Cd2+ has been shown to interfere with p53 function by inducing conformational changes in the wild-type protein (Meplan et al. 1999), but at the same time also increases p53 expression and function by PI3K-related kinase-dependent Ser15 phosphorylation (Matsuoka and Igisu 2001), studies investigating p53-dependent G2/M phase cell cycle arrest and/or death induced by Cd2+ are difficult to interpret (Xie and Shaikh 2006b; Cao et al. 2007). Studies in p53-inactivated cells have characterized mechanisms of p53-independent G2/M phase cell cycle arrest induced by Cd2+ (Bork et al. 2010; Yang et al. 2004). Yang et al. (2004) showed interference of Cd2+ (0.1–4 μM for 12–24 h in serum) with cell cycle progression in CHO K1 cells. These cells are very sensitive to Cd2+ because they do not express MTs; on the other hand, p53 is dysfunctional in CHO K1 cells. Cd2+ inhibition of cell cycle progression was global and occurred at G1/S, G2/M and during mitosis, but G2/M arrest was more sensitive to Cd2+ and independent of ROS formation (Yang et al. 2004). In contrast, p53-inactivated renal PT cells exposed to Cd2+ (100 μM for 6 h in serum) showed ROS dependence of cell cycle arrest at G2/M (Bork et al. 2010); interestingly, UCN-01, an inhibitor of the ATM/ATR downstream kinases Chk1/2, relieved G2/M transition block and increased cell death via apoptosis (and mitotic catastrophe). UCN-01 increases the cytotoxicity of chemotherapy and radiation by promoting mitotic catastrophe and is therefore used in cancer therapy to avoid that mutated premalignant or malignant cells survive and proliferate (Lapenna and Giordano 2009). In another study, Cd2+ (40 μM for 2 h in serum-free medium) induced p38-dependent prometaphase arrest of human nonsmall-cell lung carcinoma CL3 cells, which occurred through increased proteolysis of Cdc20, an activator of the anaphase-promoting complex (Yen and Yang 2010). A recent study in plant roots and leaves also investigated morphological signs of mitotic catastrophe induced by Cd2+ (1–50 μM) (Monteiro et al. 2012). Cd2+ increased ROS formation, which was associated with DNA damage, as determined by the comet assay. The tail length was maximal at 1 μM Cd2+, whereas shorter tails were found at 10 μM, suggesting formation of DNA adducts and impairment of DNA repair mechanisms. Interestingly, this correlated with a maximum of micronuclei frequency, suggesting that at this concentration, cells had an increase in mitotic catastrophe and that at 1 μM Cd2+, cells had developed strategies to repair damaged DNA by blocking the cell cycle at specific checkpoints, thus avoiding mitotic catastrophe (Monteiro et al. 2012). Hence, cells evading cell cycle arrest and mitotic catastrophe resume proliferation, which may be characterized by increased genomic instability and prone to malignant transformation.
Unfortunately, no study has been published that convincingly demonstrates interference of Cd2+- with Ca2+-dependent cell cycle regulation.
Synopsis and perspectives
By contrasting the most recent studies on Cd2+-induced cell death and survival mechanisms with state-of-the art knowledge of relevant cellular signaling pathways, this review has aimed to point out missing studies that will help to clarify the molecular mechanisms of how Cd2+ causes disease. Current understanding of many biological effects of Cd2+ and the existing knowledge of Cd2+-induced diseases are largely derived from data obtained by exposure to relatively high doses of the toxic metal, which likely lead to death by necrosis or apoptosis. But there is accumulating evidence for adverse health effects even under chronic low Cd2+ exposure conditions, which require relentless investigation. They may be the consequence of cellular reactions to noxious effects of Cd2+ and either represent secondary events intended to prevent or overcome death through activation of survival mechanisms, or primary repair and adaptive responses. With chronic low Cd2+ exposure, cellular death events can appear to be beneficial to preserve the integrity of an organ system. At these low Cd2+ concentrations, death is more likely to occur by apoptosis than by necrosis with its downside of triggering inflammatory processes. Survival, on the other hand, often occurs at the expense of faulty repair processes with cellular dysfunction and the danger of malignant transformation.
The Ca2+ and redox systems can regulate cell fate decisions through regulation of numerous functional proteins participating in cellular life-or-death decisions at the levels of signal transduction, transcriptional regulation or execution. The outcome (death or survival) depends on the integration of these pro- and antiapoptotic signals. A further level of complexity is introduced by the magnitude and duration of Cd2+ stress, which shape the dynamics of the Ca2+ and ROS signals, as well as dictate the contribution and severity of damage (functional and structural) to vital cellular organelles, which determine the reversibility of damage and cell fate (Fig. 1). Figure 2 shows that the complexity of the cellular responses elicited by Cd2+, namely the Ca2+ and redox regulation of key factors affecting cell death/survival is hierarchical, often bifurcated or cooperative and involves cross talk between various signaling pathways. This review highlights the fact that focus on one particular factor or signaling pathway is unable to provide a complete understanding of the impact of Cd2+ on cellular functions, and one should be aware that conclusions about the significance of that process for cell fate will in effect encompass multiple signaling pathways that will dictate the actual outcome for the cell.

Interactions between cell death and cell survival signaling pathways induced by Cd2+. The master regulators of Cd2+-induced signal transduction, Ca2+ and ROS, are usually the first event to take place in a cell following Cd2+ exposure. These signals are transmitted to numerous proteins in an organelle-dependent or -independent manner. Endoplasmic reticulum (ER) stress and mitochondrial dysfunction activate downstream signaling pathways involved in death or survival. The figure is not exhaustive; please refer to the text for details. The background color of the signaling molecule signifies direct activation by Ca2+ and/or ROS in addition to interactions with other signaling molecules (blue for Ca2+, orange for ROS or blue and orange for both Ca2+ and ROS). Black arrows, activation/induction; red arrows, inhibition; green arrow, cellular efflux
Of course, due to space limitations, this review could not be exhaustive and several relevant signaling pathways, factors and mechanisms (e.g., the epigenetic control of expression) that are targeted by Cd2+ and regulated by the intracellular signaling molecules Ca2+ and ROS had to be omitted. Nevertheless, a more integrated view on cellular signaling associated with Cd2+ toxicity may help to contribute to the development of preventive and novel therapeutic strategies for acute and chronic Cd2+ toxicity.
References
Abu-Bakar A, Satarug S, Marks GC, Lang MA, Moore MR (2004) Acute cadmium chloride administration induces hepatic and renal CYP2A5 mRNA, protein and activity in the mouse: involvement of transcription factor NRF2. Toxicol Lett 148(3):199–210. doi:10.1016/j.toxlet.2003.10.029
Adle DJ, Wei W, Smith N, Bies JJ, Lee J (2009) Cadmium-mediated rescue from ER-associated degradation induces expression of its exporter. Proc Natl Acad Sci USA 106(25):10189–10194. doi:10.1073/pnas.0812114106
Ak P, Levine AJ (2010) p53 and NF-kappaB: different strategies for responding to stress lead to a functional antagonism. FASEB J 24(10):3643–3652. doi:10.1096/fj.10-160549
Alam J, Stewart D, Touchard C, Boinapally S, Choi AM, Cook JL (1999) Nrf2, a Cap’n’Collar transcription factor, regulates induction of the heme oxygenase-1 gene. J Biol Chem 274(37):26071–26078
Alam J, Wicks C, Stewart D, Gong P, Touchard C, Otterbein S, Choi AM, Burow ME, Tou J (2000) Mechanism of heme oxygenase-1 gene activation by cadmium in MCF-7 mammary epithelial cells. Role of p38 kinase and Nrf2 transcription factor. J Biol Chem 275(36):27694–27702. doi:10.1074/jbc.M004729200
Appenzeller-Herzog C, Hall MN (2012) Bidirectional crosstalk between endoplasmic reticulum stress and mTOR signaling. Trends Cell Biol 22(5):274–282. doi:10.1016/j.tcb.2012.02.006
Bailey TA, Kanuga N, Romero IA, Greenwood J, Luthert PJ, Cheetham ME (2004) Oxidative stress affects the junctional integrity of retinal pigment epithelial cells. Invest Ophthalmol Vis Sci 45(2):675–684
Banakou E, Dailianis S (2010) Involvement of Na+/H+ exchanger and respiratory burst enzymes NADPH oxidase and NO synthase, in Cd-induced lipid peroxidation and DNA damage in haemocytes of mussels. Comp Biochem Physiol C Toxicol Pharmacol 152(3):346–352. doi:10.1016/j.cbpc.2010.06.001
Bassik MC, Scorrano L, Oakes SA, Pozzan T, Korsmeyer SJ (2004) Phosphorylation of BCL-2 regulates ER Ca2+ homeostasis and apoptosis. EMBO J 23(5):1207–1216. doi:10.1038/sj.emboj.7600104
Bedard K, Krause KH (2007) The NOX family of ROS-generating NADPH oxidases: physiology and pathophysiology. Physiol Rev 87(1):245–313. doi:10.1152/physrev.00044.2005
Bell SG, Vallee BL (2009) The metallothionein/thionein system: an oxidoreductive metabolic zinc link. ChemBioChem 10(1):55–62. doi:10.1002/cbic.200800511
Bellot G, Garcia-Medina R, Gounon P, Chiche J, Roux D, Pouyssegur J, Mazure NM (2009) Hypoxia-induced autophagy is mediated through hypoxia-inducible factor induction of BNIP3 and BNIP3L via their BH3 domains. Mol Cell Biol 29(10):2570–2581. doi:10.1128/MCB.00166-09
Belyaeva EA, Glazunov VV, Korotkov SM (2002) Cyclosporin A-sensitive permeability transition pore is involved in Cd(2+)-induced dysfunction of isolated rat liver mitochondria: doubts no more. Arch Biochem Biophys 405(2):252–264
Belyaeva EA, Dymkowska D, Wieckowski MR, Wojtczak L (2006) Reactive oxygen species produced by the mitochondrial respiratory chain are involved in Cd2+-induced injury of rat ascites hepatoma AS-30D cells. Biochim Biophys Acta 1757(12):1568–1574. doi:10.1016/j.bbabio.2006.09.006
Bender T, Martinou JC (2013) Where killers meet–permeabilization of the outer mitochondrial membrane during apoptosis. Cold Spring Harb Perspect Biol 5(1):a011106. doi:10.1101/cshperspect.a011106
Berridge MJ (2012) Calcium signalling remodelling and disease. Biochem Soc Trans 40(2):297–309. doi:10.1042/BST20110766
Berridge MJ, Bootman MD, Roderick HL (2003) Calcium signalling: dynamics, homeostasis and remodelling. Nat Rev Mol Cell Biol 4(7):517–529. doi:10.1038/nrm1155
Betin VM, Lane JD (2009) Caspase cleavage of Atg4D stimulates GABARAP-L1 processing and triggers mitochondrial targeting and apoptosis. J Cell Sci 122(Pt 14):2554–2566. doi:10.1242/jcs.046250
Beyersmann D, Hartwig A (2008) Carcinogenic metal compounds: recent insight into molecular and cellular mechanisms. Arch Toxicol 82(8):493–512. doi:10.1007/s00204-008-0313-y
Biagioli M, Watjen W, Beyersmann D, Zoncu R, Cappellini C, Ragghianti M, Cremisi F, Bucci S (2001) Cadmium-induced apoptosis in murine fibroblasts is suppressed by Bcl-2. Arch Toxicol 75(6):313–320
Biagioli M, Pifferi S, Ragghianti M, Bucci S, Rizzuto R, Pinton P (2008) Endoplasmic reticulum stress and alteration in calcium homeostasis are involved in cadmium-induced apoptosis. Cell Calcium 43(2):184–195
Bialik S, Kimchi A (2010) Lethal weapons: DAP-kinase, autophagy and cell death: DAP-kinase regulates autophagy. Curr Opin Cell Biol 22(2):199–205. doi:10.1016/j.ceb.2009.11.004
Bingol B, Wang CF, Arnott D, Cheng D, Peng J, Sheng M (2010) Autophosphorylated CaMKIIalpha acts as a scaffold to recruit proteasomes to dendritic spines. Cell 140(4):567–578. doi:10.1016/j.cell.2010.01.024
Bork U, Lee WK, Kuchler A, Dittmar T, Thévenod F (2010) Cadmium-induced DNA damage triggers G(2)/M arrest via chk1/2 and cdc2 in p53-deficient kidney proximal tubule cells. Am J Physiol Renal Physiol 298(2):F255–F265. doi:10.1152/ajprenal.00273.2009
Brigelius-Flohe R, Flohe L (2011) Basic principles and emerging concepts in the redox control of transcription factors. Antioxid Redox Signal 15(8):2335–2381. doi:10.1089/ars.2010.3534
Britschgi A, Bill A, Brinkhaus H, Rothwell C, Clay I, Duss S, Rebhan M, Raman P, Guy CT, Wetzel K, George E, Popa MO, Lilley S, Choudhury H, Gosling M, Wang L, Fitzgerald S, Borawski J, Baffoe J, Labow M, Gaither LA, Bentires-Alj M (2013) Calcium-activated chloride channel ANO1 promotes breast cancer progression by activating EGFR and CAMK signaling. Proc Natl Acad Sci USA 110(11):E1026–E1034. doi:10.1073/pnas.1217072110
Brodsky JL (2012) Cleaning up: ER-associated degradation to the rescue. Cell 151(6):1163–1167. doi:10.1016/j.cell.2012.11.012
Brown DI, Griendling KK (2009) Nox proteins in signal transduction. Free Radic Biol Med 47(9):1239–1253. doi:10.1016/j.freeradbiomed.2009.07.023
Burhans WC, Heintz NH (2009) The cell cycle is a redox cycle: linking phase-specific targets to cell fate. Free Radic Biol Med 47(9):1282–1293. doi:10.1016/j.freeradbiomed.2009.05.026
Burton TR, Gibson SB (2009) The role of Bcl-2 family member BNIP3 in cell death and disease: NIPping at the heels of cell death. Cell Death Differ 16(4):515–523. doi:10.1038/cdd.2008.185
Caldeira MV, Curcio M, Leal G, Salazar IL, Mele M, Santos AR, Melo CV, Pereira P, Canzoniero LM, Duarte CB (2013) Excitotoxic stimulation downregulates the ubiquitin-proteasome system through activation of NMDA receptors in cultured hippocampal neurons. Biochim Biophys Acta 1832(1):263–274. doi:10.1016/j.bbadis.2012.10.009
Campagne MV, Thibodeaux H, van Bruggen N, Cairns B, Lowe DG (2000) Increased binding activity at an antioxidant-responsive element in the metallothionein-1 promoter and rapid induction of metallothionein-1 and -2 in response to cerebral ischemia and reperfusion. J Neurosci 20(14):5200–5207
Cao F, Zhou T, Simpson D, Zhou Y, Boyer J, Chen B, Jin T, Cordeiro-Stone M, Kaufmann W (2007) p53-Dependent but ATM-independent inhibition of DNA synthesis and G2 arrest in cadmium-treated human fibroblasts. Toxicol Appl Pharmacol 218(2):174–185. doi:10.1016/j.taap.2006.10.031
Cardenas C, Foskett JK (2012) Mitochondrial Ca(2+) signals in autophagy. Cell Calcium 52(1):44–51. doi:10.1016/j.ceca.2012.03.001
Cardenas C, Miller RA, Smith I, Bui T, Molgo J, Muller M, Vais H, Cheung KH, Yang J, Parker I, Thompson CB, Birnbaum MJ, Hallows KR, Foskett JK (2010) Essential regulation of cell bioenergetics by constitutive InsP3 receptor Ca2+ transfer to mitochondria. Cell 142(2):270–283. doi:10.1016/j.cell.2010.06.007
Carrasco MA, Jaimovich E, Kemmerling U, Hidalgo C (2004) Signal transduction and gene expression regulated by calcium release from internal stores in excitable cells. Biol Res 37(4):701–712
Casalino E, Calzaretti G, Sblano C, Landriscina C (2002) Molecular inhibitory mechanisms of antioxidant enzymes in rat liver and kidney by cadmium. Toxicology 179(1–2):37–50
Castillo K, Rojas-Rivera D, Lisbona F, Caballero B, Nassif M, Court FA, Schuck S, Ibar C, Walter P, Sierralta J, Glavic A, Hetz C (2011) BAX inhibitor-1 regulates autophagy by controlling the IRE1alpha branch of the unfolded protein response. EMBO J 30(21):4465–4478. doi:10.1038/emboj.2011.318
Cereghetti GM, Scorrano L (2006) The many shapes of mitochondrial death. Oncogene 25(34):4717–4724. doi:10.1038/sj.onc.1209605
Chakraborty PK, Lee WK, Molitor M, Wolff NA, Thévenod F (2010a) Cadmium induces Wnt signaling to upregulate proliferation and survival genes in sub-confluent kidney proximal tubule cells. Mol Cancer 9:102. doi:10.1186/1476-4598-9-102
Chakraborty PK, Scharner B, Jurasovic J, Messner B, Bernhard D, Thévenod F (2010b) Chronic cadmium exposure induces transcriptional activation of the Wnt pathway and upregulation of epithelial-to-mesenchymal transition markers in mouse kidney. Toxicol Lett 198(1):69–76. doi:10.1016/j.toxlet.2010.05.007
Chang NC, Nguyen M, Germain M, Shore GC (2010) Antagonism of Beclin 1-dependent autophagy by BCL-2 at the endoplasmic reticulum requires NAF-1. EMBO J 29(3):606–618. doi:10.1038/emboj.2009.369
Chao JI, Yang JL (2001) Opposite roles of ERK and p38 mitogen-activated protein kinases in cadmium-induced genotoxicity and mitotic arrest. Chem Res Toxicol 14(9):1193–1202
Chargui A, Zekri S, Jacquillet G, Rubera I, Ilie M, Belaid A, Duranton C, Tauc M, Hofman P, Poujeol P, El May MV, Mograbi B (2011) Cadmium-induced autophagy in rat kidney: an early biomarker of subtoxic exposure. Toxicol Sci 121(1):31–42. doi:10.1093/toxsci/kfr031
Chen J, Shaikh ZA (2009) Activation of Nrf2 by cadmium and its role in protection against cadmium-induced apoptosis in rat kidney cells. Toxicol Appl Pharmacol 241(1):81–89. doi:10.1016/j.taap.2009.07.038
Chen R, Valencia I, Zhong F, McColl KS, Roderick HL, Bootman MD, Berridge MJ, Conway SJ, Holmes AB, Mignery GA, Velez P, Distelhorst CW (2004) Bcl-2 functionally interacts with inositol 1,4,5-trisphosphate receptors to regulate calcium release from the ER in response to inositol 1,4,5-trisphosphate. J Cell Biol 166(2):193–203. doi:10.1083/jcb.200309146
Chen Y, McMillan-Ward E, Kong J, Israels SJ, Gibson SB (2007) Mitochondrial electron-transport-chain inhibitors of complexes I and II induce autophagic cell death mediated by reactive oxygen species. J Cell Sci 120(Pt 23):4155–4166. doi:10.1242/jcs.011163
Chen CL, Lin CF, Chang WT, Huang WC, Teng CF, Lin YS (2008a) Ceramide induces p38 MAPK and JNK activation through a mechanism involving a thioredoxin-interacting protein-mediated pathway. Blood 111(8):4365–4374. doi:10.1182/blood-2007-08-106336
Chen L, Liu L, Huang S (2008b) Cadmium activates the mitogen-activated protein kinase (MAPK) pathway via induction of reactive oxygen species and inhibition of protein phosphatases 2A and 5. Free Radic Biol Med 45(7):1035–1044. doi:10.1016/j.freeradbiomed.2008.07.011
Chen L, Liu L, Luo Y, Huang S (2008c) MAPK and mTOR pathways are involved in cadmium-induced neuronal apoptosis. J Neurochem 105(1):251–261. doi:10.1111/j.1471-4159.2007.05133.x
Chen L, Xu B, Liu L, Luo Y, Zhou H, Chen W, Shen T, Han X, Kontos CD, Huang S (2011a) Cadmium induction of reactive oxygen species activates the mTOR pathway, leading to neuronal cell death. Free Radic Biol Med 50(5):624–632. doi:10.1016/j.freeradbiomed.2010.12.032
Chen S, He FF, Wang H, Fang Z, Shao N, Tian XJ, Liu JS, Zhu ZH, Wang YM, Wang S, Huang K, Zhang C (2011b) Calcium entry via TRPC6 mediates albumin overload-induced endoplasmic reticulum stress and apoptosis in podocytes. Cell Calcium 50(6):523–529. doi:10.1016/j.ceca.2011.08.008
Chen S, Xu Y, Xu B, Guo M, Zhang Z, Liu L, Ma H, Chen Z, Luo Y, Huang S, Chen L (2011c) CaMKII is involved in cadmium activation of MAPK and mTOR pathways leading to neuronal cell death. J Neurochem 119(5):1108–1118. doi:10.1111/j.1471-4159.2011.07493.x
Chen BP, Li M, Asaithamby A (2012) New insights into the roles of ATM and DNA-PKcs in the cellular response to oxidative stress. Cancer Lett 327(1–2):103–110. doi:10.1016/j.canlet.2011.12.004
Chiarelli R, Agnello M, Bosco L, Roccheri MC (2013) Sea urchin embryos exposed to cadmium as an experimental model for studying the relationship between autophagy and apoptosis. Marine Environ Res. doi:10.1016/j.marenvres.2013.06.001
Chipuk JE, Bouchier-Hayes L, Green DR (2006) Mitochondrial outer membrane permeabilization during apoptosis: the innocent bystander scenario. Cell Death Differ 13(8):1396–1402. doi:10.1038/sj.cdd.4401963
Chipuk JE, Moldoveanu T, Llambi F, Parsons MJ, Green DR (2010) The BCL-2 family reunion. Mol Cell 37(3):299–310. doi:10.1016/j.molcel.2010.01.025
Chiribau CB, Gaccioli F, Huang CC, Yuan CL, Hatzoglou M (2010) Molecular symbiosis of CHOP and C/EBP beta isoform LIP contributes to endoplasmic reticulum stress-induced apoptosis. Mol Cell Biol 30(14):3722–3731. doi:10.1128/MCB.01507-09
Chitnis NS, Pytel D, Bobrovnikova-Marjon E, Pant D, Zheng H, Maas NL, Frederick B, Kushner JA, Chodosh LA, Koumenis C, Fuchs SY, Diehl JA (2012) miR-211 is a prosurvival microRNA that regulates chop expression in a PERK-dependent manner. Mol Cell 48(3):353–364. doi:10.1016/j.molcel.2012.08.025
Chiu J, Dawes IW (2012) Redox control of cell proliferation. Trends Cell Biol 22(11):592–601. doi:10.1016/j.tcb.2012.08.002
Chou HC, Chan HL (2011) Proteomic analysis of potential breast cancer biomarkers. In: Done SJ (ed) Breast cancer—recent advances in biology. Imaging and Therapeutics, InTech, pp 179–202
Chun YS, Choi E, Kim GT, Choi H, Kim CH, Lee MJ, Kim MS, Park JW (2000) Cadmium blocks hypoxia-inducible factor (HIF)-1-mediated response to hypoxia by stimulating the proteasome-dependent degradation of HIF-1alpha. Eur J Biochem 267(13):4198–4204
Clapham DE (2007) Calcium signaling. Cell 131(6):1047–1058. doi:10.1016/j.cell.2007.11.028
Clevers H, Nusse R (2012) Wnt/beta-catenin signaling and disease. Cell 149(6):1192–1205. doi:10.1016/j.cell.2012.05.012
Covarrubias L, Hernandez-Garcia D, Schnabel D, Salas-Vidal E, Castro-Obregon S (2008) Function of reactive oxygen species during animal development: passive or active? Dev Biol 320(1):1–11. doi:10.1016/j.ydbio.2008.04.041
Crompton M (1999) The mitochondrial permeability transition pore and its role in cell death. Biochem J 341(Pt 2):233–249
Cross DA, Alessi DR, Cohen P, Andjelkovich M, Hemmings BA (1995) Inhibition of glycogen synthase kinase-3 by insulin mediated by protein kinase B. Nature 378(6559):785–789. doi:10.1038/378785a0
Cui ZG, Ogawa R, Piao JL, Hamazaki K, Feril LB Jr, Shimomura A, Kondo T, Inadera H (2011) Molecular mechanisms involved in the adaptive response to cadmium-induced apoptosis in human myelomonocytic lymphoma U937 cells. Toxicol In Vitro 25(8):1687–1693. doi:10.1016/j.tiv.2011.07.008
Cuypers A, Plusquin M, Remans T, Jozefczak M, Keunen E, Gielen H, Opdenakker K, Nair AR, Munters E, Artois TJ, Nawrot T, Vangronsveld J, Smeets K (2010) Cadmium stress: an oxidative challenge. Biometals 23(5):927–940. doi:10.1007/s10534-010-9329-x
Darding M, Meier P (2012) IAPs: guardians of RIPK1. Cell Death Differ 19(1):58–66. doi:10.1038/cdd.2011.163
Darios F, Lambeng N, Troadec JD, Michel PP, Ruberg M (2003) Ceramide increases mitochondrial free calcium levels via caspase 8 and Bid: role in initiation of cell death. J Neurochem 84(4):643–654
D’Autreaux B, Toledano MB (2007) ROS as signalling molecules: mechanisms that generate specificity in ROS homeostasis. Nat Rev Mol Cell Biol 8(10):813–824
De A (2011) Wnt/Ca2+ signaling pathway: a brief overview. Acta Biochim Biophys Sin (Shanghai) 43(10):745–756. doi:10.1093/abbs/gmr079
Decuypere JP, Bultynck G, Parys JB (2011a) A dual role for Ca(2+) in autophagy regulation. Cell Calcium 50(3):242–250. doi:10.1016/j.ceca.2011.04.001
Decuypere JP, Welkenhuyzen K, Luyten T, Ponsaerts R, Dewaele M, Molgo J, Agostinis P, Missiaen L, De Smedt H, Parys JB, Bultynck G (2011b) Ins(1,4,5)P3 receptor-mediated Ca2+ signaling and autophagy induction are interrelated. Autophagy 7(12):1472–1489
DeGennaro M, Hurd TR, Siekhaus DE, Biteau B, Jasper H, Lehmann R (2011) Peroxiredoxin stabilization of DE-cadherin promotes primordial germ cell adhesion. Dev Cell 20(2):233–243. doi:10.1016/j.devcel.2010.12.007
Denton D, Nicolson S, Kumar S (2012) Cell death by autophagy: facts and apparent artefacts. Cell Death Differ 19(1):87–95. doi:10.1038/cdd.2011.146
Di Gioacchino M, Petrarca C, Perrone A, Martino S, Esposito DL, Lotti LV, Mariani-Costantini R (2008) Autophagy in hematopoietic stem/progenitor cells exposed to heavy metals: biological implications and toxicological relevance. Autophagy 4(4):537–539
Ding WX, Ni HM, Gao W, Hou YF, Melan MA, Chen X, Stolz DB, Shao ZM, Yin XM (2007) Differential effects of endoplasmic reticulum stress-induced autophagy on cell survival. J Biol Chem 282(7):4702–4710. doi:10.1074/jbc.M609267200
Djakovic SN, Schwarz LA, Barylko B, DeMartino GN, Patrick GN (2009) Regulation of the proteasome by neuronal activity and calcium/calmodulin-dependent protein kinase II. J Biol Chem 284(39):26655–26665. doi:10.1074/jbc.M109.021956
Djakovic SN, Marquez-Lona EM, Jakawich SK, Wright R, Chu C, Sutton MA, Patrick GN (2012) Phosphorylation of Rpt6 regulates synaptic strength in hippocampal neurons. J Neurosci 32(15):5126–5131. doi:10.1523/JNEUROSCI.4427-11.2012
Djavaheri-Mergny M, Maiuri MC, Kroemer G (2010) Cross talk between apoptosis and autophagy by caspase-mediated cleavage of Beclin 1. Oncogene 29(12):1717–1719. doi:10.1038/onc.2009.519
Dorta DJ, Leite S, DeMarco KC, Prado IM, Rodrigues T, Mingatto FE, Uyemura SA, Santos AC, Curti C (2003) A proposed sequence of events for cadmium-induced mitochondrial impairment. J Inorg Biochem 97(3):251–257
Dremina ES, Sharov VS, Kumar K, Zaidi A, Michaelis EK, Schoneich C (2004) Anti-apoptotic protein Bcl-2 interacts with and destabilizes the sarcoplasmic/endoplasmic reticulum Ca2+-ATPase (SERCA). Biochem J 383(Pt 2):361–370. doi:10.1042/BJ20040187
Droge W (2002) Free radicals in the physiological control of cell function. Physiol Rev 82(1):47–95. doi:10.1152/physrev.00018.2001
Edinger AL, Thompson CB (2004) Death by design: apoptosis, necrosis and autophagy. Curr Opin Cell Biol 16(6):663–669. doi:10.1016/j.ceb.2004.09.011
Edwards JR, Kolman K, Lamar PC, Chandar N, Fay MJ, Prozialeck WC (2013) Effects of cadmium on the sub-cellular localization of beta-catenin and beta-catenin-regulated gene expression in NRK-52E cells. Biometals 26(1):33–42. doi:10.1007/s10534-012-9592-0
Eichler T, Ma Q, Kelly C, Mishra J, Parikh S, Ransom RF, Devarajan P, Smoyer WE (2006) Single and combination toxic metal exposures induce apoptosis in cultured murine podocytes exclusively via the extrinsic caspase 8 pathway. Toxicol Sci 90(2):392–399. doi:10.1093/toxsci/kfj106
Engel T, Sanz-Rodgriguez A, Jimenez-Mateos EM, Concannon CG, Jimenez-Pacheco A, Moran C, Mesuret G, Petit E, Delanty N, Farrell MA, O’Brien DF, Prehn JH, Lucas JJ, Henshall DC (2013) CHOP regulates the p53-MDM2 axis and is required for neuronal survival after seizures. Brain J Neurol 136(Pt 2):577–592. doi:10.1093/brain/aws337
Feissner RF, Skalska J, Gaum WE, Sheu SS (2009) Crosstalk signaling between mitochondrial Ca2+ and ROS. Front Biosci 14:1197–1218
Ferrari D, Pinton P, Campanella M, Callegari MG, Pizzirani C, Rimessi A, Di Virgilio F, Pozzan T, Rizzuto R (2011) Functional and structural alterations in the endoplasmic reticulum and mitochondria during apoptosis triggered by C2-ceramide and CD95/APO-1/FAS receptor stimulation. Biochem Biophys Res Commun 391(1):575–581. doi:10.1016/j.bbrc.2009.11.101
Ferraro PM, Costanzi S, Naticchia A, Sturniolo A, Gambaro G (2010) Low level exposure to cadmium increases the risk of chronic kidney disease: analysis of the NHANES 1999–2006. BMC Public Health 10:304. doi:10.1186/1471-2458-10-304
Figueiredo-Pereira ME, Cohen G (1999) The ubiquitin/proteasome pathway: friend or foe in zinc-, cadmium-, and H2O2-induced neuronal oxidative stress. Mol Biol Rep 26(1–2):65–69
Filipic M (2012) Mechanisms of cadmium induced genomic instability. Mutat Res 733(1–2):69–77. doi:10.1016/j.mrfmmm.2011.09.002
Fischer H, Koenig U, Eckhart L, Tschachler E (2002) Human caspase 12 has acquired deleterious mutations. Biochem Biophys Res Commun 293(2):722–726. doi:10.1016/S0006-291X(02)00289-9
Foster KG, Fingar DC (2010) Mammalian target of rapamycin (mTOR): conducting the cellular signaling symphony. J Biol Chem 285(19):14071–14077. doi:10.1074/jbc.R109.094003
Fouad AA, Jresat I (2011) Protective effect of telmisartan against cadmium-induced nephrotoxicity in mice. Life Sci 89(1–2):29–35. doi:10.1016/j.lfs.2011.04.019
Franklin RA, Rodriguez-Mora OG, Lahair MM, McCubrey JA (2006) Activation of the calcium/calmodulin-dependent protein kinases as a consequence of oxidative stress. Antioxid Redox Signal 8(9–10):1807–1817. doi:10.1089/ars.2006.8.1807
Freitas M, Fernandes E (2011) Zinc, cadmium and nickel increase the activation of NF-kappaB and the release of cytokines from THP-1 monocytic cells. Metallomics 3(11):1238–1243. doi:10.1039/c1mt00050k
Fruehauf JP, Meyskens FL Jr (2007) Reactive oxygen species: a breath of life or death? Clin Cancer Res 13(3):789–794. doi:10.1158/1078-0432.CCR-06-2082
Fujiki K, Inamura H, Matsuoka M (2013) Phosphorylation of FOXO3a by PI3K/Akt pathway in HK-2 renal proximal tubular epithelial cells exposed to cadmium. Arch Toxicol. doi:10.1007/s00204-013-1077-6
Funato Y, Miki H (2010) Redox regulation of Wnt signalling via nucleoredoxin. Free Radic Res 44(4):379–388. doi:10.3109/10715761003610745
Funato Y, Michiue T, Asashima M, Miki H (2006) The thioredoxin-related redox-regulating protein nucleoredoxin inhibits Wnt-beta-catenin signalling through dishevelled. Nat Cell Biol 8(5):501–508. doi:10.1038/ncb1405
Galluzzi L, Vanden Berghe T, Vanlangenakker N, Buettner S, Eisenberg T, Vandenabeele P, Madeo F, Kroemer G (2011) Programmed necrosis from molecules to health and disease. Int Rev Cell Mol Biol 289:1–35. doi:10.1016/B978-0-12-386039-2.00001-8
Galluzzi L, Vitale I, Abrams JM, Alnemri ES, Baehrecke EH, Blagosklonny MV, Dawson TM, Dawson VL, El-Deiry WS, Fulda S, Gottlieb E, Green DR, Hengartner MO, Kepp O, Knight RA, Kumar S, Lipton SA, Lu X, Madeo F, Malorni W, Mehlen P, Nunez G, Peter ME, Piacentini M, Rubinsztein DC, Shi Y, Simon HU, Vandenabeele P, White E, Yuan J, Zhivotovsky B, Melino G, Kroemer G (2012) Molecular definitions of cell death subroutines: recommendations of the nomenclature committee on cell death. Cell Death Differ 19(1):107–120. doi:10.1038/cdd.2011.96
Ghosh S, Hayden MS (2012) Celebrating 25 years of NF-kappaB research. Immunol Rev 246(1):5–13. doi:10.1111/j.1600-065X.2012.01111.x
Ghosh G, Wang VY, Huang DB, Fusco A (2012) NF-kappaB regulation: lessons from structures. Immunol Rev 246(1):36–58. doi:10.1111/j.1600-065X.2012.01097.x
Gil-Parrado S, Fernandez-Montalvan A, Assfalg-Machleidt I, Popp O, Bestvater F, Holloschi A, Knoch TA, Auerswald EA, Welsh K, Reed JC, Fritz H, Fuentes-Prior P, Spiess E, Salvesen GS, Machleidt W (2002) Ionomycin-activated calpain triggers apoptosis. A probable role for Bcl-2 family members. J Biol Chem 277(30):27217–27226. doi:10.1074/jbc.M202945200
Glickman MH, Ciechanover A (2002) The ubiquitin-proteasome proteolytic pathway: destruction for the sake of construction. Physiol Rev 82(2):373–428. doi:10.1152/physrev.00027.2001
Golstein P, Kroemer G (2007) Cell death by necrosis: towards a molecular definition. Trends Biochem Sci 32(1):37–43. doi:10.1016/j.tibs.2006.11.001
Gomez-Vicente V, Donovan M, Cotter TG (2005) Multiple death pathways in retina-derived 661W cells following growth factor deprivation: crosstalk between caspases and calpains. Cell Death Differ 12(7):796–804. doi:10.1038/sj.cdd.4401621
Gorlach A, Klappa P, Kietzmann T (2006) The endoplasmic reticulum: folding, calcium homeostasis, signaling, and redox control. Antioxid Redox Signal 8(9–10):1391–1418. doi:10.1089/ars.2006.8.1391
Grimm S (2012) The ER-mitochondria interface: the social network of cell death. Biochim Biophys Acta 1823(2):327–334. doi:10.1016/j.bbamcr.2011.11.018
Grosch S, Schiffmann S, Geisslinger G (2012) Chain length-specific properties of ceramides. Prog Lipid Res 51(1):50–62. doi:10.1016/j.plipres.2011.11.001
Gulbins E, Li PL (2006) Physiological and pathophysiological aspects of ceramide. Am J Physiol Regul Integr Comp Physiol 290(1):R11–R26
Guntaka SR, Samak G, Seth A, LaRusso NF, Rao R (2011) Epidermal growth factor protects the apical junctional complexes from hydrogen peroxide in bile duct epithelium. Lab Invest 91(9):1396–1409. doi:10.1038/labinvest.2011.73
Habeebu SS, Liu J, Klaassen CD (1998) Cadmium-induced apoptosis in mouse liver. Toxicol Appl Pharmacol 149(2):203–209. doi:10.1006/taap.1997.8334
Hader C, Hadnagy W, Seemayer NH (1996) A rapid method for detection of nongenotoxic carcinogens of environmental pollutants using synchronized V79 cells and flow cytometry. Toxicol Lett 88(1–3):99–108
Hampton RY, Sommer T (2012) Finding the will and the way of ERAD substrate retrotranslocation. Curr Opin Cell Biol 24(4):460–466. doi:10.1016/j.ceb.2012.05.010
Han J, Back SH, Hur J, Lin YH, Gildersleeve R, Shan J, Yuan CL, Krokowski D, Wang S, Hatzoglou M, Kilberg MS, Sartor MA, Kaufman RJ (2013) ER-stress-induced transcriptional regulation increases protein synthesis leading to cell death. Nat Cell Biol 15(5):481–490. doi:10.1038/ncb2738
Hanahan D, Weinberg RA (2011) Hallmarks of cancer: the next generation. Cell 144(5):646–674. doi:10.1016/j.cell.2011.02.013
Hannun YA, Obeid LM (2008) Principles of bioactive lipid signalling: lessons from sphingolipids. Nat Rev Mol Cell Biol 9(2):139–150. doi:10.1038/nrm2329
Hansen JM, Zhang H, Jones DP (2006) Differential oxidation of thioredoxin-1, thioredoxin-2, and glutathione by metal ions. Free Radic Biol Med 40(1):138–145. doi:10.1016/j.freeradbiomed.2005.09.023
Hanson CJ, Bootman MD, Distelhorst CW, Maraldi T, Roderick HL (2008a) The cellular concentration of Bcl-2 determines its pro- or anti-apoptotic effect. Cell Calcium 44(3):243–258. doi:10.1016/j.ceca.2007.11.014
Hanson CJ, Bootman MD, Distelhorst CW, Wojcikiewicz RJ, Roderick HL (2008b) Bcl-2 suppresses Ca2+ release through inositol 1,4,5-trisphosphate receptors and inhibits Ca2+ uptake by mitochondria without affecting ER calcium store content. Cell Calcium 44(3):324–338. doi:10.1016/j.ceca.2008.01.003
Haq F, Mahoney M, Koropatnick J (2003) Signaling events for metallothionein induction. Mutat Res 533(1–2):211–226
Hart BA, Lee CH, Shukla GS, Shukla A, Osier M, Eneman JD, Chiu JF (1999) Characterization of cadmium-induced apoptosis in rat lung epithelial cells: evidence for the participation of oxidant stress. Toxicology 133(1):43–58
Hartwig A (2013a) Cadmium and cancer. Met Ions Life Sci 11:491–507. doi:10.1007/978-94-007-5179-8_15
Hartwig A (2013b) Metal interaction with redox regulation: an integrating concept in metal carcinogenesis? Free Radic Biol Med 55:63–72. doi:10.1016/j.freeradbiomed.2012.11.009
Hartzell HC, Qu Z, Yu K, Xiao Q, Chien LT (2008) Molecular physiology of bestrophins: multifunctional membrane proteins linked to best disease and other retinopathies. Physiol Rev 88(2):639–672. doi:10.1152/physrev.00022.2007
Hatai T, Matsuzawa A, Inoshita S, Mochida Y, Kuroda T, Sakamaki K, Kuida K, Yonehara S, Ichijo H, Takeda K (2000) Execution of apoptosis signal-regulating kinase 1 (ASK1)-induced apoptosis by the mitochondria-dependent caspase activation. J Biol Chem 275(34):26576–26581. doi:10.1074/jbc.M003412200
Hattori K, Naguro I, Runchel C, Ichijo H (2009) The roles of ASK family proteins in stress responses and diseases. Cell Commun Signal 7:9. doi:10.1186/1478-811X-7-9
He CH, Gong P, Hu B, Stewart D, Choi ME, Choi AM, Alam J (2001) Identification of activating transcription factor 4 (ATF4) as an Nrf2-interacting protein. Implication for heme oxygenase-1 gene regulation. J Biol Chem 276(24):20858–20865. doi:10.1074/jbc.M101198200
He X, Chen MG, Ma Q (2008) Activation of Nrf2 in defense against cadmium-induced oxidative stress. Chem Res Toxicol 21(7):1375–1383. doi:10.1021/tx800019a
Hers I, Vincent EE, Tavare JM (2011) Akt signalling in health and disease. Cell Signal 23(10):1515–1527. doi:10.1016/j.cellsig.2011.05.004
Hetz C (2012) The unfolded protein response: controlling cell fate decisions under ER stress and beyond. Nat Rev Mol Cell Biol 13(2):89–102. doi:10.1038/nrm3270
Hidalgo C, Donoso P (2008) Crosstalk between calcium and redox signaling: from molecular mechanisms to health implications. Antioxid Redox Signal 10(7):1275–1312. doi:10.1089/ars.2007.1886
Higa A, Chevet E (2012) Redox signaling loops in the unfolded protein response. Cell Signal 24(8):1548–1555. doi:10.1016/j.cellsig.2012.03.011
Hiramatsu N, Kasai A, Du S, Takeda M, Hayakawa K, Okamura M, Yao J, Kitamura M (2007) Rapid, transient induction of ER stress in the liver and kidney after acute exposure to heavy metal: evidence from transgenic sensor mice. FEBS Lett 581(10):2055–2059. doi:10.1016/j.febslet.2007.04.040
Hitomi J, Katayama T, Eguchi Y, Kudo T, Taniguchi M, Koyama Y, Manabe T, Yamagishi S, Bando Y, Imaizumi K, Tsujimoto Y, Tohyama M (2004) Involvement of caspase-4 in endoplasmic reticulum stress-induced apoptosis and Abeta-induced cell death. J Cell Biol 165(3):347–356. doi:10.1083/jcb.200310015
Hoyer-Hansen M, Bastholm L, Szyniarowski P, Campanella M, Szabadkai G, Farkas T, Bianchi K, Fehrenbacher N, Elling F, Rizzuto R, Mathiasen IS, Jaattela M (2007) Control of macroautophagy by calcium, calmodulin-dependent kinase kinase-beta, and Bcl-2. Mol Cell 25(2):193–205. doi:10.1016/j.molcel.2006.12.009
Hsiao CJ, Stapleton SR (2004) Characterization of Cd-induced molecular events prior to cellular damage in primary rat hepatocytes in culture: activation of the stress activated signal protein JNK and transcription factor AP-1. J Biochem Mol Toxicol 18(3):133–142. doi:10.1002/jbt.20018
Hsu TS, Yang PM, Tsai JS, Lin LY (2009) Attenuation of cadmium-induced necrotic cell death by necrostatin-1: potential necrostatin-1 acting sites. Toxicol Appl Pharmacol 235(2):153–162. doi:10.1016/j.taap.2008.12.012
Huang YH, Shih CM, Huang CJ, Lin CM, Chou CM, Tsai ML, Liu TP, Chiu JF, Chen CT (2006) Effects of cadmium on structure and enzymatic activity of Cu, Zn-SOD and oxidative status in neural cells. J Cell Biochem 98(3):577–589. doi:10.1002/jcb.20772
Huang J, Canadien V, Lam GY, Steinberg BE, Dinauer MC, Magalhaes MA, Glogauer M, Grinstein S, Brumell JH (2009) Activation of antibacterial autophagy by NADPH oxidases. Proc Natl Acad Sci USA 106(15):6226–6231. doi:10.1073/pnas.0811045106
Huang J, Lam GY, Brumell JH (2011) Autophagy signaling through reactive oxygen species. Antioxid Redox Signal 14(11):2215–2231. doi:10.1089/ars.2010.3554
Ichijo H, Nishida E, Irie K, ten Dijke P, Saitoh M, Moriguchi T, Takagi M, Matsumoto K, Miyazono K, Gotoh Y (1997) Induction of apoptosis by ASK1, a mammalian MAPKKK that activates SAPK/JNK and p38 signaling pathways. Science 275(5296):90–94
Ikediobi CO, Badisa VL, Ayuk-Takem LT, Latinwo LM, West J (2004) Response of antioxidant enzymes and redox metabolites to cadmium-induced oxidative stress in CRL-1439 normal rat liver cells. Int J Mol Med 14(1):87–92
Iriyama T, Takeda K, Nakamura H, Morimoto Y, Kuroiwa T, Mizukami J, Umeda T, Noguchi T, Naguro I, Nishitoh H, Saegusa K, Tobiume K, Homma T, Shimada Y, Tsuda H, Aiko S, Imoto I, Inazawa J, Chida K, Kamei Y, Kozuma S, Taketani Y, Matsuzawa A, Ichijo H (2009) ASK1 and ASK2 differentially regulate the counteracting roles of apoptosis and inflammation in tumorigenesis. EMBO J 28(7):843–853. doi:10.1038/emboj.2009.32
Ishido M, Homma-Takeda S, Tohyama C, Suzuki T (1998) Apoptosis in rat renal proximal tubular cells induced by cadmium. J Toxicol Environ Health A 55(1):1–12
Ishido M, Ohtsubo R, Adachi T, Kunimoto M (2002) Attenuation of both apoptotic and necrotic actions of cadmium by Bcl-2. Environ Health Perspect 110(1):37–42
Jackson SP, Bartek J (2009) The DNA-damage response in human biology and disease. Nature 461(7267):1071–1078. doi:10.1038/nature08467
Janssen-Heininger YM, Mossman BT, Heintz NH, Forman HJ, Kalyanaraman B, Finkel T, Stamler JS, Rhee SG, van der Vliet A (2008) Redox-based regulation of signal transduction: principles, pitfalls, and promises. Free Radic Biol Med 45(1):1–17. doi:10.1016/j.freeradbiomed.2008.03.011
Jarup L, Akesson A (2009) Current status of cadmium as an environmental health problem. Toxicol Appl Pharmacol 238(3):201–208. doi:10.1016/j.taap.2009.04.020
Jauhiainen A, Thomsen C, Strombom L, Grundevik P, Andersson C, Danielsson A, Andersson MK, Nerman O, Rorkvist L, Stahlberg A, Aman P (2012) Distinct cytoplasmic and nuclear functions of the stress induced protein DDIT3/CHOP/GADD153. PLoS ONE 7(4):e33208. doi:10.1371/journal.pone.0033208
Jeon HK, Jin HS, Lee DH, Choi WS, Moon CK, Oh YJ, Lee TH (2004) Proteome analysis associated with cadmium adaptation in U937 cells: identification of calbindin-D28k as a secondary cadmium-responsive protein that confers resistance to cadmium-induced apoptosis. J Biol Chem 279(30):31575–31583. doi:10.1074/jbc.M400823200
Jeong W, Chang TS, Boja ES, Fales HM, Rhee SG (2004) Roles of TRP14, a thioredoxin-related protein in tumor necrosis factor-alpha signaling pathways. J Biol Chem 279(5):3151–3159. doi:10.1074/jbc.M307959200
Ji YL, Wang H, Zhao XF, Wang Q, Zhang C, Zhang Y, Zhao M, Chen YH, Meng XH, Xu DX (2011) Crosstalk between endoplasmic reticulum stress and mitochondrial pathway mediates cadmium-induced germ cell apoptosis in testes. Toxicol Sci 124(2):446–459. doi:10.1093/toxsci/kfr232
Ji YL, Wang Z, Wang H, Zhang C, Zhang Y, Zhao M, Chen YH, Meng XH, Xu DX (2012) Ascorbic acid protects against cadmium-induced endoplasmic reticulum stress and germ cell apoptosis in testes. Reprod Toxicol 34(3):357–363. doi:10.1016/j.reprotox.2012.04.011
Jiang L, Liu Y, Ma MM, Tang YB, Zhou JG, Guan YY (2013) Mitochondria dependent pathway is involved in the protective effect of bestrophin-3 on hydrogen peroxide-induced apoptosis in basilar artery smooth muscle cells. Apoptosis 18(5):556–565. doi:10.1007/s10495-013-0828-4
Jing Y, Liu LZ, Jiang Y, Zhu Y, Guo NL, Barnett J, Rojanasakul Y, Agani F, Jiang BH (2012) Cadmium increases HIF-1 and VEGF expression through ROS, ERK, and AKT signaling pathways and induces malignant transformation of human bronchial epithelial cells. Toxicol Sci 125(1):10–19. doi:10.1093/toxsci/kfr256
Jung Y, Kim H, Min SH, Rhee SG, Jeong W (2008) Dynein light chain LC8 negatively regulates NF-kappaB through the redox-dependent interaction with IkappaBalpha. J Biol Chem 283(35):23863–23871. doi:10.1074/jbc.M803072200
Jungmann J, Reins HA, Schobert C, Jentsch S (1993) Resistance to cadmium mediated by ubiquitin-dependent proteolysis. Nature 361(6410):369–371. doi:10.1038/361369a0
Kajla S, Mondol AS, Nagasawa A, Zhang Y, Kato M, Matsuno K, Yabe-Nishimura C, Kamata T (2012) A crucial role for Nox 1 in redox-dependent regulation of Wnt-beta-catenin signaling. FASEB J 26(5):2049–2059. doi:10.1096/fj.11-196360
Kalai M, Lamkanfi M, Denecker G, Boogmans M, Lippens S, Meeus A, Declercq W, Vandenabeele P (2003) Regulation of the expression and processing of caspase-12. J Cell Biol 162(3):457–467. doi:10.1083/jcb.200303157
Karin M, Shaulian E (2001) AP-1: linking hydrogen peroxide and oxidative stress to the control of cell proliferation and death. IUBMB Life 52(1–2):17–24. doi:10.1080/15216540252774711
Kato H, Katoh R, Kitamura M (2013) Dual regulation of cadmium-induced apoptosis by mTORC1 through selective induction of IRE1 branches in unfolded protein response. PLoS ONE 8(5):e64344. doi:10.1371/journal.pone.0064344
Kerr JF, Wyllie AH, Currie AR (1972) Apoptosis: a basic biological phenomenon with wide-ranging implications in tissue kinetics. Br J Cancer 26(4):239–257
Khan NM, Sandur SK, Checker R, Sharma D, Poduval TB, Sainis KB (2011) Pro-oxidants ameliorate radiation-induced apoptosis through activation of the calcium-ERK1/2-Nrf2 pathway. Free Radic Biol Med 51(1):115–128. doi:10.1016/j.freeradbiomed.2011.03.037
Kim J, Kim SH, Johnson VJ, Sharma RP (2005a) Extracellular signal-regulated kinase-signaling-dependent G2/M arrest and cell death in murine macrophages by cadmium. Environ Toxicol Chem 24(12):3069–3077
Kim SD, Moon CK, Eun SY, Ryu PD, Jo SA (2005b) Identification of ASK1, MKK4, JNK, c-Jun, and caspase-3 as a signaling cascade involved in cadmium-induced neuronal cell apoptosis. Biochem Biophys Res Commun 328(1):326–334. doi:10.1016/j.bbrc.2004.11.173
Kim AN, Jeon WK, Lee JJ, Kim BC (2010) Up-regulation of heme oxygenase-1 expression through CaMKII-ERK1/2-Nrf2 signaling mediates the anti-inflammatory effect of bisdemethoxycurcumin in LPS-stimulated macrophages. Free Radic Biol Med 49(3):323–331. doi:10.1016/j.freeradbiomed.2010.04.015
Kim JH, Xu EY, Sacks DB, Lee J, Shu L, Xia B, Kong AN (2012) Identification and functional studies of a new Nrf2 partner IQGAP1: a critical role in the stability and transactivation of Nrf2. Antioxid Redox Signal. doi:10.1089/ars.2012.4586
Kim S, Cheon HS, Kim SY, Juhnn YS, Kim YY (2013) Cadmium induces neuronal cell death through reactive oxygen species activated by GADD153. BMC cell biology 14:4. doi:10.1186/1471-2121-14-4
Kitamura M (2011) Control of NF-kappaB and inflammation by the unfolded protein response. Int Rev Immunol 30(1):4–15. doi:10.3109/08830185.2010.522281
Kitamura M, Hiramatsu N (2010) The oxidative stress: endoplasmic reticulum stress axis in cadmium toxicity. Biometals 23(5):941–950. doi:10.1007/s10534-010-9296-2
Kiviluoto S, Vervliet T, Ivanova H, Decuypere JP, De Smedt H, Missiaen L, Bultynck G, Parys JB (2013) Regulation of inositol 1,4,5-trisphosphate receptors during endoplasmic reticulum stress. Biochim Biophys Acta 1833(7):1612–1624. doi:10.1016/j.bbamcr.2013.01.026
Klaassen CD, Liu J (1998) Induction of metallothionein as an adaptive mechanism affecting the magnitude and progression of toxicological injury. Environ Health Perspect 106(Suppl 1):297–300
Klaassen CD, Liu J, Diwan BA (2009) Metallothionein protection of cadmium toxicity. Toxicol Appl Pharmacol 238(3):215–220. doi:10.1016/j.taap.2009.03.026
Komoike Y, Inamura H, Matsuoka M (2012) Effects of salubrinal on cadmium-induced apoptosis in HK-2 human renal proximal tubular cells. Arch Toxicol 86(1):37–44. doi:10.1007/s00204-011-0742-x
Krishna S, Low IC, Pervaiz S (2011) Regulation of mitochondrial metabolism: yet another facet in the biology of the oncoprotein Bcl-2. Biochem J 435(3):545–551. doi:10.1042/BJ20101996
Kroemer G, Galluzzi L, Brenner C (2007) Mitochondrial membrane permeabilization in cell death. Physiol Rev 87(1):99–163. doi:10.1152/physrev.00013.2006
Kunzelmann K, Kongsuphol P, Chootip K, Toledo C, Martins JR, Almaca J, Tian Y, Witzgall R, Ousingsawat J, Schreiber R (2011) Role of the Ca2+-activated Cl- channels bestrophin and anoctamin in epithelial cells. Biol Chem 392(1–2):125–134. doi:10.1515/BC.2011.010
Kuranaga E, Miura M (2007) Nonapoptotic functions of caspases: caspases as regulatory molecules for immunity and cell-fate determination. Trends Cell Biol 17(3):135–144. doi:10.1016/j.tcb.2007.01.001
Kyriakis JM, Avruch J (2001) Mammalian mitogen-activated protein kinase signal transduction pathways activated by stress and inflammation. Physiol Rev 81(2):807–869
Lam D, Kosta A, Luciani MF, Golstein P (2008) The inositol 1,4,5-trisphosphate receptor is required to signal autophagic cell death. Mol Biol Cell 19(2):691–700. doi:10.1091/mbc.E07-08-0823
Lamkanfi M, Kalai M, Vandenabeele P (2004) Caspase-12: an overview. Cell Death Differ 11(4):365–368. doi:10.1038/sj.cdd.4401364
Lapenna S, Giordano A (2009) Cell cycle kinases as therapeutic targets for cancer. Nat Rev Drug Discov 8(7):547–566. doi:10.1038/nrd2907
Lawal AO, Ellis EM (2011) Nrf2-mediated adaptive response to cadmium-induced toxicity involves protein kinase C delta in human 1321N1 astrocytoma cells. Environ Toxicol Pharmacol 32(1):54–62. doi:10.1016/j.etap.2011.03.010
Lawal AO, Ellis EM (2012) Phospholipase C mediates cadmium-dependent apoptosis in HEK 293 cells. Basic Clin Pharmacol Toxicol 110(6):510–517. doi:10.1111/j.1742-7843.2011.00843.x
Lee J, Lim KT (2011) Inhibitory effect of plant-originated glycoprotein (27 kDa) on expression of matrix metalloproteinase-9 in cadmium chloride-induced BNL CL.2 cells. J Trace Elem Med Biol 25(4):239–246. doi:10.1016/j.jtemb.2011.08.142
Lee WK, Thévenod F (2006) A role for mitochondrial aquaporins in cellular life-and-death decisions? Am J Physiol Cell Physiol 291(2):C195–C202
Lee WK, Thévenod F (2008) Novel roles for ceramides, calpains and caspases in kidney proximal tubule cell apoptosis: lessons from in vitro cadmium toxicity studies. Biochem Pharmacol 76(11):1323–1332. doi:10.1016/j.bcp.2008.07.004
Lee WK, Bork U, Gholamrezaei F, Thévenod F (2005a) Cd2+-induced cytochrome c release in apoptotic proximal tubule cells: role of mitochondrial permeability transition pore and Ca2+ uniporter. Am J Physiol Renal Physiol 288(1):F27–F39
Lee WK, Spielmann M, Bork U, Thévenod F (2005b) Cd2+-induced swelling-contraction dynamics in isolated kidney cortex mitochondria: role of Ca2+ uniporter, K+ cycling, and protonmotive force. Am J Physiol Cell Physiol 289(3):C656–C664
Lee WK, Abouhamed M, Thévenod F (2006) Caspase-dependent and -independent pathways for cadmium-induced apoptosis in cultured kidney proximal tubule cells. Am J Physiol Renal Physiol 291(4):F823–F832. doi:10.1152/ajprenal.00276.2005
Lee WK, Torchalski B, Thévenod F (2007) Cadmium-induced ceramide formation triggers calpain-dependent apoptosis in cultured kidney proximal tubule cells. Am J Physiol Cell Physiol 293(3):C839–C847
Lee WK, Torchalski B, Kohistani N, Thévenod F (2011) ABCB1 protects kidney proximal tubule cells against cadmium-induced apoptosis: roles of cadmium and ceramide transport. Toxicol Sci 121(2):343–356. doi:10.1093/toxsci/kfr071
Lee WK, Chakraborty PK, Roussa E, Wolff NA, Thévenod F (2012) ERK1/2-dependent bestrophin-3 expression prevents ER-stress-induced cell death in renal epithelial cells by reducing CHOP. Biochim Biophys Acta 1823(10):1864–1876. doi:10.1016/j.bbamcr.2012.06.003
Lei K, Davis RJ (2003) JNK phosphorylation of Bim-related members of the Bcl2 family induces Bax-dependent apoptosis. Proc Natl Acad Sci USA 100(5):2432–2437. doi:10.1073/pnas.0438011100
Lei K, Nimnual A, Zong WX, Kennedy NJ, Flavell RA, Thompson CB, Bar-Sagi D, Davis RJ (2002) The Bax subfamily of Bcl2-related proteins is essential for apoptotic signal transduction by c-Jun NH(2)-terminal kinase. Mol Cell Biol 22(13):4929–4942
Leiser SF, Miller RA (2010) Nrf2 signaling, a mechanism for cellular stress resistance in long-lived mice. Mol Cell Biol 30(3):871–884. doi:10.1128/MCB.01145-09
Lemarie A, Lagadic-Gossmann D, Morzadec C, Allain N, Fardel O, Vernhet L (2004) Cadmium induces caspase-independent apoptosis in liver Hep3B cells: role for calcium in signaling oxidative stress-related impairment of mitochondria and relocation of endonuclease G and apoptosis-inducing factor. Free Radic Biol Med 36(12):1517–1531
Leslie NR (2006) The redox regulation of PI 3-kinase-dependent signaling. Antioxid Redox Signal 8(9–10):1765–1774. doi:10.1089/ars.2006.8.1765
Leverrier P, Montigny C, Garrigos M, Champeil P (2007) Metal binding to ligands: cadmium complexes with glutathione revisited. Anal Biochem 371(2):215–228. doi:10.1016/j.ab.2007.07.015
Li Y, Lim SC (2007) Cadmium-induced apoptosis of hepatocytes is not associated with death receptor-related caspase-dependent pathways in the rat. Environ Toxicol Pharmacol 24(3):231–238. doi:10.1016/j.etap.2007.05.010
Li M, Kondo T, Zhao QL, Li FJ, Tanabe K, Arai Y, Zhou ZC, Kasuya M (2000) Apoptosis induced by cadmium in human lymphoma U937 cells through Ca2+-calpain and caspase-mitochondria-dependent pathways. J Biol Chem 275(50):39702–39709
Li M, Xia T, Jiang CS, Li LJ, Fu JL, Zhou ZC (2003) Cadmium directly induced the opening of membrane permeability pore of mitochondria which possibly involved in cadmium-triggered apoptosis. Toxicology 194(1–2):19–33
Li Z, Arnaud L, Rockwell P, Figueiredo-Pereira ME (2004) A single amino acid substitution in a proteasome subunit triggers aggregation of ubiquitinated proteins in stressed neuronal cells. J Neurochem 90(1):19–28. doi:10.1111/j.1471-4159.2004.02456.x
Li G, Mongillo M, Chin KT, Harding H, Ron D, Marks AR, Tabas I (2009) Role of ERO1-alpha-mediated stimulation of inositol 1,4,5-triphosphate receptor activity in endoplasmic reticulum stress-induced apoptosis. J Cell Biol 186(6):783–792. doi:10.1083/jcb.200904060
Li L, Ishdorj G, Gibson SB (2012) Reactive oxygen species regulation of autophagy in cancer: implications for cancer treatment. Free Radic Biol Med 53(7):1399–1410. doi:10.1016/j.freeradbiomed.2012.07.011
Liao Y, Fung TS, Huang M, Fang SG, Zhong Y, Liu DX (2013) Upregulation of CHOP/GADD153 during coronavirus infectious bronchitis virus infection modulates apoptosis by restricting activation of the extracellular signal-regulated kinase pathway. J Virol 87(14):8124–8134. doi:10.1128/JVI.00626-13
Lim SC, Hahm KS, Lee SH, Oh SH (2010) Autophagy involvement in cadmium resistance through induction of multidrug resistance-associated protein and counterbalance of endoplasmic reticulum stress WI38 lung epithelial fibroblast cells. Toxicology 276(1):18–26. doi:10.1016/j.tox.2010.06.010
Lipford JR, Deshaies RJ (2003) Diverse roles for ubiquitin-dependent proteolysis in transcriptional activation. Nat Cell Biol 5(10):845–850. doi:10.1038/ncb1003-845
Liu J, Lin A (2005) Role of JNK activation in apoptosis: a double-edged sword. Cell Res 15(1):36–42. doi:10.1038/sj.cr.7290262
Liu Y, Templeton DM (2008) Initiation of caspase-independent death in mouse mesangial cells by Cd2+: involvement of p38 kinase and CaMK-II. J Cell Physiol 217(2):307–318. doi:10.1002/jcp.21499
Liu F, Inageda K, Nishitai G, Matsuoka M (2006) Cadmium induces the expression of Grp78, an endoplasmic reticulum molecular chaperone, in LLC-PK1 renal epithelial cells. Environ Health Perspect 114(6):859–864
Liu J, Qu W, Kadiiska MB (2009) Role of oxidative stress in cadmium toxicity and carcinogenesis. Toxicol Appl Pharmacol 238(3):209–214. doi:10.1016/j.taap.2009.01.029
Livnat-Levanon N, Glickman MH (2011) Ubiquitin-proteasome system and mitochondria—reciprocity. Biochim Biophys Acta 1809(2):80–87. doi:10.1016/j.bbagrm.2010.07.005
Lizotte J, Abed E, Signor C, Malu DT, Cuevas J, Kevorkova O, Sanchez-Dardon J, Satoskar A, Scorza T, Jumarie C, Moreau R (2012) Expression of macrophage migration inhibitory factor by osteoblastic cells: protection against cadmium toxicity. Toxicol Lett 215(3):167–173. doi:10.1016/j.toxlet.2012.10.006
Lopez E, Figueroa S, Oset-Gasque MJ, Gonzalez MP (2003) Apoptosis and necrosis: two distinct events induced by cadmium in cortical neurons in culture. Br J Pharmacol 138(5):901–911. doi:10.1038/sj.bjp.0705111
Lopez E, Arce C, Oset-Gasque MJ, Canadas S, Gonzalez MP (2006) Cadmium induces reactive oxygen species generation and lipid peroxidation in cortical neurons in culture. Free Radic Biol Med 40(6):940–951. doi:10.1016/j.freeradbiomed.2005.10.062
Lorz C, Justo P, Sanz A, Subira D, Egido J, Ortiz A (2004) Paracetamol-induced renal tubular injury: a role for ER stress. J Am Soc Nephrol 15(2):380–389
Lu SC (2013) Glutathione synthesis. Biochim Biophys Acta 1830(5):3143–3153. doi:10.1016/j.bbagen.2012.09.008
Macian F (2005) NFAT proteins: key regulators of T-cell development and function. Nat Rev Immunol 5(6):472–484. doi:10.1038/nri1632
Macian F, Lopez-Rodriguez C, Rao A (2001) Partners in transcription: NFAT and AP-1. Oncogene 20(19):2476–2489. doi:10.1038/sj.onc.1204386
MacKinnon Y, Kapron CM (2010) Reduction in cadmium-induced toxicity and c-Jun N-terminal kinase activation by glutathione in cultured mouse embryonic cells. Birth Defects Res A Clin Mol Teratol 88(9):707–714. doi:10.1002/bdra.20703
Madden DT, Egger L, Bredesen DE (2007) A calpain-like protease inhibits autophagic cell death. Autophagy 3(5):519–522
Malhotra JD, Kaufman RJ (2007) Endoplasmic reticulum stress and oxidative stress: a vicious cycle or a double-edged sword? Antioxid Redox Signal 9(12):2277–2293. doi:10.1089/ars.2007.1782
Manning BD, Cantley LC (2007) AKT/PKB signaling: navigating downstream. Cell 129(7):1261–1274. doi:10.1016/j.cell.2007.06.009
Mao WP, Ye JL, Guan ZB, Zhao JM, Zhang C, Zhang NN, Jiang P, Tian T (2007) Cadmium induces apoptosis in human embryonic kidney (HEK) 293 cells by caspase-dependent and -independent pathways acting on mitochondria. Toxicol In Vitro 21(3):343–354. doi:10.1016/j.tiv.2006.09.004
Margariti A, Li H, Chen T, Martin D, Vizcay-Barrena G, Alam S, Karamariti E, Xiao Q, Zampetaki A, Zhang Z, Wang W, Jiang Z, Gao C, Ma B, Chen YG, Cockerill G, Hu Y, Xu Q, Zeng L (2013) XBP1 mRNA splicing triggers an autophagic response in endothelial cells through BECLIN-1 transcriptional activation. J Biol Chem 288(2):859–872. doi:10.1074/jbc.M112.412783
Martinez Flores K, Uribe Marin BC, Souza Arroyo V, Bucio Ortiz L, Lopez Reyes A, Gomez-Quiroz LE, Rojas del Castillo E, Gutierrez Ruiz MC (2013) Hepatocytes display a compensatory survival response against cadmium toxicity by a mechanism mediated by EGFR and Src. Toxicol In Vitro 27(3):1031–1042. doi:10.1016/j.tiv.2013.01.017
Matchkov VV, Larsen P, Bouzinova EV, Rojek A, Boedtkjer DM, Golubinskaya V, Pedersen FS, Aalkjaer C, Nilsson H (2008) Bestrophin-3 (vitelliform macular dystrophy 2-like 3 protein) is essential for the cGMP-dependent calcium-activated chloride conductance in vascular smooth muscle cells. Circ Res 103(8):864–872. doi:10.1161/CIRCRESAHA.108.178517
Matsuoka M, Igisu H (2001) Cadmium induces phosphorylation of p53 at serine 15 in MCF-7 cells. Biochem Biophys Res Commun 282(5):1120–1125. doi:10.1006/bbrc.2001.4700
Matsuzawa A, Ichijo H (2005) Stress-responsive protein kinases in redox-regulated apoptosis signaling. Antioxid Redox Signal 7(3–4):472–481. doi:10.1089/ars.2005.7.472
McCubrey JA, Steelman LS, Chappell WH, Abrams SL, Wong EW, Chang F, Lehmann B, Terrian DM, Milella M, Tafuri A, Stivala F, Libra M, Basecke J, Evangelisti C, Martelli AM, Franklin RA (2007) Roles of the Raf/MEK/ERK pathway in cell growth, malignant transformation and drug resistance. Biochim Biophys Acta 1773(8):1263–1284. doi:10.1016/j.bbamcr.2006.10.001
McCullough KD, Martindale JL, Klotz LO, Aw TY, Holbrook NJ (2001) Gadd153 sensitizes cells to endoplasmic reticulum stress by down-regulating Bcl2 and perturbing the cellular redox state. Mol Cell Biol 21(4):1249–1259. doi:10.1128/MCB.21.4.1249-1259.2001
Medicherla B, Goldberg AL (2008) Heat shock and oxygen radicals stimulate ubiquitin-dependent degradation mainly of newly synthesized proteins. J Cell Biol 182(4):663–673. doi:10.1083/jcb.200803022
Meffert MK, Baltimore D (2005) Physiological functions for brain NF-kappaB. Trends Neurosci 28(1):37–43. doi:10.1016/j.tins.2004.11.002
Mekahli D, Bultynck G, Parys JB, De Smedt H, Missiaen L (2011) Endoplasmic-reticulum calcium depletion and disease. Cold Spring Harb Perspect Biol 3(6). doi:10.1101/cshperspect.a004317
Meldolesi J, Pozzan T (1998) The endoplasmic reticulum Ca2+ store: a view from the lumen. Trends Biochem Sci 23(1):10–14
Meplan C, Mann K, Hainaut P (1999) Cadmium induces conformational modifications of wild-type p53 and suppresses p53 response to DNA damage in cultured cells. J Biol Chem 274(44):31663–31670
Mihailidou C, Papazian I, Papavassiliou AG, Kiaris H (2010) CHOP-dependent regulation of p21/waf1 during ER stress. Cell Physiol Biochem Int J Exp Cell Physiol Biochem Pharmacol 25(6):761–766. doi:10.1159/000315096
Misra UK, Gawdi G, Akabani G, Pizzo SV (2002) Cadmium-induced DNA synthesis and cell proliferation in macrophages: the role of intracellular calcium and signal transduction mechanisms. Cell Signal 14(4):327–340
Misra UK, Gawdi G, Pizzo SV (2003) Induction of mitogenic signalling in the 1LN prostate cell line on exposure to submicromolar concentrations of cadmium+. Cell Signal 15(11):1059–1070
Mizushima N, Komatsu M (2011) Autophagy: renovation of cells and tissues. Cell 147(4):728–741. doi:10.1016/j.cell.2011.10.026
Moenner M, Pluquet O, Bouchecareilh M, Chevet E (2007) Integrated endoplasmic reticulum stress responses in cancer. Cancer Res 67(22):10631–10634. doi:10.1158/0008-5472.CAN-07-1705
Monteiro C, Santos C, Pinho S, Oliveira H, Pedrosa T, Dias MC (2012) Cadmium-induced cyto- and genotoxicity are organ-dependent in lettuce. Chem Res Toxicol 25(7):1423–1434. doi:10.1021/tx300039t
Morgan MJ, Liu ZG (2011) Crosstalk of reactive oxygen species and NF-kappaB signaling. Cell Res 21(1):103–115. doi:10.1038/cr.2010.178
Mullen TD, Obeid LM (2012) Ceramide and apoptosis: exploring the enigmatic connections between sphingolipid metabolism and programmed cell death. Anticancer Agents Med Chem 12(4):340–363
Muratani M, Tansey WP (2003) How the ubiquitin-proteasome system controls transcription. Nat Rev Mol Cell Biol 4(3):192–201. doi:10.1038/nrm1049
Naguro I, Umeda T, Kobayashi Y, Maruyama J, Hattori K, Shimizu Y, Kataoka K, Kim-Mitsuyama S, Uchida S, Vandewalle A, Noguchi T, Nishitoh H, Matsuzawa A, Takeda K, Ichijo H (2012) ASK3 responds to osmotic stress and regulates blood pressure by suppressing WNK1-SPAK/OSR1 signaling in the kidney. Nat Commun 3:1285. doi:10.1038/ncomms2283
Nakagawa T, Yuan J (2000) Cross-talk between two cysteine protease families. Activation of caspase-12 by calpain in apoptosis. J Cell Biol 150(4):887–894
Nakagawa T, Zhu H, Morishima N, Li E, Xu J, Yankner BA, Yuan J (2000) Caspase-12 mediates endoplasmic-reticulum-specific apoptosis and cytotoxicity by amyloid-beta. Nature 403(6765):98–103. doi:10.1038/47513
Nawrot TS, Staessen JA, Roels HA, Munters E, Cuypers A, Richart T, Ruttens A, Smeets K, Clijsters H, Vangronsveld J (2010) Cadmium exposure in the population: from health risks to strategies of prevention. Biometals 23(5):769–782. doi:10.1007/s10534-010-9343-z
Nayak MK, Kumar K, Dash D (2011) Regulation of proteasome activity in activated human platelets. Cell Calcium 49(4):226–232. doi:10.1016/j.ceca.2011.02.005
Nguyen KC, Willmore WG, Tayabali AF (2013) Cadmium telluride quantum dots cause oxidative stress leading to extrinsic and intrinsic apoptosis in hepatocellular carcinoma HepG2 cells. Toxicology 306:114–123. doi:10.1016/j.tox.2013.02.010
Ni M, Zhang Y, Lee AS (2011) Beyond the endoplasmic reticulum: atypical GRP78 in cell viability, signalling and therapeutic targeting. Biochem J 434(2):181–188. doi:10.1042/BJ20101569
Nicotera P, Melino G (2004) Regulation of the apoptosis-necrosis switch. Oncogene 23(16):2757–2765. doi:10.1038/sj.onc.1207559
Nicotera P, Leist M, Ferrando-May E (1998) Intracellular ATP, a switch in the decision between apoptosis and necrosis. Toxicol Lett 102–103:139–142
Niehrs C (2012) The complex world of WNT receptor signalling. Nat Rev Mol Cell Biol 13(12):767–779. doi:10.1038/nrm3470
Nishitoh H, Saitoh M, Mochida Y, Takeda K, Nakano H, Rothe M, Miyazono K, Ichijo H (1998) ASK1 is essential for JNK/SAPK activation by TRAF2. Mol Cell 2(3):389–395
Nishitoh H, Matsuzawa A, Tobiume K, Saegusa K, Takeda K, Inoue K, Hori S, Kakizuka A, Ichijo H (2002) ASK1 is essential for endoplasmic reticulum stress-induced neuronal cell death triggered by expanded polyglutamine repeats. Genes Dev 16(11):1345–1355. doi:10.1101/gad.992302
Nitta M, Kobayashi O, Honda S, Hirota T, Kuninaka S, Marumoto T, Ushio Y, Saya H (2004) Spindle checkpoint function is required for mitotic catastrophe induced by DNA-damaging agents. Oncogene 23(39):6548–6558. doi:10.1038/sj.onc.1207873
Niture SK, Jaiswal AK (2012) Nrf2 protein up-regulates antiapoptotic protein Bcl-2 and prevents cellular apoptosis. J Biol Chem. 2287(13):9873-9886. doi:10.1074/jbc.M111.312694
Niture SK, Jaiswal AK (2013) Nrf2-induced antiapoptotic Bcl-xL protein enhances cell survival and drug resistance. Free Radic Biol Med 57:119-131. doi:10.1016/j.freeradbiomed.2012.12.014
Novgorodov SA, Chudakova DA, Wheeler BW, Bielawski J, Kindy MS, Obeid LM, Gudz TI (2011) Developmentally regulated ceramide synthase 6 increases mitochondrial Ca2+ loading capacity and promotes apoptosis. J Biol Chem 286(6):4644–4658. doi:10.1074/jbc.M110.164392
O’Brien P, Salacinski HJ (1998) Evidence that the reactions of cadmium in the presence of metallothionein can produce hydroxyl radicals. Arch Toxicol 72(11):690–700
O’Driscoll KE, Hatton WJ, Burkin HR, Leblanc N, Britton FC (2008) Expression, localization, and functional properties of Bestrophin 3 channel isolated from mouse heart. Am J Physiol Cell Physiol 295(6):C1610–C1624. doi:10.1152/ajpcell.00461.2008
Ogata M, Hino S, Saito A, Morikawa K, Kondo S, Kanemoto S, Murakami T, Taniguchi M, Tanii I, Yoshinaga K, Shiosaka S, Hammarback JA, Urano F, Imaizumi K (2006) Autophagy is activated for cell survival after endoplasmic reticulum stress. Mol Cell Biol 26(24):9220–9231. doi:10.1128/MCB.01453-06
Oh SH, Lee BH, Lim SC (2004) Cadmium induces apoptotic cell death in WI 38 cells via caspase-dependent Bid cleavage and calpain-mediated mitochondrial Bax cleavage by Bcl-2-independent pathway. Biochem Pharmacol 68(9):1845–1855
Okada T, Yoshida H, Akazawa R, Negishi M, Mori K (2002) Distinct roles of activating transcription factor 6 (ATF6) and double-stranded RNA-activated protein kinase-like endoplasmic reticulum kinase (PERK) in transcription during the mammalian unfolded protein response. Biochem J 366(Pt 2):585–594. doi:10.1042/BJ20020391
Onuma Y, Haramoto Y, Nejigane S, Takahashi S, Asashima M (2009) Bestrophin genes are expressed in Xenopus development. Biochem Biophys Res Commun 384(3):290–295. doi:10.1016/j.bbrc.2009.04.117
Orrenius S, Zhivotovsky B, Nicotera P (2003) Regulation of cell death: the calcium-apoptosis link. Nat Rev Mol Cell Biol 4(7):552–565. doi:10.1038/nrm1150
Palam LR, Baird TD, Wek RC (2011) Phosphorylation of eIF2 facilitates ribosomal bypass of an inhibitory upstream ORF to enhance CHOP translation. J Biol Chem 286(13):10939–10949. doi:10.1074/jbc.M110.216093
Papadakis ES, Finegan KG, Wang X, Robinson AC, Guo C, Kayahara M, Tournier C (2006) The regulation of Bax by c-Jun N-terminal protein kinase (JNK) is a prerequisite to the mitochondrial-induced apoptotic pathway. FEBS Lett 580(5):1320–1326
Paredes RM, Bollo M, Holstein D, Lechleiter JD (2013) Luminal Ca2+ depletion during the unfolded protein response in Xenopus oocytes: cause and consequence. Cell Calcium 53(4):286–296. doi:10.1016/j.ceca.2013.01.002
Park J, Kim I, Oh YJ, Lee K, Han PL, Choi EJ (1997) Activation of c-Jun N-terminal kinase antagonizes an anti-apoptotic action of Bcl-2. J Biol Chem 272(27):16725–16728
Park CS, Kim OS, Yun SM, Jo SA, Jo I, Koh YH (2008) Presenilin 1/gamma-secretase is associated with cadmium-induced E-cadherin cleavage and COX-2 gene expression in T47D breast cancer cells. Toxicol Sci 106(2):413–422. doi:10.1093/toxsci/kfn197
Parys JB, Decuypere JP, Bultynck G (2012) Role of the inositol 1,4,5-trisphosphate receptor/Ca2+-release channel in autophagy. Cell Commun Signal 10(1):17. doi:10.1186/1478-811X-10-17
Pehar M, Jonas MC, Hare TM, Puglielli L (2012) SLC33A1/AT-1 protein regulates the induction of autophagy downstream of IRE1/XBP1 pathway. J Biol Chem 287(35):29921–29930. doi:10.1074/jbc.M112.363911
Pena LB, Pasquini LA, Tomaro ML, Gallego SM (2007) 20S proteasome and accumulation of oxidized and ubiquitinated proteins in maize leaves subjected to cadmium stress. Phytochemistry 68(8):1139–1146. doi:10.1016/j.phytochem.2007.02.022
Peng TI, Jou MJ (2010) Oxidative stress caused by mitochondrial calcium overload. Ann N Y Acad Sci 1201:183–188. doi:10.1111/j.1749-6632.2010.05634.x
Peng Z, Peng L, Fan Y, Zandi E, Shertzer HG, Xia Y (2007) A critical role for IkappaB kinase beta in metallothionein-1 expression and protection against arsenic toxicity. J Biol Chem 282(29):21487–21496. doi:10.1074/jbc.M702510200
Permenter MG, Lewis JA, Jackson DA (2011) Exposure to nickel, chromium, or cadmium causes distinct changes in the gene expression patterns of a rat liver derived cell line. PLoS ONE 6(11):e27730. doi:10.1371/journal.pone.0027730
Picaud T, Desbois A (2006) Interaction of glutathione reductase with heavy metal: the binding of Hg(II) or Cd(II) to the reduced enzyme affects both the redox dithiol pair and the flavin. Biochemistry 45(51):15829–15837. doi:10.1021/bi061304m
Pinton P, Ferrari D, Rapizzi E, Di Virgilio F, Pozzan T, Rizzuto R (2001) The Ca2+ concentration of the endoplasmic reticulum is a key determinant of ceramide-induced apoptosis: significance for the molecular mechanism of Bcl-2 action. EMBO J 20(11):2690–2701
Poppe M, Reimertz C, Munstermann G, Kogel D, Prehn JH (2002) Ceramide-induced apoptosis of D283 medulloblastoma cells requires mitochondrial respiratory chain activity but occurs independently of caspases and is not sensitive to Bcl-xL overexpression. J Neurochem 82(3):482–494
Pourahmad J, O’Brien PJ, Jokar F, Daraei B (2003) Carcinogenic metal induced sites of reactive oxygen species formation in hepatocytes. Toxicol In Vitro 17(5–6):803–810
Qu W, Diwan BA, Reece JM, Bortner CD, Pi J, Liu J, Waalkes MP (2005) Cadmium-induced malignant transformation in rat liver cells: role of aberrant oncogene expression and minimal role of oxidative stress. Int J Cancer 114(3):346–355. doi:10.1002/ijc.20736
Qu W, Fuquay R, Sakurai T, Waalkes MP (2006) Acquisition of apoptotic resistance in cadmium-induced malignant transformation: specific perturbation of JNK signal transduction pathway and associated metallothionein overexpression. Mol Carcinog 45(8):561–571. doi:10.1002/mc.20185
Qu W, Ke H, Pi J, Broderick D, French JE, Webber MM, Waalkes MP (2007) Acquisition of apoptotic resistance in cadmium-transformed human prostate epithelial cells: Bcl-2 overexpression blocks the activation of JNK signal transduction pathway. Environ Health Perspect 115(7):1094–1100. doi:10.1289/ehp.10075
Qu W, Pi J, Waalkes MP (2013) Metallothionein blocks oxidative DNA damage in vitro. Arch Toxicol 87(2):311–321. doi:10.1007/s00204-012-0927-y
Rao RV, Hermel E, Castro-Obregon S, del Rio G, Ellerby LM, Ellerby HM, Bredesen DE (2001) Coupling endoplasmic reticulum stress to the cell death program. Mechanism of caspase activation. J Biol Chem 276(36):33869–33874. doi:10.1074/jbc.M102225200
Rao RV, Peel A, Logvinova A, del Rio G, Hermel E, Yokota T, Goldsmith PC, Ellerby LM, Ellerby HM, Bredesen DE (2002) Coupling endoplasmic reticulum stress to the cell death program: role of the ER chaperone GRP78. FEBS Lett 514(2–3):122–128
Rao RV, Ellerby HM, Bredesen DE (2004) Coupling endoplasmic reticulum stress to the cell death program. Cell Death Differ 11(4):372–380. doi:10.1038/sj.cdd.4401378
Ravid T, Hochstrasser M (2008) Diversity of degradation signals in the ubiquitin-proteasome system. Nat Rev Mol Cell Biol 9(9):679–690. doi:10.1038/nrm2468
Ray PD, Huang BW, Tsuji Y (2012) Reactive oxygen species (ROS) homeostasis and redox regulation in cellular signaling. Cell Signal 24(5):981–990. doi:10.1016/j.cellsig.2012.01.008
Rickert RC, Jellusova J, Miletic AV (2011) Signaling by the tumor necrosis factor receptor superfamily in B-cell biology and disease. Immunol Rev 244(1):115–133. doi:10.1111/j.1600-065X.2011.01067.x
Riedl SJ, Shi Y (2004) Molecular mechanisms of caspase regulation during apoptosis. Nat Rev Mol Cell Biol 5(11):897–907. doi:10.1038/nrm1496
Rizvi F, Heimann T, Herrnreiter A, O’Brien WJ (2011) Mitochondrial dysfunction links ceramide activated HRK expression and cell death. PLoS ONE 6(3):e18137. doi:10.1371/journal.pone.0018137
Rizzuto R, Pinton P, Ferrari D, Chami M, Szabadkai G, Magalhaes PJ, Di Virgilio F, Pozzan T (2003) Calcium and apoptosis: facts and hypotheses. Oncogene 22(53):8619–8627. doi:10.1038/sj.onc.1207105
Roderick HL, Cook SJ (2008) Ca2+ signalling checkpoints in cancer: remodelling Ca2+ for cancer cell proliferation and survival. Nat Rev Cancer 8(5):361–375. doi:10.1038/nrc2374
Ron D, Walter P (2007) Signal integration in the endoplasmic reticulum unfolded protein response. Nat Rev Mol Cell Biol 8(7):519–529. doi:10.1038/nrm2199
Runchel C, Matsuzawa A, Ichijo H (2011) Mitogen-activated protein kinases in mammalian oxidative stress responses. Antioxid Redox Signal 15(1):205–218. doi:10.1089/ars.2010.3733
Ruvolo PP (2003) Intracellular signal transduction pathways activated by ceramide and its metabolites. Pharmacol Res 47(5):383–392
Sabolic I, Breljak D, Skarica M, Herak-Kramberger CM (2010) Role of metallothionein in cadmium traffic and toxicity in kidneys and other mammalian organs. Biometals 23(5):897–926. doi:10.1007/s10534-010-9351-z
Saitoh M, Nishitoh H, Fujii M, Takeda K, Tobiume K, Sawada Y, Kawabata M, Miyazono K, Ichijo H (1998) Mammalian thioredoxin is a direct inhibitor of apoptosis signal-regulating kinase (ASK) 1. EMBO J 17(9):2596–2606. doi:10.1093/emboj/17.9.2596
Sakaki K, Wu J, Kaufman RJ (2008) Protein kinase Ctheta is required for autophagy in response to stress in the endoplasmic reticulum. J Biol Chem 283(22):15370–15380. doi:10.1074/jbc.M710209200
Sakamoto K, Iwasaki K, Sugiyama H, Tsuji Y (2009) Role of the tumor suppressor PTEN in antioxidant responsive element-mediated transcription and associated histone modifications. Mol Biol Cell 20(6):1606–1617. doi:10.1091/mbc.E08-07-0762
Sakurai A, Nishimoto M, Himeno S, Imura N, Tsujimoto M, Kunimoto M, Hara S (2005) Transcriptional regulation of thioredoxin reductase 1 expression by cadmium in vascular endothelial cells: role of NF-E2-related factor-2. J Cell Physiol 203(3):529–537. doi:10.1002/jcp.20246
Sancho P, Fernandez C, Yuste VJ, Amran D, Ramos AM, de Blas E, Susin SA, Aller P (2006) Regulation of apoptosis/necrosis execution in cadmium-treated human promonocytic cells under different forms of oxidative stress. Apoptosis 11(5):673–686
Sano R, Hou YC, Hedvat M, Correa RG, Shu CW, Krajewska M, Diaz PW, Tamble CM, Quarato G, Gottlieb RA, Yamaguchi M, Nizet V, Dahl R, Thomas DD, Tait SW, Green DR, Fisher PB, Matsuzawa S, Reed JC (2012) Endoplasmic reticulum protein BI-1 regulates Ca(2)(+)-mediated bioenergetics to promote autophagy. Genes Dev 26(10):1041–1054. doi:10.1101/gad.184325.111
Santos CX, Tanaka LY, Wosniak J, Laurindo FR (2009) Mechanisms and implications of reactive oxygen species generation during the unfolded protein response: roles of endoplasmic reticulum oxidoreductases, mitochondrial electron transport, and NADPH oxidase. Antioxid Redox Signal 11(10):2409–2427. doi:10.1089/ARS.2009.2625
Saydam N, Adams TK, Steiner F, Schaffner W, Freedman JH (2002) Regulation of metallothionein transcription by the metal-responsive transcription factor MTF-1: identification of signal transduction cascades that control metal-inducible transcription. J Biol Chem 277(23):20438–20445. doi:10.1074/jbc.M110631200
Scherz-Shouval R, Elazar Z (2011) Regulation of autophagy by ROS: physiology and pathology. Trends Biochem Sci 36(1):30–38. doi:10.1016/j.tibs.2010.07.007
Scorrano L, Oakes SA, Opferman JT, Cheng EH, Sorcinelli MD, Pozzan T, Korsmeyer SJ (2003) BAX and BAK regulation of endoplasmic reticulum Ca2+: a control point for apoptosis. Science 300(5616):135–139. doi:10.1126/science.1081208
Sedelnikova OA, Redon CE, Dickey JS, Nakamura AJ, Georgakilas AG, Bonner WM (2010) Role of oxidatively induced DNA lesions in human pathogenesis. Mutat Res 704(1–3):152–159. doi:10.1016/j.mrrev.2009.12.005
Sena LA, Chandel NS (2012) Physiological roles of mitochondrial reactive oxygen species. Mol Cell 48(2):158–167. doi:10.1016/j.molcel.2012.09.025
Senkal CE, Ponnusamy S, Manevich Y, Meyers-Needham M, Saddoughi SA, Mukhopadyay A, Dent P, Bielawski J, Ogretmen B (2011) Alteration of ceramide synthase 6/C16-ceramide induces activating transcription factor 6-mediated endoplasmic reticulum (ER) stress and apoptosis via perturbation of cellular Ca2+ and ER/Golgi membrane network. J Biol Chem 286(49):42446–42458. doi:10.1074/jbc.M111.287383
Shang F, Taylor A (2011) Ubiquitin-proteasome pathway and cellular responses to oxidative stress. Free Radic Biol Med 51(1):5–16. doi:10.1016/j.freeradbiomed.2011.03.031
Shaulian E, Karin M (2002) AP-1 as a regulator of cell life and death. Nat Cell Biol 4(5):E131–E136. doi:10.1038/ncb0502-e131
Shiloh Y, Ziv Y (2013) The ATM protein kinase: regulating the cellular response to genotoxic stress, and more. Nat Rev Mol Cell Biol 14(4):197–210. doi:10.1038/nrm3546
Shimizu S, Konishi A, Nishida Y, Mizuta T, Nishina H, Yamamoto A, Tsujimoto Y (2010) Involvement of JNK in the regulation of autophagic cell death. Oncogene 29(14):2070–2082. doi:10.1038/onc.2009.487
Shimoda R, Achanzar WE, Qu W, Nagamine T, Takagi H, Mori M, Waalkes MP (2003) Metallothionein is a potential negative regulator of apoptosis. Toxicol Sci 73(2):294–300. doi:10.1093/toxsci/kfg095
Shiraishi N, Waalkes MP (1994) Enhancement of metallothionein gene expression in male Wistar (WF/NCr) rats by treatment with calmodulin inhibitors: potential role of calcium regulatory pathways in metallothionein induction. Toxicol Appl Pharmacol 125(1):97–103. doi:10.1006/taap.1994.1053
Siemen D, Ziemer M (2013) What is the nature of the mitochondrial permeability transition pore and what is it not? IUBMB Life 65(3):255–262. doi:10.1002/iub.1130
Singh KP, Kumari R, Pevey C, Jackson D, DuMond JW (2009) Long duration exposure to cadmium leads to increased cell survival, decreased DNA repair capacity, and genomic instability in mouse testicular Leydig cells. Cancer Lett 279(1):84–92. doi:10.1016/j.canlet.2009.01.023
Sinha K, Das J, Pal PB, Sil PC (2013) Oxidative stress: the mitochondria-dependent and mitochondria-independent pathways of apoptosis. Arch Toxicol. doi:10.1007/s00204-013-1034-4
Slusarski DC, Pelegri F (2007) Calcium signaling in vertebrate embryonic patterning and morphogenesis. Dev Biol 307(1):1–13. doi:10.1016/j.ydbio.2007.04.043
Somji S, Garrett SH, Sens MA, Sens DA (2006) The unique N-terminal sequence of metallothionein-3 is required to regulate the choice between apoptotic or necrotic cell death of human proximal tubule cells exposed to Cd+ 2. Toxicol Sci 90(2):369–376. doi:10.1093/toxsci/kfj089
Son YO, Lee JC, Hitron JA, Pan J, Zhang Z, Shi X (2010) Cadmium induces intracellular Ca2+- and H2O2-dependent apoptosis through JNK- and p53-mediated pathways in skin epidermal cell line. Toxicol Sci 113(1):127–137. doi:10.1093/toxsci/kfp259
Son YO, Wang L, Poyil P, Budhraja A, Hitron JA, Zhang Z, Lee JC, Shi X (2012) Cadmium induces carcinogenesis in BEAS-2B cells through ROS-dependent activation of PI3K/AKT/GSK-3beta/beta-catenin signaling. Toxicol Appl Pharmacol 264(2):153–160. doi:10.1016/j.taap.2012.07.028
Song G, Ouyang G, Bao S (2005) The activation of Akt/PKB signaling pathway and cell survival. J Cell Mol Med 9(1):59–71
Souza V, Escobar Mdel C, Bucio L, Hernandez E, Gomez-Quiroz LE, Gutierrez Ruiz MC (2009) NADPH oxidase and ERK1/2 are involved in cadmium induced-STAT3 activation in HepG2 cells. Toxicol Lett 187(3):180–186. doi:10.1016/j.toxlet.2009.02.021
Stewart D, Killeen E, Naquin R, Alam S, Alam J (2003) Degradation of transcription factor Nrf2 via the ubiquitin-proteasome pathway and stabilization by cadmium. J Biol Chem 278(4):2396–2402. doi:10.1074/jbc.M209195200
Storr SJ, Carragher NO, Frame MC, Parr T, Martin SG (2011) The calpain system and cancer. Nat Rev Cancer 11(5):364–374. doi:10.1038/nrc3050
Sugawara H, Kurosaki M, Takata M, Kurosaki T (1997) Genetic evidence for involvement of type 1, type 2 and type 3 inositol 1,4,5-trisphosphate receptors in signal transduction through the B-cell antigen receptor. EMBO J 16(11):3078–3088. doi:10.1093/emboj/16.11.3078
Suzuki H, Tashiro S, Sun J, Doi H, Satomi S, Igarashi K (2003) Cadmium induces nuclear export of Bach1, a transcriptional repressor of heme oxygenase-1 gene. J Biol Chem 278(49):49246–49253. doi:10.1074/jbc.M306764200
Szegezdi E, Logue SE, Gorman AM, Samali A (2006) Mediators of endoplasmic reticulum stress-induced apoptosis. EMBO Rep 7(9):880–885. doi:10.1038/sj.embor.7400779
Tabas I, Ron D (2011) Integrating the mechanisms of apoptosis induced by endoplasmic reticulum stress. Nat Cell Biol 13(3):184–190. doi:10.1038/ncb0311-184
Taguchi K, Motohashi H, Yamamoto M (2011) Molecular mechanisms of the Keap1-Nrf2 pathway in stress response and cancer evolution. Genes Cells 16(2):123–140. doi:10.1111/j.1365-2443.2010.01473.x
Tait SW, Green DR (2010) Mitochondria and cell death: outer membrane permeabilization and beyond. Nat Rev Mol Cell Biol 11(9):621–632. doi:10.1038/nrm2952
Takeda K, Matsuzawa A, Nishitoh H, Tobiume K, Kishida S, Ninomiya-Tsuji J, Matsumoto K, Ichijo H (2004) Involvement of ASK1 in Ca2+-induced p38 MAP kinase activation. EMBO Rep 5(2):161–166. doi:10.1038/sj.embor.7400072
Takeda K, Naguro I, Nishitoh H, Matsuzawa A, Ichijo H (2011) Apoptosis signaling kinases: from stress response to health outcomes. Antioxid Redox Signal 15(3):719–761. doi:10.1089/ars.2010.3392
Tano JY, Lee RH, Vazquez G (2012) Involvement of calmodulin and calmodulin kinase II in tumor necrosis factor alpha-induced survival of bone marrow derived macrophages. Biochem Biophys Res Commun 427(1):178–184. doi:10.1016/j.bbrc.2012.09.038
Tchounwou PB, Ishaque AB, Schneider J (2001) Cytotoxicity and transcriptional activation of stress genes in human liver carcinoma cells (HepG2) exposed to cadmium chloride. Mol Cell Biochem 222(1–2):21–28
Tell G, Quadrifoglio F, Tiribelli C, Kelley MR (2009) The many functions of APE1/Ref-1: not only a DNA repair enzyme. Antioxid Redox Signal 11(3):601–620. doi:10.1089/ars.2008.2194
Templeton DM, Liu Y (2010) Multiple roles of cadmium in cell death and survival. Chem Biol Interact 188(2):267–275. doi:10.1016/j.cbi.2010.03.040
Tenev T, Bianchi K, Darding M, Broemer M, Langlais C, Wallberg F, Zachariou A, Lopez J, MacFarlane M, Cain K, Meier P (2011) The Ripoptosome, a signaling platform that assembles in response to genotoxic stress and loss of IAPs. Mol Cell 43(3):432–448. doi:10.1016/j.molcel.2011.06.006
Thévenod F (2003) Nephrotoxicity and the proximal tubule. Insights from cadmium. Nephron Physiol 93(4):87–93
Thévenod F (2009) Cadmium and cellular signaling cascades: to be or not to be? Toxicol Appl Pharmacol 238(3):221–239. doi:10.1016/j.taap.2009.01.013
Thévenod F, Chakraborty PK (2010) The role of Wnt/beta-catenin signaling in renal carcinogenesis: lessons from cadmium toxicity studies. Curr Mol Med 10(4):387–404
Thévenod F, Friedmann JM (1999) Cadmium-mediated oxidative stress in kidney proximal tubule cells induces degradation of Na+/K(+)-ATPase through proteasomal and endo-/lysosomal proteolytic pathways. FASEB J 13(13):1751–1761
Thévenod F, Lee WK (2013) Toxicology of cadmium and its damage to Mammalian organs. Met Ions Life Sci 11:415–490. doi:10.1007/978-94-007-5179-8_14
Thévenod F, Friedmann JM, Katsen AD, Hauser IA (2000) Up-regulation of multidrug resistance P-glycoprotein via nuclear factor-kappaB activation protects kidney proximal tubule cells from cadmium- and reactive oxygen species-induced apoptosis. J Biol Chem 275(3):1887–1896
Thévenod F, Wolff NA, Bork U, Lee WK, Abouhamed M (2007) Cadmium induces nuclear translocation of beta-catenin and increases expression of c-myc and Abcb1a in kidney proximal tubule cells. Biometals 20(5):807–820
Thijssen S, Cuypers A, Maringwa J, Smeets K, Horemans N, Lambrichts I, Van Kerkhove E (2007) Low cadmium exposure triggers a biphasic oxidative stress response in mice kidneys. Toxicology 236(1–2):29–41
Tirone F, Radu L, Craescu CT, Cox JA (2010) Identification of the binding site for the regulatory calcium-binding domain in the catalytic domain of NOX5. Biochemistry 49(4):761–771. doi:10.1021/bi901846y
Tokumoto M, Fujiwara Y, Shimada A, Hasegawa T, Seko Y, Nagase H, Satoh M (2011) Cadmium toxicity is caused by accumulation of p53 through the down-regulation of Ube2d family genes in vitro and in vivo. J Toxicol Sci 36(2):191–200
Tomar D, Singh R, Singh AK, Pandya CD, Singh R (2012) TRIM13 regulates ER stress induced autophagy and clonogenic ability of the cells. Biochim Biophys Acta 1823(2):316–326. doi:10.1016/j.bbamcr.2011.11.015
Trachootham D, Lu W, Ogasawara MA, Nilsa RD, Huang P (2008) Redox regulation of cell survival. Antioxid Redox Signal 10(8):1343–1374. doi:10.1089/ars.2007.1957
Tsang KY, Chan D, Bateman JF, Cheah KS (2010) In vivo cellular adaptation to ER stress: survival strategies with double-edged consequences. J Cell Sci 123(Pt 13):2145–2154. doi:10.1242/jcs.068833
Tsirigotis M, Zhang M, Chiu RK, Wouters BG, Gray DA (2001) Sensitivity of mammalian cells expressing mutant ubiquitin to protein-damaging agents. J Biol Chem 276(49):46073–46078. doi:10.1074/jbc.M109023200
Tvermoes BE, Bird GS, Freedman JH (2011) Cadmium induces transcription independently of intracellular calcium mobilization. PLoS ONE 6(6):e20542. doi:10.1371/journal.pone.0020542
Ullman E, Fan Y, Stawowczyk M, Chen HM, Yue Z, Zong WX (2008) Autophagy promotes necrosis in apoptosis-deficient cells in response to ER stress. Cell Death Differ 15(2):422–425. doi:10.1038/sj.cdd.4402234
Venugopal R, Jaiswal AK (1998) Nrf2 and Nrf1 in association with Jun proteins regulate antioxidant response element-mediated expression and coordinated induction of genes encoding detoxifying enzymes. Oncogene 17(24):3145–3156. doi:10.1038/sj.onc.1202237
Verfaillie T, Rubio N, Garg AD, Bultynck G, Rizzuto R, Decuypere JP, Piette J, Linehan C, Gupta S, Samali A, Agostinis P (2012) PERK is required at the ER-mitochondrial contact sites to convey apoptosis after ROS-based ER stress. Cell Death Differ 19(11):1880–1891. doi:10.1038/cdd.2012.74
Vicencio JM, Ortiz C, Criollo A, Jones AW, Kepp O, Galluzzi L, Joza N, Vitale I, Morselli E, Tailler M, Castedo M, Maiuri MC, Molgo J, Szabadkai G, Lavandero S, Kroemer G (2009) The inositol 1,4,5-trisphosphate receptor regulates autophagy through its interaction with Beclin 1. Cell Death Differ 16(7):1006–1017. doi:10.1038/cdd.2009.34
Vitale I, Galluzzi L, Castedo M, Kroemer G (2011) Mitotic catastrophe: a mechanism for avoiding genomic instability. Nat Rev Mol Cell Biol 12(6):385–392. doi:10.1038/nrm3115
Vogelstein B, Lane D, Levine AJ (2000) Surfing the p53 network. Nature 408(6810):307–310. doi:10.1038/35042675
Waisberg M, Joseph P, Hale B, Beyersmann D (2003) Molecular and cellular mechanisms of cadmium carcinogenesis. Toxicology 192(2–3):95–117
Wang XZ, Ron D (1996) Stress-induced phosphorylation and activation of the transcription factor CHOP (GADD153) by p38 MAP Kinase. Science 272(5266):1347–1349
Wang HG, Pathan N, Ethell IM, Krajewski S, Yamaguchi Y, Shibasaki F, McKeon F, Bobo T, Franke TF, Reed JC (1999) Ca2+-induced apoptosis through calcineurin dephosphorylation of BAD. Science 284(5412):339–343
Wang Y, Fang J, Leonard SS, Rao KM (2004) Cadmium inhibits the electron transfer chain and induces reactive oxygen species. Free Radic Biol Med 36(11):1434–1443
Wang S, Tang M, Pei B, Xiao X, Wang J, Hang H, Wu L (2008a) Cadmium-induced germline apoptosis in Caenorhabditis elegans: the roles of HUS1, p53, and MAPK signaling pathways. Toxicol Sci 102(2):345–351. doi:10.1093/toxsci/kfm220
Wang SH, Shih YL, Ko WC, Wei YH, Shih CM (2008b) Cadmium-induced autophagy and apoptosis are mediated by a calcium signaling pathway. Cell Mol Life Sci 65(22):3640–3652. doi:10.1007/s00018-008-8383-9
Wang SH, Shih YL, Lee CC, Chen WL, Lin CJ, Lin YS, Wu KH, Shih CM (2009) The role of endoplasmic reticulum in cadmium-induced mesangial cell apoptosis. Chem Biol Interact 181(1):45–51. doi:10.1016/j.cbi.2009.05.004
Wang Z, Wang H, Xu ZM, Ji YL, Chen YH, Zhang ZH, Zhang C, Meng XH, Zhao M, Xu DX (2012) Cadmium-induced teratogenicity: association with ROS-mediated endoplasmic reticulum stress in placenta. Toxicol Appl Pharmacol 259(2):236–247. doi:10.1016/j.taap.2012.01.001
Ward CL, Omura S, Kopito RR (1995) Degradation of CFTR by the ubiquitin-proteasome pathway. Cell 83(1):121–127
Wiegert JS, Bading H (2011) Activity-dependent calcium signaling and ERK-MAP kinases in neurons: a link to structural plasticity of the nucleus and gene transcription regulation. Cell Calcium 49(5):296–305. doi:10.1016/j.ceca.2010.11.009
Williams RJ (2002) The fundamental nature of life as a chemical system: the part played by inorganic elements. J Inorg Biochem 88(3–4):241–250
Winterbourn CC, Hampton MB (2008) Thiol chemistry and specificity in redox signaling. Free Radic Biol Med 45(5):549–561. doi:10.1016/j.freeradbiomed.2008.05.004
Won JS, Singh I (2006) Sphingolipid signaling and redox regulation. Free Radic Biol Med 40(11):1875–1888. doi:10.1016/j.freeradbiomed.2006.01.035
Wu KC, Liu JJ, Klaassen CD (2012) Nrf2 activation prevents cadmium-induced acute liver injury. Toxicol Appl Pharmacol 263(1):14–20. doi:10.1016/j.taap.2012.05.017
Xiao W, Liu Y, Templeton DM (2009) Pleiotropic effects of cadmium in mesangial cells. Toxicol Appl Pharmacol 238(3):315–326. doi:10.1016/j.taap.2009.02.005
Xiao Q, Hartzell HC, Yu K (2010) Bestrophins and retinopathies. Pflugers Arch 460(2):559–569. doi:10.1007/s00424-010-0821-5
Xie J, Shaikh ZA (2006a) Cadmium-induced apoptosis in rat kidney epithelial cells involves decrease in nuclear factor-kappa B activity. Toxicol Sci 91(1):299–308. doi:10.1093/toxsci/kfj131
Xie J, Shaikh ZA (2006b) Cadmium induces cell cycle arrest in rat kidney epithelial cells in G2/M phase. Toxicology 224(1–2):56–65. doi:10.1016/j.tox.2006.04.026
Xu W, Liu L, Charles IG, Moncada S (2004) Nitric oxide induces coupling of mitochondrial signalling with the endoplasmic reticulum stress response. Nat Cell Biol 6(11):1129–1134. doi:10.1038/ncb1188
Yacoub A, Hamed HA, Allegood J, Mitchell C, Spiegel S, Lesniak MS, Ogretmen B, Dash R, Sarkar D, Broaddus WC, Grant S, Curiel DT, Fisher PB, Dent P (2010) PERK-dependent regulation of ceramide synthase 6 and thioredoxin play a key role in mda-7/IL-24-induced killing of primary human glioblastoma multiforme cells. Cancer Res 70(3):1120–1129. doi:10.1158/0008-5472.CAN-09-4043
Yang Z, Klionsky DJ (2010) Mammalian autophagy: core molecular machinery and signaling regulation. Curr Opin Cell Biol 22(2):124–131. doi:10.1016/j.ceb.2009.11.014
Yang PM, Chiu SJ, Lin KA, Lin LY (2004) Effect of cadmium on cell cycle progression in Chinese hamster ovary cells. Chem Biol Interact 149(2–3):125–136
Yang PM, Chen HC, Tsai JS, Lin LY (2007a) Cadmium induces Ca2+-dependent necrotic cell death through calpain-triggered mitochondrial depolarization and reactive oxygen species-mediated inhibition of nuclear factor-kappaB activity. Chem Res Toxicol 20(3):406–415. doi:10.1021/tx060144c
Yang S, Misner BJ, Chiu RJ, Meyskens FL Jr (2007b) Redox effector factor-1, combined with reactive oxygen species, plays an important role in the transformation of JB6 cells. Carcinogenesis 28(11):2382–2390. doi:10.1093/carcin/bgm128
Yang Z, Yang S, Qian SY, Hong JS, Kadiiska MB, Tennant RW, Waalkes MP, Liu J (2007c) Cadmium-induced toxicity in rat primary mid-brain neuroglia cultures: role of oxidative stress from microglia. Toxicol Sci 98(2):488–494. doi:10.1093/toxsci/kfm106
Yang S, Misner B, Chiu R, Meyskens FL Jr (2008) Common and distinct mechanisms of different redox-active carcinogens involved in the transformation of mouse JB6P+ cells. Mol Carcinog 47(7):485–491. doi:10.1002/mc.20410
Yano S, Tokumitsu H, Soderling TR (1998) Calcium promotes cell survival through CaM-K kinase activation of the protein-kinase-B pathway. Nature 396(6711):584–587. doi:10.1038/25147
Yazaki M, Kashiwagi K, Aritake K, Urade Y, Fujimori K (2012) Rapid degradation of cyclooxygenase-1 and hematopoietic prostaglandin D synthase through ubiquitin-proteasome system in response to intracellular calcium level. Mol Biol Cell 23(1):12–21. doi:10.1091/mbc.E11-07-0623
Yen AH, Yang JL (2010) Cdc20 proteolysis requires p38 MAPK signaling and Cdh1-independent APC/C ubiquitination during spindle assembly checkpoint activation by cadmium. J Cell Physiol 223(2):327–334. doi:10.1002/jcp.22038
Yokouchi M, Hiramatsu N, Hayakawa K, Kasai A, Takano Y, Yao J, Kitamura M (2007) Atypical, bidirectional regulation of cadmium-induced apoptosis via distinct signaling of unfolded protein response. Cell Death Differ 14(8):1467–1474. doi:10.1038/sj.cdd.4402154
Yokouchi M, Hiramatsu N, Hayakawa K, Okamura M, Du S, Kasai A, Takano Y, Shitamura A, Shimada T, Yao J, Kitamura M (2008) Involvement of selective reactive oxygen species upstream of proapoptotic branches of unfolded protein response. J Biol Chem 283(7):4252–4260. doi:10.1074/jbc.M705951200
Yoon S, Woo SU, Kang JH, Kim K, Kwon MH, Park S, Shin HJ, Gwak HS, Chwae YJ (2010) STAT3 transcriptional factor activated by reactive oxygen species induces IL6 in starvation-induced autophagy of cancer cells. Autophagy 6(8):1125–1138
Youle RJ, Strasser A (2008) The BCL-2 protein family: opposing activities that mediate cell death. Nat Rev Mol Cell Biol 9(1):47–59. doi:10.1038/nrm2308
Yousefi S, Perozzo R, Schmid I, Ziemiecki A, Schaffner T, Scapozza L, Brunner T, Simon HU (2006) Calpain-mediated cleavage of Atg5 switches autophagy to apoptosis. Nat Cell Biol 8(10):1124–1132. doi:10.1038/ncb1482
Yu X, Hong S, Faustman EM (2008) Cadmium-induced activation of stress signaling pathways, disruption of ubiquitin-dependent protein degradation and apoptosis in primary rat Sertoli cell-gonocyte cocultures. Toxicol Sci 104(2):385–396. doi:10.1093/toxsci/kfn087
Yu X, Sidhu JS, Hong S, Robinson JF, Ponce RA, Faustman EM (2011) Cadmium induced p53-dependent activation of stress signaling, accumulation of ubiquitinated proteins, and apoptosis in mouse embryonic fibroblast cells. Toxicol Sci 120(2):403–412. doi:10.1093/toxsci/kfr010
Zhang K, Kaufman RJ (2006) Protein folding in the endoplasmic reticulum and the unfolded protein response. Handb Exp Pharmacol 172:69–91
Zhang GH, Yamaguchi M, Kimura S, Higham S, Kraus-Friedmann N (1990) Effects of heavy metal on rat liver microsomal Ca2(+)-ATPase and Ca2+ sequestering. Relation to SH groups. J Biol Chem 265(4):2184–2189
Zhao Y, Hu J, Miao G, Qu L, Wang Z, Li G, Lv P, Ma D, Chen Y (2013) Transmembrane protein 208: a novel ER-localized protein that regulates autophagy and ER stress. PLoS ONE 8(5):e64228. doi:10.1371/journal.pone.0064228
Zhivotovsky B, Orrenius S (2011) Calcium and cell death mechanisms: a perspective from the cell death community. Cell Calcium 50(3):211–221. doi:10.1016/j.ceca.2011.03.003
Zigdon H, Kogot-Levin A, Park JW, Goldschmidt R, Kelly S, Merrill AH Jr, Scherz A, Pewzner-Jung Y, Saada A, Futerman AH (2013) Ablation of ceramide synthase 2 causes chronic oxidative stress due to disruption of the mitochondrial respiratory chain. J Biol Chem 288(7):4947–4956. doi:10.1074/jbc.M112.402719
Zoncu R, Efeyan A, Sabatini DM (2011) mTOR: from growth signal integration to cancer, diabetes and ageing. Nat Rev Mol Cell Biol 12(1):21–35. doi:10.1038/nrm3025
Acknowledgments
We apologize to the many scientists whose work we were not able to credit due to space restrictions. We thank the current and previous members of the laboratory for many stimulating ideas, and Ann Cuypers, Andrea Hartwig, Wolfgang Maret and Jean-Marc Moulis for helpful discussions. Research in the laboratory is funded by The German Research Foundation (DFG) (grants FT345/8-1 to FT345/11-1), the Westermann-Westerdorp Foundation and the Centre for Biomedical Research and Training (ZBAF) at the University of Witten/Herdecke.
Author information
Authors and Affiliations
Corresponding authors
Rights and permissions
About this article
Cite this article
Thévenod, F., Lee, WK. Cadmium and cellular signaling cascades: interactions between cell death and survival pathways. Arch Toxicol 87, 1743–1786 (2013). https://doi.org/10.1007/s00204-013-1110-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00204-013-1110-9