
Abstract
Cell death maintains cell morphology and homeostasis during development by removing damaged or obsolete cells. The concentration of metal ions whithin cells is regulated by various intracellular transporters and repositories to maintain dynamic balance. External or internal stimuli might increase the concentration of metal ions, which results in ions overloading. Abnormal accumulation of large amounts of metal ions can lead to disruption of various signaling in the cell, which in turn can produce toxic effects and lead to the occurrence of different types of cell deaths. In order to further study the occurrence and development of metal ions overloading induced cell death, this paper reviewed the regulation of Ca2+, Fe3+, Cu2+ and Zn2+ metal ions, and the internal mechanism of cell death induced by overloading. Furthermore, we found that different metal ions possess a synergistic and competitive relationship in the regulation of cell death. And the enhanced level of oxidative stress was present in all the processes of cell death due to metal ions overloading, which possibly due to the combination of factors. Therefore, this review offers a theoretical foundation for the investigation of the toxic effects of metal ions, and presents innovative insights for targeted regulation and therapeutic intervention.
Graphical Abstract

Highlights
• Metal ions overloading disrupts homeostasis, which in turn affects the regulation of cell death.
• Metal ions overloading can cause cell death via reactive oxygen species (ROS).
• Different metal ions have synergistic and competitive relationships for regulating cell death.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Abnormality of Cell death is closely related to many diseases. Cell death can be both programmed and non-programmed. Programmed cell death(PCD) is a complex and precise, genetically controlled cellular process, and includes apoptosis, autophagy, pyroptosis, necroptosis, ferroptosis and cuprotosis. Diverse modes of cell death often involve various of signaling cascades and molecular mechanisms with unique morphological and biochemical features. We have found that iron ions were involved in radiation-induced cell death, especially ferroptosis (Afroze et al. 2003; Ahn et al. 2014). Meanwhile, several studies have found that calcium overloading is strongly associated with apoptosis and ferroptosis (Alhillawi et al. 2021; Arosio et al. 2009). It has also been demonstrated that disruption of intracellular calcium homeostasis gives rise to endoplasmic reticulum stress and activation of the unfolded protein response (UPR), which can result in apoptosis (Baev 2022). The question was then raised whether the disruption of homeostasis by metal ions overloading could affect the regulation of cell death.
Common intracellular metal ions include calcium (Ca2+), ferric (Fe3+), ferrous (Fe2+), copper (Cu2+), zinc (Zn2+), sodium (Na+), potassium (K+), etc. Under normal circumstances, the concentration of metal ions is maintained dynamic balance under the regulation of various transporter proteins and storage libraries. But when subjected to radiation, tumors, or accidental injuries, which induce high intracellular concentration of metal ions and cause ionic overloading, a large number of metal ions abnormally accumulates and binds to inappropriate receptor sites, which will lead to signaling disorders and produce toxic effects (Bagur et al. 2017; Bai et al. 2022; Balashova et al. 2020; B'Chir et al. 2014). In recent years, increasing evidence suggested that Fe3+ and Cu2+overloading could cause a toxic stress response ultimately leading to cell death (Berezhnov et al. 2008; Bernardi and Bernardi 2013).
This paper elucidated the roles and mechanisms of Ca2+, Fe3+, Cu2+ and Zn2+ overloading in inducing cell death, which might fulfil theoretical basis for the study of the toxic effects of metal ions and provided novel insights for targeted regulation and therapeutic intervention.
Calcium overloading and cell death
Occurrence of calcium overloading
Intracellular calcium homeostasis plays a crucial role in determining cellular function and survival. Mitochondria and endoplasmic reticulum, as the major calcium storage systems in the cytoplasm, play key roles in regulating intracellular Ca2+ signaling and maintaining intracellular calcium homeostasis. As an critical second messenger, Ca2+plays a significant role in regulating signaling pathways associated with cell differentiation, growth and proliferation. The occurrence and progression of numerous diseases are intimately linked to calcium overloading (Blaby-Haas et al. 2014). Calcium overloading is a significant increase in intracellular Ca2+ concentration caused by a variety of triggers, resulting in structural or functional impairments in the cell. The mechanisms of generation include massive inward flow of extracellular Ca2+, release from intracellular calcium stores, including the endoplasmic reticulum, mitochondria, and sarcoplasmic reticulum, as well as compromised Ca2+recycling and outward migration (Boal et al. 2009). When exogenous stimuli or other deleterious factors, such as membrane depolarization, disruption of mitochondrial structure and function, activation of calcium channels, activation of extracellular signaling molecules or intracellular messengers, can cause dysfunction of the calcium homeostasis system and disturbances in calcium distribution, leading to a sustained increase in intracellular Ca2+concentration and placing the cell in a state of calcium overloading (Bonaventura et al. 2015; Bossy-Wetzel et al. 2004; Bostanci et al. 2014).
Calcium overloading and apoptosis
When intracellular calcium overloading occurs, Ca2+ activates Ca2+-dependent phospholipases, which can cause membrane phospholipolysis, and increased intracytoplasmic Ca2+can contribute to the overproduction of ROS and cause membrane lipid peroxidation, thereby resulting in the disruption of the structure and function of the cell membrane (Brady et al. 2014; Britton et al. 2016; Camaschella et al. 2020). Meanwhile, free fatty acids, prostaglandins, leukotrienes, and lysophospholipids produced during the decomposition process can have toxic effects on cells (Cappetta et al. 2021; Cave et al. 2006). When calcium overloading akes place within cells, mitochondria take up Ca2+from the cytoplasm. This, in turn induces the opening of the permeability transition pore (PTP) and disrupts the mitochondrial membrane potential. The PTP, a group of protein complexes located between the inner and outer membranes of the mitochondrion, is a nonspecific channel that plays a crucial position in cell survival and apoptosis (Cen et al. 2018). When the PTP opens, large amounts of Ca2+ are released from calcium pools in the mitochondria, inducing apoptosis via caspase-dependent or caspase-independent pathways. This is accompanied by the release of cytochrome C from mitochondria, the activation of caspases, DNA fragmentation, nuclear fragmentation, and ultimately cell death. When intracellular calcium is overloaded, Ca2+can also bind to PTP, leading to mitochondrial swelling and dysfunction, causing apoptosis (Chen et al. 2014; Chen et al. 2020).
Apoptotic are strongly associated with apoptosis regulators in addition to the disruption of mitochondrial membrane potential. The anti-apoptotic members of the BCL-2 family play a pivotal role in apoptosis. BCL-x can interact with the calcium channel IP3R on the ER to effectively limit Ca2+transfer from the endoplasmic reticulum to mitochondria (Chen et al. 2020). The BCL2 family apoptosis regulator BOK is capable of triggering apoptosis in the absence of BAX and BAK. It has been demonstrated that BOK binds to IP3R and provides protection against protein hydrolysis. However, more evidence is requisite to elucidate whether BOK modifies Ca2+efflux from the ER via IP3R (Chen et al. 2021; Cheng et al. 2022; Chu 2024).
Studies have shown that calpain is a Ca2+-dependent cysteine protease that mediates the occurrence of apoptosis and programmed cell death (PCD) when cells undergo calcium overloading (Collins et al. 2010; Davaanyam et al. 2023; de Oliveira Otto et al. 2011). Calcium overloading can cause endoplasmic reticulum stress through calcium-dependent proteases, which disrupt calcium homeostasis in the endoplasmic reticulum, activate its mediated apoptotic pathway, promote the release of apoptotic proteins, and then trigger apoptosis (de Romaña et al. 2011; Dhaouadi et al. 2023; Dixon et al. 2012). Ca2+also directly activates calpain, which promotes the cleavage of fodrin, a component of the cytoskeleton, thereby inducing cell death (Doguer et al. 2018; Dong et al. 2023; Eckenrode et al. 2010).
Calcium overloading and autophagy
The disruption of the endoplasmic reticulum environment, the restriction of protein production, and the imbalance of calcium homeostasis during periods of stress cause increased aggregation of unfolded or misfolded proteins, which is known as the endoplasmic reticulum stress response. Studies have shown that ER stress can potentially trigger autophagy (Eom et al. 2016; Fang 2017), and the endoplasmic reticulum activates PERK, ATF6, and IRE1 upon stress. eIF2a-involved phosphorylation of PERK enhances translation of downstream ATF4 mRNA, and activated PERK can also phosphorylate NRF2. ATF4 and NRF2 are translocated to the nucleus and activate the transcription factor CHOP, subsequently inducing a cascade of downstream genes associated with autophagy, apoptosis, amino acid metabolism, and others (Feng et al. 2022; Feng et al. 2022). Moreover, endoplasmic reticulum stress also induces the opening of IP3R channels, releasing a large amount of Ca2+into the cytoplasm, which activates kinases involved in autophagy, such as CAMKK2, DAPK, etc. CAMKK2 can activate the downstream AMPK, which can inhibit the autophagy induced by mTOR, while DAPK can stimulate the phosphorylation of Beclin 1 to activate autophagy (Filetti et al. 2018; Fonseca-Nunes et al. 2014; Forrester et al. 2018).
Calcium overloading and cancer treatment
An imbalance in calcium homeostasis is directly linked to the progression and transformation of many cancers, etc. Therefore, more and more researches focus on calcium overloading and cell death for tumor therapy, including various nanomaterials, radiotherapy drugs, and biotargeted therapies. Nano-ionic HAPNs have demonstrated the ability to induce calcium overloading in tumor cells by inhibiting the calcium pump PMCA and activating tumor cell calpain. This ultimately leads to specific apoptosis of cancer cells, thereby playing a significant role in cancer therapy (Frey et al. 2014).
Nanomaterial CaO2 can enter the cell can simultaneously generate ROS and rapidly release Ca2+, causing calcium channel imbalance induced calcium overloading, oxidative stress leading to death. It is widely used in the treatment of liver cancer, breast cancer, lung cancer and colorectal cancer (Fukuda et al. 1998). However, due to the chemical properties of CaO2 itself will cause certain side effects to the organism, so the current CaO2-based treatment is mainly nanocomposites. Nanoparticles made of CaO2 integrated with the antibody GPC3Ab are guided to tumor cells by GPC3Ab, and CaO2 releases Ca2+ and H2O2to induce apoptosis of cancer cells, and this therapy can be targeted for the treatment of hepatocellular carcinoma (Fusakio et al. 2016). In addition to this, calcium-based nanomaterials such as CaCO3, CaP, CaH2, CaF2, etc. can exert a role in tumor localization by induction of calcium overloading, mitochondrial dysfunction, and oxidative stress in the application of tumor therapy (Gaetke et al. 2014) (Fig.1).

Mechanism of Ca2+ overload-induced cell death. Cytoplasmic calcium overload causes a burst of intracellular ROS, which, on the one hand, acts directly on the cell membrane, causing membrane lipid peroxidation, resulting in cell membrane rupture and causing death, and on the other hand, stimulates the activation of mitochondria and endoplasmic reticulum to activate downstream factors that regulate cell death to induce various kinds of death
Figure 1 is about here
Iron overloading and cell death
Occurrence of iron overloading
Iron is essential for cellular survival and is a basic component of hemoglobin, myoglobin and iron-sulfur proteins, which are mainly responsible for cellular respiration, oxygen transport, DNA synthesis and other key functions in biology, with complex storage and transport mechanisms. The liver is the main site of secretion of iron-regulated proteins, which mainly expresses hepcidin, TfR 2, and hemojuvelin(HJV) and other proteins. Hepcidin plays a crucial role in maintaining the balance of iron metabolism by regulating the absorption of intracellular iron through binding to ferroportin(FPN) on the cell membrane. It further controls serum iron concentration to prevent the formation of iron overloading (Gao et al. 2021; Genoud et al. 2017).
Iron metabolism homeostasis is regulated by several factors (Guha et al. 2010). Under physiological conditions, the metabolism of iron is influenced by the absorption of dietary iron. Within the diet, non-heme iron is absorbed and utilized by duodenal epithelial cells via the conversion of Fe3+ to Fe2+ through the ferric reductase STEAP3. Fe2+ can be transferred to the cytoplasm by means of divalent metal transporter protein 1 (DMT1), and Fe2+ aggregation leads to the destabilization of iron pools in the cytoplasm and mitochondria. Since Fe2+ is a highly redox-active iron, it can induce redox reactions. Poly(rC)-binding proteins (PCBPs) in the cytoplasm can transport Fe2+ in combination with Fe2+, storing Fe2+as Ferritin (Guo et al. 2022; Guo et al. 2022; Guo et al. 2022).
The expression of ferritin is intricately regulated by levels of iron and the presence of oxidative damage (Guo et al. 2023). When endogenous iron-regulating proteins are deficient or exogenous iron intake is excessive, iron metabolic homeostasis is disrupted and iron overloading occurs. Subsequently, large amounts of ROS and cytotoxicity are generated, even triggering the cells to undergo ferroptosis leading to dysfunction.
Iron overloading and ferroptosis
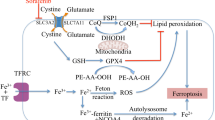
Excessive intracellular Fe2+induces ferroptosis by generating substantial amounts of ROS through Fenton reactions on various intracellular lipid membranes. It has been found that nuclear receptor coactivator 4 (NCOA4) is capable of interacting with ferritin and facilitating its autophagic degradation, followed by the release of a considerable amount of free Fe. This free Fe can activate sideroflexin 1 (SFXN1) on the inner mitochondrial membrane to translocate free iron to the mitochondria, thereby inducing mitochondrial iron overloading, outbreak of ROS and ferroptosis (Gupte and Mumper 2009). It was demonstrated that HMGB1 mediates the up-regulation of hepcidin, which subsequently governs intracellular iron accumulation and induces ferroptosis (Han et al. 2022). It has been reported that the iron chelator desferrioxamine (DFO) inhibited ferroptosis (He et al. 2021), on the contrary, increasing intracellular iron ions promoted ferroptosis through the ferroptosis inducer erastin. Moreover, inhibition of transferrin TFRC on the cell membrane, which consequently restrains the entry of iron ions into the cell, also inhibited erastin-induced ferroptosis (Horn 2021; Høyer-Hansen et al. 2007).
Heme oxygenase (HO-1) is an antioxidant enzyme and one of the important sources of intracellular iron, which mainly promotes the decomposition of heme into ferrous and other products. HO-1 is upregulated when oxidative stress and cellular injury occur, while Nrf2 also promotes HO-1 promoter activity. Upregulation of HO-1 can cause heme degradation, release a large amount of free and unstable ferrous ions Fe2+, and cause intracellular iron overloading. These Fe2+can catalyze the Fenton reaction, causing to the generation of a substantial quantity of lipid peroxides and ROS. This process results in the oxidation of cell membrane and organelles. Subsequently, the damaged mitochondria release more heme and ROS, thus initiating a vicious ferroptosis cycle (Hwang et al. 2008; Ishida et al. 2013).
Iron overloading and apoptosis, necroptosis
When the accumulation of iron ions exceeds the amount excreted, it can have some deleterious effects on tissues, including the skeleton. It has been proved that iron overloading is a risk factor for the occurrence of osteoporosis, and it has been found that excess iron ions have toxic effects on bone cells, and that iron overloading increases oxidative stress, cell cycle block, promotes apoptosis in bone cells, affects osteoclast differentiation, and destroys bone cell morphology and function (James et al. 2017; Karim et al. 2022). Furthermore, iron overloading has been demonstrated to trigger the opening of the mitochondrial permeability transition pore (mPTP), which mediates the RIPK1/RIPK3/MLKL pathway and necroptosis induction (Kim and Koh 2002; Kimura et al. 2016). The liver serves as an important iron storage site, and iron overloading-induced oxidative stress, lipid peroxidation, and DNA damage can cause a great deal of acute and chronic damage to liver tissue. Numerous studies have demonstrated that long-term iron overloading is correlated with a variety of liver diseases, including hepatitis, hepatic fibrosis, cirrhosis and tumor development (Kohzadi et al. 2017; Lee et al. 2018; Leidgens et al. 2013). In addition, the heart is one of the organs where iron overloading injury is prevalent. In the situation where there are excessive unstable iron ions in cardiomyocytes, transferrin is inhibited, thereby inducing ROS production and cardiomyocyte apoptosis through mitochondria-mediated activation of caspase-3-dependent caspases (Li 2020).
Iron overloading and autophagy
Numerous studies have demonstrated that iron overloading inhibits cell proliferative vigor and induces osteoblast apoptosis. Recent studies have demonstrated that iron overloading induces apoptosis in osteoblasts while activating autophagy to exert a cytoprotective role (Li et al. 2020). It has also been demonstrated that iron overloading induces the generation of ROS, impairs the PI3K/AKT and Jak/Stat3 signaling pathways, and activates p38 MAPK, thereby stimulating autophagy in osteoblasts (Li 2024). In addition, iron overloading can impair the proliferation of endometrial stromal cells while activating their protective PAP1/SIRT1 autophagy signaling (Lin et al. 2017). In addition to protective autophagy, iron overloading can activate LC3II/I up-regulation in cells, inducing hepatocytes to generate a large number of autophagic vesicles, undergo excessive autophagy and apoptosis, and thus cause some liver injury (Lin 2019) (Fig. 2).

Mechanism of Fe3+ overload-induced cell death. Cytoplasmic iron overload activates ROS and initiates oxidative stress, causing cellular ferroptosis, apoptosis and autophagy
Copper overloading and cell death
Occurrence of copper overloading
Cu is involved in the synthesis of various enzymes for instance copper/zinc superoxide dismutase (Cu/ZnSOD or SOD1), cytochrome c oxidase, and mitogen-activated protein kinase MEK1 (Liu et al. 2020). Copper is a vital catalytic cofactor, playing a critical function in redox reactions and various biological processes. These processes include the neutralization of free radicals, angiogenesis, cell respiration, and proliferation (Liu et al. 2021).
Under normal conditions, the maintenance of copper homeostasis is dependent on the processes of intestinal absorption, bile excretion, and intrahepatic storage. Cu absorbed by the small intestine can be transported to the liver by plasma proteins, albumin, and transluthrins. The liver, as the primary catcher, distributor, and excreta receiver of copper, is also the core role in regulating copper homeostasis (Liu et al. 2022; Llambi et al. 2016). During cellular uptake of copper, Cu2+ is initially reduced to Cu+ by reductase. Subsequently, the Cu+ enters the cytoplasm through copper transport protein 1 (CTR1) on the cell membrane. Thereafter, Cu+is bound and transported to specific target proteins by glutathione (GSH) or copper molecule chaperone proteins, CCS, COX17, and ATOX1, to fulfill their functions (Llanos and Mercer 2002; Łopatniuk and Witkowski 2011; Ma et al. 2022). Excess intracellular cuprous ions (Cu+) are usually chelated by metallothioneins (MT1 and MT2) or stored in lysosomes as reservoirs to maintain copper homeostasis and avoid disorders (Mao and Huang 2013; Marmolejo-Garza et al. 2023).
When copper homeostasis is disrupted, it may induce the development of various diseases. Typical hereditary illnesses of impaired copper metabolism include Wilson's disease and Menkes syndrome. Mutations in ATP7A, the gene encoding the transmembrane copper-transporting adenine nucleoside triphosphate (ATP) enzyme, are the main causative agent of Menkes syndrome. The pathogenesis of Wilson's disease is due to ATP7B gene mutation leading to copper transport dysfunction or abnormal protein transport, resulting in abnormal accumulation of copper in certain tissues (Marreiro 2017; McAlary et al. 2022). Moreover, copper imbalance has been implicated in the mechanism of pathogenesis of Alzheimer's disease, Parkinson's disease, and Huntington's disease (Miao et al. 2023; Mohr and Weiss 2019).
Copper overloading and cuprotosis
The most significant reason for the cytotoxicity caused by copper lies in the potent pro-oxidation of Cu+, which is implicated in ROS generation, triggering free radical-induced oxidative stress damage and ultimately resulting in cell death. Moreover, ROS destroy organic macromolecules, including proteins, nucleic acids, and lipids, and can also regulate a variety of cellular signaling pathways, activate pro-tumor signals, and promote tumor proliferation. In addition, it has been found that copper death, when Cu2+ is over-accumulated in cells, intracellular Cu2+ is converted to the more oxidizing Cu+ by the reductase FDX1. On the one hand, reduction generates Cu+, which hinders the synthesis of Fe-S cluster proteins in mitochondria. On the other hand, FDX1 also governs intracellular protein thiooctanoylation, a conserved post-translational modification of lysines. The excess Cu2+in the cell binds to the lipoacylated protein DLAT, which induces the ester acylated protein to aggregate. At the same time, inhibition of Fe-S cluster proteins synthesis induces proteotoxic stress and cell death (Bernardi and Bernardi 2013).
Copper overloading and ferroptosis
Copper, as an essential ligand for Cu/Zn superoxide dismutase, plays a vital position in the antioxidant process. There is evidence that copper ions interact with iron ions and can modulate the onset of iron death. Copper has been shown to inhibit ferroptosis and protect neuronal cells to ameliorate neurodegeneration. However, the role of copper metabolism in regulating ferroptosis requires further support from additional evidence. Excessive copper accumulation disrupts mitochondrial respiration, targeting its antioxidant and iron death-inhibiting effects, and thus copper chelators have applications in tumor therapy (Muckenthaler et al. 2017; Muhamed 2014; Nemeth et al. 2004).
Copper overloading and apoptosis, autophagy
It is known from existing studies that copper overloading can lead to toxic effects, including neurotoxicity, hematotoxicity, reproductive toxicity, and hepatotoxicity, triggering oxidative stress, inflammatory responses, and DNA damage (Noh et al. 1999; Ogata et al. 2006). Some animal experiments have verified that copper overloading augments the expression of autophagy-related proteins including Beclin 1, ATG12, ATG5, while concurrently decreasing the expression of P62 in hepatocytes. Meanwhile, copper overloading can also increase the expression of ULK1, activate the AMPK pathway and initiate autophagy (Pals et al. 2003). In studies of reproductive toxicity, copper overloading induces oxidative stress damage in spermatogonia and triggers autophagy while also activating apoptosis (Pantopoulos et al. 2012). It has also been found that copper overloading activates the autophagic response when ATP7B is defective, while protecting hepatocytes from copper-induced apoptosis (Park et al. 1999). In addition, copper overloading can also cause the increase of necroptosis, and there is a negative regulatory effect between autophagy and necroptosis (Peacock 2010). More evidence is needed to study the mechanism of copper overloading regulating necroptosis.
Copper overloading and clinical diseases
When massive amounts of copper accumulate in the liver, the intracellular copper transporter ATP7B is primarily utilized to excrete excess copper ions through the bile. When ATP7B is inactivated by mutations and biliary excretion is impaired, pathological intracellular copper accumulation results. Accumulation of copper in the liver results in liver dysfunction and cirrhosis, while accumulation of copper ions in the brain leads to neurodegeneration, Wilson's disease (WD). When copper ions accumulate in lysosomes and exceed their loading capacity, they cause the lysosomes to rupture, releasing acidic contents into the cytoplasm, thereby causing cellular damage and ultimately cell death (Pei et al. 2022; Polishchuk et al. 2019).
Alternatively, elevated levels of intracellular copper can activate crucial enzymes for oxidative phosphorylation within tumors. This activation promotes the production of ATP, thereby accelerating the rapid proliferation of cancer cells and the progression of associated tumors (Portbury 2017). Numerous studies have shown that elevated copper levels within cells are correlated with the emergence and advancement of cancers, such as breast, lung, stomach, and bladder cancers (Robinson et al. 2010; Roemhild et al. 2021; Rouschop et al. 2010; Rudolf and Cervinka 2010; Schulman et al. 2013). When cells undergo copper overloading, Cu+ in the cytoplasm would stimulate RAS/MAPK signaling induced by FGF-, which activates ERK1/2 phosphorylation and regulates transcription factor activity. In addition, Cu+can also bind MEK1 kinase, and then MEK1/2 phosphorylation and then activate ERK1/2 phosphorylation, regulate the expression of various genes, mediate BRAF kinase mutations, and induce tumor growth, such as melanoma, thyroid cancer, hair cell leukemia, etc. (Liu et al. 2020; Sensi et al. 2018) (Fig. 3).

Mechanism of Cu2+ overload-induced cell death. Cytoplasmic occurrence of copper overload combined with esteroylated protein aggregation can induce cuprotosis, and oxidizing Cu+ can induce a burst of ROS, causing ferroptosis, autophagy, and apoptosis
Zinc overloading
Occurrence of zinc overloading
Zinc constitutes a component of numerous significant enzymes, is implicated in the metabolism of proteins, lipids and carbohydrates, and assumes an essential role in maintaining the integrity of red blood cells as well as in the hematopoietic process, which is a key element for growth and development (Shanmughapriya 2023). Zinc is also implicated in the synthesis of antioxidant enzymes as well as in the synthesis, storage, and release of insulin, suggesting that zinc ions occupy a crucial position in the progression of chronic inflammation, type 2 diabetes, and atherosclerosis (Shen et al. 2016; Shi et al. 2021).
The variation of free zinc concentration in organisms is very limited and is stringently regulated primarily by metallothioneins, zinc transporter proteins and zinc-iron regulatory proteins in order to sustain zinc homeostasis within the organism (Smaili et al. 2000). And it has been discovered that during the early stages of intracellular zinc increase, there is an initial accumulation of zinc in mitochondria and lysosomes. This serves to mitigate the potential toxic effects of free zinc. (Smaili 2003; Smith and Schnellmann 2012). Exogenous Zn2+ mainly passes through the intestinal tract, where it is absorbed by enterocytes and transported intracellularly via Zn2+ transporter proteins. It has been confirmed that SLC39A/ZIP and SLC30A/ZnT of the Zn2+ transporter protein family are respectively involved in the transportation of zinc ions into and out of cells and various organelles. ZnT is mainly responsible for transporting zinc ions into the intracellular compartment, and ZIP can transport excess zinc ions out of the compartment when the concentration of Zn2+increases to maintain the dynamic balance of intracellular zinc ions (Song et al. 2022). Zinc functions as a potent metallothionein inducer, which binds to metallothioneins to safeguard cells against oxidative stress (Southon et al. 2020).
Zinc overloading and cell death
The homeostatic balance of zinc ions metabolism is essential for the maintenance of normal physiological functions. Zinc deficiency can cause multi-system damage, impacting the proliferation and differentiation of immune cells, resulting in diminished immunity and a higher likelihood of developing immune diseases such as rheumatoid arthritis (Stremmel et al. 2019). Zinc overloading has been confirmed by numerous studies to be strongly correlated with neuronal degenerative diseases, such as Parkinson's disease (PD) and Alzheimer's disease (Szabo 2021; Tanaka et al. 2019; Tang et al. 2023). In pathological conditions or in response to specific stimuli, elevated intracellular free zinc levels can activate kinases such as protein kinase C (PKC) and extracellular regulated protein kinase (Erk1/2). The activation of PKC can subsequently enhance the activity of NADPH oxidases, leading to increased intracellular oxidative stress and the generation of large quantities of oxygen free radicals. These oxidative stresses can further activate PARP-1, leading to NAD+and ATP depletion, which in turn induces cell necrosis (Tavender et al. 2010; Tian et al. 2020; Torti et al. 2018). In addition, overloading of intracellular zinc in neuronal cells can also lead to the activation of neuronal-type nitric oxide synthase (nNOS), resulting in an elevation of nitric oxide (NO). However, NO can combine with superoxide anion radicals to create the strong oxidizing agent peroxynitrite (ONOO-). The mitochondrial membrane permeability transition pore (mPTP) is subsequently induced to open, the cytochrome C is released, resulting in the bursting of ROS, and in neuronal cells the development of apoptosis is inducedisis (Tsvetkov et al. 2022). It has been found that zinc overloading can increase the levels of ROS and LDH, lead to decreased mitochondrial membrane potential, activate the PINK1/Parkin pathway, and induce mitochondrial autophagy, and lead to mitochondrial dysfunction (Turski et al. 2012).
Lysosomes, as another important intracellular reservoir of zinc in addition to mitochondria, can also promote cell death when intracellular zinc ions are abnormally elevated. Overloading of intracytoplasmic Zn2+ and opening of ZnT, a zinc transporter protein on the lysosomal membrane, led to a massive influx of Zn2+into lysosomal vesicles, inducing lysosomal membrane permeabilization (LMP) and release of large amounts of histone proteases, thereby inducing cell death (Vásquez-Vivar et al. 1998). On the other hand, it has been demonstrated that intracellular zinc overloading can also activate caspase 3 through activation of Liver kinase B1 (LKB1), which triggers an increase in downstream AMPK, Bim proteins, and consequently caspase-dependent apoptosis (Wallhaus et al. 1998). At present, the molecular mechanism of zinc ion overloading-induced homeostatic disorders in organelles, which triggers related diseases, still requires further detailed studies (Fig. 4).

Mechanism of Zn2+ overload-induced cell death. Cytoplasmic zinc overload can stimulate mitochondria, lysosomes, and the nucleus to activate downstream autophagy- and apoptosis-related cytokines thereby causing cell death
Interactions between ionic overloads
In the process of studying ferroptosis, in addition to the direct activation of iron ion channels, the elevated Fe2+ levels can be also caused indirectly by activating the Ca2+/ CAM signaling pathway, which can promote the occurrence of ferroptosis (Wang et al. 2018). The presence of Cu2+has also been observed to enhance the initiation of iron overloading-induced ferroptosis by elevating the levels of GPX4 autophagic degradation (Wang et al. 2021). Strong evidence confirms that copper and iron act antagonistically to each other, with copper metabolism enhancing the release of stored iron through the production of copper cyanoproteins when iron is deficient, and iron loading when it occurs, which is also closely associated with copper deficiency (Wang et al. 2022; Wu et al. 2019). Moreover, high concentrations of copper, directly or indirectly, can reduce Ca2+influx thereby impairing myocardial contractile mechanisms (Xie et al. 2019). Some animal experiments have confirmed that when iron overloading occurs in mice, both serum and hepatocytes increase Zn2+levels, suggesting an association between the metabolism of different metallic elements (Xu et al. 2021; Xue et al. 2023). The interaction between Cu2+ and Zn2+ is evidenced by the observation that an excessive amount of Cu2+ can amplify cell death induced by Zn2+through the activation of oxidative stress and endoplasmic reticulum stress responses (Yang et al. 2022). Moreover, the composition of superoxide dismutase (SOD) is dependent on Cu and Zn, which is effective in scavenging superoxide anion radicals and protecting cells from oxidative damage when its conformation is stable. But when SOD is mutated, it can lead to protein misfolding by disrupting disulfide bonds, which ultimately leads to cell death (Yang et al. 2023). Zn2+overloading has likewise been found to promote ferroptosis (Zalckvar et al. 2009). All of these studies indicate that ferroptosis is not only associated with iron overloading, but also with imbalances in the levels of other ions.
As research continues to continue, more and more researchers are focusing on the range of abnormal cellular activities triggered by intracellular metal ions overloading. They are also exploring various mechanisms to enhance the prognosis and survival time of patients being treated for different diseases in clinic settings, in conjunction with conventional therapeutic tools such as drugs, nanomaterials or ionizing radiation (Zhang and Yang 2018; Zhang et al. 2009; Zhang et al. 2015).
In addition to Ca2+, Fe3+, Cu2+, and Zn2+ mentioned in this article, there are many other ions as well as disruptions of inter-ion homeostasis that are strongly associated with the progression of a variety of diseases. It has been discovered that in the event of heart failure, the Na+/H+ Exchanger is activated, resulting in Na+overloading within the cardiomyocytes, accompanied by an elevation in the concentration of calcium ions. This in turn, exacerbates the damage and dysfunction of the cardiomyocytes (Zhang et al. 2020). In addition, the Mitochondrial Calcium Uniporter (MCU) is regulated by Mg2+ in the mitochondrial matrix in addition to mediating mitochondrial Ca2+ uptake, and it has been demonstrated that calcium overloading occurs with a decrease in mitochondrial matrix Mg2+, as well as with the opening of the mPTP and an increase in MCU activity (Zhang et al. 2022). Utilizing the ion storage reservoirs in the cytoplasm or organelles and the exchanges and translocations between ions that are constantly occurring, it is highly likely that higher therapeutic efficacy will be achieved by administering treatments such as ion chelators and ion channel inhibitors when treating a variety of inflammatory and systemic disorders in the clinical setting (Fig.5).

Mechanism of Ca2+\Fe3+\Cu2+\Zn2+ overload-induced cell death. The four metal ions have mutually promoting or inhibiting effects on each other, and ROS act as an integral part in the process of ion overload-induced death of different cells
Furthermore, the process of cell death induced by various different ion overloads demands the activation of diverse downstream signaling pathways mediated by ROS and oxidative stress. Whether different ions induced ROS generation in the same or similar manner still deserves further in-depth study. ROS are well-common by-products of a large number of enzymatic reactions, including superoxide radical anion (O2−), hydrogen peroxide (H2O2), hydroxyl radical (−OH), peroxyl hydroxyl radical (OOH−), singlet oxygen (1O2), and hydroperoxyl radical (ROO−). ROS is currently known to be produced by the cytoplasm, mitochondria, ER, and peroxisomes. Cytoplasmic ROS are mainly produced by NADPH oxidase (NOX) and nitric oxide synthase (NOS), and O2−produced also participate in death mechanisms like glycolysis and oxidative phosphorylation, pentose phosphate pathway, and autophagy (Zhang et al. 2023; Zhou et al. 2014). Mitochondrial ROS are by-products generated through electron transfer between complexes I-III in the mitochondrial respiratory chain. When cellular stress results in the accumulation of anomalously high levels of mitochondrial ROS, ATP synthesis, glycolysis, fatty acid synthesis pathways, and cell death are all affected (Zhou et al. 2015). While ER is highly sensitive to the redox state, ROS generation in the ER relies on the pathways implicating protein disulfide isomerase (PDI) and endoplasmic reticulum oxidoreductase 1(ERO1), ultimately resulting to the production of H2O2. When oxidative stress occurs in the endoplasmic reticulum, it induces UPR and aggravates the oxidative stress of ER. And oxidative stress of ER can also affect mitochondrial function by inducing mitochondrial Ca2+ uptake and mitochondrial ROS production, affecting Ca2+release and mitochondrial respiration (Zhou et al. 2021). H2O2production by peroxisomes relies on enzymatic reactions and electron transfer exchanges by many of oxidizing enzymes. It was confirmed that induction of AMPK activity and regulation of downstream mTOR and ULK1 in the peroxisomal ROS reaction, followed by targeting of peroxisomes to autophagic vesicles, activates autophagy (Zhou et al. 2022; Zhou et al. 2022).
Conclusions
In summary, the four metal ions, Ca2+, Fe3+, Cu2+, and Zn2+, when overloaded, cause the onset of one or more cell deaths through the activation of one or more key downstream molecules as well as signaling pathways. These biological processes, however, all entail, to a greater or lesser degree, the participation of significant intracellular ion storage compartments such as mitochondria, or lysosomes. More importantly, the occurrence of oxidative stress and ROS after overloading of different metal ions in the cell is a strong confirmation that the process of cellular life activities is an intricate and complex regulatory network with mutual influence. In this review, we describe the basic transport process of four metal ions, Ca2+, Fe3+, Cu2+, and Zn2+, as well as the mechanism by which cell death is induced after overloading occurs, and will provide compelling evidence and clinical references for further studies on the toxic effects of metal ions.
Availability of data and materials
No datasets were generated or analysed during the current study.
Abbreviations
- PTP:
-
Permeability transition pore
- IP3R:
-
Inositol 1,4,5-triphosphate receptor
- ER:
-
Endoplasmic reticulum
- UPR:
-
Unfolded protein response
- PCD:
-
Programmed cell death
- TfR2:
-
Transferrin receptor 2
- HJV:
-
Hemojuvelin
- DMT1:
-
Divalent metal transporter protein 1
- NCOA4:
-
Nuclear receptor coactivator 4
- SFXN1:
-
Sideroflexin 1
- DFO:
-
Desferrioxamine
- HO-1:
-
Heme oxygenase 1
- Nrf2:
-
Nuclear factor erythroid 2-related factor 2
- SOD:
-
Superoxide dismutase
- GSH:
-
Glutathione
- TCA:
-
Tricarboxylic acid cycle
- PKC:
-
Protein kinase C
- nNOS:
-
Neuronal-type nitric oxide synthase
- LMP:
-
Lysosomal membrane permeabilization
- LKB1:
-
Liver kinase B1
- MCU:
-
Mitochondrial Calcium Uniporter
- PDI:
-
Protein disulfide isomerase
- ERO1:
-
Endoplasmic reticulum oxidoreductase 1
References
Afroze T, Yang LL, Wang C, Gros R, Kalair W, Hoque AN, et al. Calcineurin-independent regulation of plasma membrane Ca2+ ATPase-4 in the vascular smooth muscle cell cycle. Am J Physiol Cell Physiol. 2003;285(1):C88-95.
Ahn BI, Kim MJ, Koo HS, Seo N, Joo NS, Kim YS. Serum zinc concentration is inversely associated with insulin resistance but not related with metabolic syndrome in nondiabetic Korean adults. Biol Trace Elem Res. 2014;160(2):169–75.
Alhillawi ZH, Al-Hakeim HK, Moustafa SR, Maes M. Increased zinc and albumin but lowered copper in children with transfusion-dependent thalassemia. J Trace Elem Med Biol. 2021;65:126713.
Arosio P, Ingrassia R, Cavadini P. Ferritins: a family of molecules for iron storage, antioxidation and more. Biochim Biophys Acta. 2009;1790(7):589–99.
Baev AY, Vinokurov AY, Novikova IN, Dremin VV, Potapova EV, Abramov AY. Interaction of Mitochondrial Calcium and ROS in Neurodegeneration. Cells. 2022;11(4):706.
Bagur R, Hajnóczky G. Intracellular Ca(2+) Sensing: Its Role in Calcium Homeostasis and Signaling. Mol Cell. 2017;66(6):780–8.
Bai S, Lan Y, Fu S, Cheng H, Lu Z, Liu G. Connecting Calcium-Based Nanomaterials and Cancer: From Diagnosis to Therapy. Nanomicro Lett. 2022;14(1):145.
Balashova MS, Tuluzanovskaya IG, Glotov OS, Glotov AS, Barbitoff YA, Fedyakov MA, et al. The spectrum of pathogenic variants of the ATP7B gene in Wilson disease in the Russian Federation. J Trace Elem Med Biol. 2020;59:126420.
B’Chir W, Chaveroux C, Carraro V, Averous J, Maurin AC, Jousse C, et al. Dual role for CHOP in the crosstalk between autophagy and apoptosis to determine cell fate in response to amino acid deprivation. Cell Signal. 2014;26(7):1385–91.
Berezhnov AV, Fedotova EI, Nenov MN, Kokoz IuM, Zinchenko VP, Dynnik VV. Destabilization of the cytosolic calcium level and cardiomyocyte death in the presence of long-chain fatty acid derivatives. Biofizika. 2008;53(6):1025–32.
Bernardi P. The mitochondrial permeability transition pore: a mystery solved? Front Physiol. 2013;4:95.
Blaby-Haas CE, Merchant SS. Lysosome-related organelles as mediators of metal homeostasis. J Biol Chem. 2014;289(41):28129–36.
Boal AK, Rosenzweig AC. Structural biology of copper trafficking. Chem Rev. 2009;109(10):4760–79.
Bonaventura P, Benedetti G, Albarède F, Miossec P. Zinc and its role in immunity and inflammation. Autoimmun Rev. 2015;14(4):277–85.
Bossy-Wetzel E, Talantova MV, Lee WD, Schölzke MN, Harrop A, Mathews E, et al. Crosstalk between nitric oxide and zinc pathways to neuronal cell death involving mitochondrial dysfunction and p38-activated K+ channels. Neuron. 2004;41(3):351–65.
Bostanci Z, Alam S, Soybel DI, Kelleher SL. Prolactin receptor attenuation induces zinc pool redistribution through ZnT2 and decreases invasion in MDA-MB-453 breast cancer cells. Exp Cell Res. 2014;321(2):190–200.
Brady DC, Crowe MS, Turski ML, Hobbs GA, Yao X, Chaikuad A, et al. Copper is required for oncogenic BRAF signalling and tumorigenesis. Nature. 2014;509(7501):492–6.
Britton LJ, Subramaniam VN, Crawford DH. Iron and non-alcoholic fatty liver disease. World J Gastroenterol. 2016;22(36):8112–22.
Camaschella C, Nai A, Silvestri L. Iron metabolism and iron disorders revisited in the hepcidin era. Haematologica. 2020;105(2):260–72.
Cappetta D, De Angelis A, Bellocchio G, Telesca M, Cianflone E, Torella D, et al. Sodium-Glucose Cotransporter 2 Inhibitors and Heart Failure: A Bedside-to-Bench Journey. Front Cardiovasc Med. 2021;8:810791.
Cave AC, Brewer AC, Narayanapanicker A, Ray R, Grieve DJ, Walker S, et al. NADPH oxidases in cardiovascular health and disease. Antioxid Redox Signal. 2006;8(5–6):691–728.
Cen WJ, Feng Y, Li SS, Huang LW, Zhang T, Zhang W, et al. Iron overload induces G1 phase arrest and autophagy in murine preosteoblast cells. J Cell Physiol. 2018;233(9):6779–89.
Chen MP, Cabantchik ZI, Chan S, Chan GC, Cheung YF. Iron overload and apoptosis of HL-1 cardiomyocytes: effects of calcium channel blockade. PLoS ONE. 2014;9(11):e112915.
Chen J, Jiang Y, Shi H, Peng Y, Fan X, Li C. The molecular mechanisms of copper metabolism and its roles in human diseases. Pflugers Arch. 2020a;472(10):1415–29.
Chen P, Wu Q, Feng J, Yan L, Sun Y, Liu S, et al. Erianin, a novel dibenzyl compound in Dendrobium extract, inhibits lung cancer cell growth and migration via calcium/calmodulin-dependent ferroptosis. Signal Transduct Target Ther. 2020b;5(1):51.
Chen PH, Wu J, Xu Y, Ding CC, Mestre AA, Lin CC, et al. Zinc transporter ZIP7 is a novel determinant of ferroptosis. Cell Death Dis. 2021;12(2):198.
Cheng Y, Qu W, Li J, Jia B, Song Y, Wang L, et al. Ferristatin II, an Iron Uptake Inhibitor, Exerts Neuroprotection against Traumatic Brain Injury via Suppressing Ferroptosis. ACS Chem Neurosci. 2022;13(5):664–75.
Chu X, Hou HY, Duan MD, Zhang YJ, Zhu YY, Liu Y, et al. Tumor microenvironment specific regulation Ca-Fe-nanospheres for ferroptosis-promoted domino synergistic therapy and tumor immune response. Small (Weinheim an der Bergstrasse, Germany). 2024:e2312141.
Collins JF, Prohaska JR, Knutson MD. Metabolic crossroads of iron and copper. Nutr Rev. 2010;68(3):133–47.
Davaanyam D, Lee H, Seol SI, Oh SA, Kim SW, Lee JK. HMGB1 induces hepcidin upregulation in astrocytes and causes an acute iron surge and subsequent ferroptosis in the postischemic brain. Exp Mol Med. 2023;55(11):2402–16.
de Oliveira Otto MC, Alonso A, Lee DH, Delclos GL, Jenny NS, Jiang R, et al. Dietary micronutrient intakes are associated with markers of inflammation but not with markers of subclinical atherosclerosis. J Nutr. 2011;141(8):1508–15.
de Romaña DL, Olivares M, Uauy R, Araya M. Risks and benefits of copper in light of new insights of copper homeostasis. J Trace Elem Med Biol. 2011;25(1):3–13.
Dhaouadi N, Vitto VAM, Pinton P, Galluzzi L, Marchi S. Ca(2+) signaling and cell death. Cell Calcium. 2023;113:102759.
Dixon SJ, Lemberg KM, Lamprecht MR, Skouta R, Zaitsev EM, Gleason CE, et al. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell. 2012;149(5):1060–72.
Doguer C, Ha JH, Collins JF. Intersection of Iron and Copper Metabolism in the Mammalian Intestine and Liver. Compr Physiol. 2018;8(4):1433–61.
Dong X, Zang C, Sun Y, Zhang S, Liu C, Qian J. Hydroxyapatite nanoparticles induced calcium overload-initiated cancer cell-specific apoptosis through inhibition of PMCA and activation of calpain. Journal of Materials Chemistry B. 2023;11(32):7609–22.
Eckenrode EF, Yang J, Velmurugan GV, Foskett JK, White C. Apoptosis protection by Mcl-1 and Bcl-2 modulation of inositol 1,4,5-trisphosphate receptor-dependent Ca2+ signaling. J Biol Chem. 2010;285(18):13678–84.
Eom JW, Lee JM, Koh JY, Kim YH. AMP-activated protein kinase contributes to zinc-induced neuronal death via activation by LKB1 and induction of Bim in mouse cortical cultures. Mol Brain. 2016;9:14.
Fang R, Wu R, Du H, Jin M, Liu Y, Lei G, et al. Pneumolysin-Dependent Calpain Activation and Interleukin-1α Secretion in Macrophages Infected with Streptococcus pneumoniae. Infect Immun. 2017;85(9):e00201-17.
Feng H, Liu Y, Gan Y, Li M, Liu R, Liang Z, et al. AdipoR1 Regulates Ionizing Radiation-Induced Ferroptosis in HCC cells through Nrf2/xCT Pathway. Oxid Med Cell Longev. 2022a;2022:8091464.
Feng X, Hu W, Hong Y, Ruan L, Hu Y, Liu D. Taurine Ameliorates Iron Overload-Induced Hepatocyte Injury via the Bcl-2/VDAC1-Mediated Mitochondrial Apoptosis Pathway. Oxid Med Cell Longev. 2022b;2022:4135752.
Filetti FM, Vassallo DV, Fioresi M, Simões MR. Reactive oxygen species impair the excitation-contraction coupling of papillary muscles after acute exposure to a high copper concentration. Toxicol in Vitro. 2018;51:106–13.
Fonseca-Nunes A, Jakszyn P, Agudo A. Iron and cancer risk–a systematic review and meta-analysis of the epidemiological evidence. Cancer Epidemiology, Biomarkers & Prevention : a Publication of the American Association for Cancer Research, Cosponsored by the American Society of Preventive Oncology. 2014;23(1):12–31.
Forrester SJ, Kikuchi DS, Hernandes MS, Xu Q, Griendling KK. Reactive Oxygen Species in Metabolic and Inflammatory Signaling. Circ Res. 2018;122(6):877–902.
Frey AG, Nandal A, Park JH, Smith PM, Yabe T, Ryu MS, et al. Iron chaperones PCBP1 and PCBP2 mediate the metallation of the dinuclear iron enzyme deoxyhypusine hydroxylase. Proc Natl Acad Sci USA. 2014;111(22):8031–6.
Fukuda S, Harada K, Kunimatsu M, Sakabe T, Yoshida K. Postischemic reperfusion induces alpha-fodrin proteolysis by m-calpain in the synaptosome and nucleus in rat brain. J Neurochem. 1998;70(6):2526–32.
Fusakio ME, Willy JA, Wang Y, Mirek ET, Al Baghdadi RJ, Adams CM, et al. Transcription factor ATF4 directs basal and stress-induced gene expression in the unfolded protein response and cholesterol metabolism in the liver. Mol Biol Cell. 2016;27(9):1536–51.
Gaetke LM, Chow-Johnson HS, Chow CK. Copper: toxicological relevance and mechanisms. Arch Toxicol. 2014;88(11):1929–38.
Gao W, Huang Z, Duan J, Nice EC, Lin J, Huang C. Elesclomol induces copper-dependent ferroptosis in colorectal cancer cells via degradation of ATP7A. Mol Oncol. 2021;15(12):3527–44.
Genoud S, Roberts BR, Gunn AP, Halliday GM, Lewis SJG, Ball HJ, et al. Subcellular compartmentalisation of copper, iron, manganese, and zinc in the Parkinson’s disease brain. Metallomics : Integrated Biometal Science. 2017;9(10):1447–55.
Guha P, Dey A, Dhyani MV, Sen R, Chatterjee M, Chattopadhyay S, et al. Calpain and caspase orchestrated death signal to accomplish apoptosis induced by resveratrol and its novel analog hydroxystilbene-1 [correction of hydroxstilbene-1] in cancer cells. J Pharmacol Exp Ther. 2010;334(2):381–94.
Guo Z, Lin J, Sun K, Guo J, Yao X, Wang G, et al. Deferoxamine Alleviates Osteoarthritis by Inhibiting Chondrocyte Ferroptosis and Activating the Nrf2 Pathway. Front Pharmacol. 2022a;13:791376.
Guo H, Wang Y, Cui H, Ouyang Y, Yang T, Liu C, et al. Copper Induces Spleen Damage Through Modulation of Oxidative Stress, Apoptosis, DNA Damage, and Inflammation. Biol Trace Elem Res. 2022b;200(2):669–77.
Guo H, Ouyang Y, Yin H, Cui H, Deng H, Liu H, et al. Induction of autophagy via the ROS-dependent AMPK-mTOR pathway protects copper-induced spermatogenesis disorder. Redox Biol. 2022c;49:102227.
Guo D, Dai X, Liu K, Liu Y, Wu J, Wang K, et al. A Self-Reinforcing Nanoplatform for Highly Effective Synergistic Targeted Combinatary Calcium-Overload and Photodynamic Therapy of Cancer. Adv Healthc Mater. 2023;12(12):e2202424.
Gupte A, Mumper RJ. Elevated copper and oxidative stress in cancer cells as a target for cancer treatment. Cancer Treat Rev. 2009;35(1):32–46.
Han S, Lin F, Qi Y, Liu C, Zhou L, Xia Y, et al. HO-1 Contributes to Luteolin-Triggered Ferroptosis in Clear Cell Renal Cell Carcinoma via Increasing the Labile Iron Pool and Promoting Lipid Peroxidation. Oxid Med Cell Longev. 2022;2022:3846217.
He J, Fu LH, Qi C, Lin J, Huang P. Metal peroxides for cancer treatment. Bioactive Materials. 2021;6(9):2698–710.
Horn N, Wittung-Stafshede P. ATP7A-Regulated Enzyme Metalation and Trafficking in the Menkes Disease Puzzle. Biomedicines. 2021;9(4):391.
Høyer-Hansen M, Bastholm L, Szyniarowski P, Campanella M, Szabadkai G, Farkas T, et al. Control of macroautophagy by calcium, calmodulin-dependent kinase kinase-beta, and Bcl-2. Mol Cell. 2007;25(2):193–205.
Hwang JJ, Lee SJ, Kim TY, Cho JH, Koh JY. Zinc and 4-hydroxy-2-nonenal mediate lysosomal membrane permeabilization induced by H2O2 in cultured hippocampal neurons. J Neurosci. 2008;28(12):3114–22.
Ishida S, Andreux P, Poitry-Yamate C, Auwerx J, Hanahan D. Bioavailable copper modulates oxidative phosphorylation and growth of tumors. Proc Natl Acad Sci U S A. 2013;110(48):19507–12.
James SA, Churches QI, de Jonge MD, Birchall IE, Streltsov V, McColl G, et al. Iron, Copper, and Zinc Concentration in Aβ Plaques in the APP/PS1 Mouse Model of Alzheimer’s Disease Correlates with Metal Levels in the Surrounding Neuropil. ACS Chem Neurosci. 2017;8(3):629–37.
Karim A, Bajbouj K, Shafarin J, Qaisar R, Hall AC, Hamad M. Iron Overload Induces Oxidative Stress, Cell Cycle Arrest and Apoptosis in Chondrocytes. Front Cell Dev Biol. 2022;10:821014.
Kim YH, Koh JY. The role of NADPH oxidase and neuronal nitric oxide synthase in zinc-induced poly(ADP-ribose) polymerase activation and cell death in cortical culture. Exp Neurol. 2002;177(2):407–18.
Kimura T, Kambe T. The Functions of Metallothionein and ZIP and ZnT Transporters: An Overview and Perspective. Int J Mol Sci. 2016;17(3):336.
Kohzadi S, Sheikhesmaili F, Rahehagh R, Parhizkar B, Ghaderi E, Loqmani H, et al. Evaluation of trace element concentration in cancerous and non-cancerous tissues of human stomach. Chemosphere. 2017;184:747–52.
Lee JH, Amarsanaa K, Wu J, Jeon SC, Cui Y, Jung SC, et al. Nobiletin attenuates neurotoxic mitochondrial calcium overload through K(+) influx and ΔΨ(m) across mitochondrial inner membrane. The Korean Journal of Physiology & Pharmacology : Official Journal of the Korean Physiological Society and the Korean Society of Pharmacology. 2018;22(3):311–9.
Leidgens S, Bullough KZ, Shi H, Li F, Shakoury-Elizeh M, Yabe T, et al. Each member of the poly-r(C)-binding protein 1 (PCBP) family exhibits iron chaperone activity toward ferritin. J Biol Chem. 2013;288(24):17791–802.
Li Y. Copper homeostasis: Emerging target for cancer treatment. IUBMB Life. 2020;72(9):1900–8.
Li N, Wang W, Zhou H, Wu Q, Duan M, Liu C, et al. Ferritinophagy-mediated ferroptosis is involved in sepsis-induced cardiac injury. Free Radic Biol Med. 2020;160:303–18.
Li F, Guan Z, Gao Y, Bai Y, Zhan X, Ji X, et al. ER stress promotes mitochondrial calcium overload and activates the ROS/NLRP3 axis to mediate fatty liver ischemic injury. Hepatol Commun. 2024;8(4):e0399.
Lin X, Zhao Y, Li S. Astaxanthin attenuates glutamate-induced apoptosis via inhibition of calcium influx and endoplasmic reticulum stress. Eur J Pharmacol. 2017;806:43–51.
Lin Y, Jiang M, Chen W, Zhao T, Wei Y. Cancer and ER stress: Mutual crosstalk between autophagy, oxidative stress and inflammatory response. Biomed Pharmacother. 2019;118:109249.
Liu H, Guo H, Jian Z, Cui H, Fang J, Zuo Z, et al. Copper Induces Oxidative Stress and Apoptosis in the Mouse Liver. Oxid Med Cell Longev. 2020;2020:1359164.
Liu H, Deng H, Cui H, Jian Z, Guo H, Fang J, et al. Copper induces hepatocyte autophagy via the mammalian targets of the rapamycin signaling pathway in mice. Ecotoxicol Environ Saf. 2021;208:111656.
Liu L, Zhang C, Qu S, Liu R, Chen H, Liang Z, et al. ESR1 inhibits ionizing radiation-induced ferroptosis in breast cancer cells via the NEDD4L/CD71 pathway. Arch Biochem Biophys. 2022;725:109299.
Llambi F, Wang YM, Victor B, Yang M, Schneider DM, Gingras S, et al. BOK Is a Non-canonical BCL-2 Family Effector of Apoptosis Regulated by ER-Associated Degradation. Cell. 2016;165(2):421–33.
Llanos RM, Mercer JF. The molecular basis of copper homeostasis copper-related disorders. DNA Cell Biol. 2002;21(4):259–70.
Łopatniuk P, Witkowski JM. Conventional calpains and programmed cell death. Acta Biochim Pol. 2011;58(3):287–96.
Ma J, Wang A, Zhang H, Liu B, Geng Y, Xu Y, et al. Iron overload induced osteocytes apoptosis and led to bone loss in Hepcidin(-/-) mice through increasing sclerostin and RANKL/OPG. Bone. 2022;164:116511.
Mao S, Huang S. Zinc and copper levels in bladder cancer: a systematic review and meta-analysis. Biol Trace Elem Res. 2013;153(1–3):5–10.
Marmolejo-Garza A, Krabbendam IE, Luu MDA, Brouwer F, Trombetta-Lima M, Unal O, et al. Negative modulation of mitochondrial calcium uniporter complex protects neurons against ferroptosis. Cell Death Dis. 2023;14(11):772.
Marreiro DD, Cruz KJ, Morais JB, Beserra JB, Severo JS, de Oliveira AR. Zinc and Oxidative Stress: Current Mechanisms. Antioxidants (Basel, Switzerland). 2017;6(2):24.
McAlary L, Shephard VK, Wright GSA, Yerbury JJ. A copper chaperone-mimetic polytherapy for SOD1-associated amyotrophic lateral sclerosis. J Biol Chem. 2022;298(3):101612.
Miao Z, Miao Z, Feng S, Xu S. Chlorpyrifos-mediated mitochondrial calcium overload induces EPC cell apoptosis via ROS/AMPK/ULK1. Fish Shellfish Immunol. 2023;141:109053.
Mohr I, Weiss KH. Biochemical Markers for the Diagnosis and Monitoring of Wilson Disease. The Clinical Biochemist Reviews. 2019;40(2):59–77.
Muckenthaler MU, Rivella S, Hentze MW, Galy B. A Red Carpet for Iron Metabolism. Cell. 2017;168(3):344–61.
Muhamed PK, Vadstrup S. [Zinc is the most important trace element]. Ugeskr Laeger. 2014;176(5):V11120654.
Nemeth E, Tuttle MS, Powelson J, Vaughn MB, Donovan A, Ward DM, et al. Hepcidin regulates cellular iron efflux by binding to ferroportin and inducing its internalization. Science (new York, NY). 2004;306(5704):2090–3.
Noh KM, Kim YH, Koh JY. Mediation by membrane protein kinase C of zinc-induced oxidative neuronal injury in mouse cortical cultures. J Neurochem. 1999;72(4):1609–16.
Ogata M, Hino S, Saito A, Morikawa K, Kondo S, Kanemoto S, et al. Autophagy is activated for cell survival after endoplasmic reticulum stress. Mol Cell Biol. 2006;26(24):9220–31.
Pals P, Van Everbroeck B, Grubben B, Viaene MK, Dom R, van der Linden C, et al. Case-control study of environmental risk factors for Parkinson’s disease in Belgium. Eur J Epidemiol. 2003;18(12):1133–42.
Pantopoulos K, Porwal SK, Tartakoff A, Devireddy L. Mechanisms of mammalian iron homeostasis. Biochemistry. 2012;51(29):5705–24.
Park JA, Koh JY. Induction of an immediate early gene egr-1 by zinc through extracellular signal-regulated kinase activation in cortical culture: its role in zinc-induced neuronal death. J Neurochem. 1999;73(2):450–6.
Peacock M. Calcium metabolism in health and disease. Clinical Journal of the American Society of Nephrology : CJASN. 2010;5(Suppl 1):S23-30.
Pei Z, Qin Y, Fu X, Yang F, Huo F, Liang X, et al. Inhibition of ferroptosis and iron accumulation alleviates pulmonary fibrosis in a bleomycin model. Redox Biol. 2022;57:102509.
Polishchuk EV, Merolla A, Lichtmannegger J, Romano A, Indrieri A, Ilyechova EY, et al. Activation of Autophagy, Observed in Liver Tissues From Patients With Wilson Disease and From ATP7B-Deficient Animals. Protects Hepatocytes from Copper-Induced Apoptosis Gastroenterology. 2019;156(4):1173-89.e5.
Portbury SD, Adlard PA. Zinc Signal in Brain Diseases. Int J Mol Sci. 2017;18(12):2506.
Robinson NJ, Winge DR. Copper metallochaperones. Annu Rev Biochem. 2010;79:537–62.
Roemhild K, von Maltzahn F, Weiskirchen R, Knüchel R, von Stillfried S, Lammers T. Iron metabolism: pathophysiology and pharmacology. Trends Pharmacol Sci. 2021;42(8):640–56.
Rouschop KM, van den Beucken T, Dubois L, Niessen H, Bussink J, Savelkouls K, et al. The unfolded protein response protects human tumor cells during hypoxia through regulation of the autophagy genes MAP1LC3B and ATG5. J Clin Investig. 2010;120(1):127–41.
Rudolf E, Cervinka M. Zinc pyrithione induces cellular stress signaling and apoptosis in Hep-2 cervical tumor cells: the role of mitochondria and lysosomes. Biometals : an International Journal on the Role of Metal Ions in Biology, Biochemistry, and Medicine. 2010;23(2):339–54.
Schulman JJ, Wright FA, Kaufmann T, Wojcikiewicz RJH. The Bcl-2 protein family member Bok binds to the coupling domain of inositol 1,4,5-trisphosphate receptors and protects them from proteolytic cleavage. J Biol Chem. 2013;288(35):25340–9.
Sensi SL, Granzotto A, Siotto M, Squitti R. Copper and Zinc Dysregulation in Alzheimer’s Disease. Trends Pharmacol Sci. 2018;39(12):1049–63.
Shanmughapriya S, Ponnusamy T, Velusamy P, Kumar A, Morris D, Zhang X, et al. Mitochondrial magnesium is the cationic rheostat for MCU-mediated mitochondrial Ca2+ uptake. Research square. 2023.
Shen L, Wen N, Xia M, Zhang YU, Liu W, Xu YE, et al. Calcium efflux from the endoplasmic reticulum regulates cisplatin-induced apoptosis in human cervical cancer HeLa cells. Oncol Lett. 2016;11(4):2411–9.
Shi H, Yu Y, Wang Y, Liu X, Yu Y, Li M, et al. Inhibition of Calpain Alleviates Apoptosis in Coxsackievirus B3-induced Acute Virus Myocarditis Through Suppressing Endoplasmic Reticulum Stress. Int Heart J. 2021;62(4):900–9.
Smaili SS, Hsu YT, Youle RJ, Russell JT. Mitochondria in Ca2+ signaling and apoptosis. J Bioenerg Biomembr. 2000;32(1):35–46.
Smaili SS, Hsu YT, Carvalho AC, Rosenstock TR, Sharpe JC, Youle RJ. Mitochondria, calcium and pro-apoptotic proteins as mediators in cell death signaling. Braz J Med Biol Res. 2003;36(2):183–90.
Smith MA, Schnellmann RG. Calpains, mitochondria, and apoptosis. Cardiovasc Res. 2012;96(1):32–7.
Song S, Zhang M, Xie P, Wang S, Wang Y. Comprehensive analysis of cuproptosis-related genes and tumor microenvironment infiltration characterization in breast cancer. Front Immunol. 2022;13:978909.
Southon A, Szostak K, Acevedo KM, Dent KA, Volitakis I, Belaidi AA, et al. Cu(II) (atsm) inhibits ferroptosis: Implications for treatment of neurodegenerative disease. Br J Pharmacol. 2020;177(3):656–67.
Stremmel W, Merle U, Weiskirchen R. Clinical features of Wilson disease. Ann Transl Med. 2019;7(Suppl 2):S61.
Szabo R, Bodolea C, Mocan T. Iron, Copper, and Zinc Homeostasis: Physiology, Physiopathology, and Nanomediated Applications. Nanomaterials (Basel, Switzerland). 2021;11(11):2958.
Tanaka KI, Kasai M, Shimoda M, Shimizu A, Kubota M, Kawahara M. Nickel Enhances Zinc-Induced Neuronal Cell Death by Priming the Endoplasmic Reticulum Stress Response. Oxid Med Cell Longev. 2019;2019:9693726.
Tang S, Liang C, Hou W, Hu Z, Chen X, Zhao J, et al. ATP7B R778L mutant hepatocytes resist copper toxicity by activating autophagy and inhibiting necroptosis. Cell Death Discovery. 2023;9(1):344.
Tavender TJ, Springate JJ, Bulleid NJ. Recycling of peroxiredoxin IV provides a novel pathway for disulphide formation in the endoplasmic reticulum. EMBO J. 2010;29(24):4185–97.
Tian Q, Qin B, Gu Y, Zhou L, Chen S, Zhang S, et al. ROS-Mediated Necroptosis Is Involved in Iron Overload-Induced Osteoblastic Cell Death. Oxid Med Cell Longev. 2020;2020:1295382.
Torti SV, Manz DH, Paul BT, Blanchette-Farra N, Torti FM. Iron and Cancer. Annu Rev Nutr. 2018;38:97–125.
Tsvetkov P, Coy S, Petrova B, Dreishpoon M, Verma A, Abdusamad M, et al. Copper induces cell death by targeting lipoylated TCA cycle proteins. Science (new York, NY). 2022;375(6586):1254–61.
Turski ML, Brady DC, Kim HJ, Kim BE, Nose Y, Counter CM, et al. A novel role for copper in Ras/mitogen-activated protein kinase signaling. Mol Cell Biol. 2012;32(7):1284–95.
Vásquez-Vivar J, Kalyanaraman B, Martásek P, Hogg N, Masters BS, Karoui H, et al. Superoxide generation by endothelial nitric oxide synthase: the influence of cofactors. Proc Natl Acad Sci U S A. 1998;95(16):9220–5.
Wallhaus TR, Lacy J, Whang J, Green MA, Nickles RJ, Stone CK. Human biodistribution and dosimetry of the PET perfusion agent copper-62-PTSM. Journal of Nuclear Medicine : Official Publication, Society of Nuclear Medicine. 1998;39(11):1958–64.
Wang X, Flores SR, Ha JH, Doguer C, Woloshun RR, Xiang P, et al. Intestinal DMT1 Is Essential for Optimal Assimilation of Dietary Copper in Male and Female Mice with Iron-Deficiency Anemia. J Nutr. 2018;148(8):1244–52.
Wang M, Wang R, Sun H, Sun G, Sun X. Ginsenoside Rb1 ameliorates cardiotoxicity triggered by aconitine via inhibiting calcium overload and pyroptosis. Phytomedicine. 2021;83:153468.
Wang S, Li W, Zhang P, Wang Z, Ma X, Liu C, et al. Mechanical overloading induces GPX4-regulated chondrocyte ferroptosis in osteoarthritis via Piezo1 channel facilitated calcium influx. J Adv Res. 2022;41:63–75.
Wu J, Chen YJ, Dobbs N, Sakai T, Liou J, Miner JJ, et al. STING-mediated disruption of calcium homeostasis chronically activates ER stress and primes T cell death. J Exp Med. 2019;216(4):867–83.
Xie D, Zhou P, Liu L, Jiang W, Xie H, Zhang L, et al. Protective Effect of Astragaloside IV on Hepatic Injury Induced by Iron Overload. Biomed Res Int. 2019;2019:3103946.
Xu G, Li X, Zhu Z, Wang H, Bai X. Iron Overload Induces Apoptosis and Cytoprotective Autophagy Regulated by ROS Generation in Mc3t3-E1 Cells. Biol Trace Elem Res. 2021;199(10):3781–92.
Xue Q, Yan D, Chen X, Li X, Kang R, Klionsky DJ, et al. Copper-dependent autophagic degradation of GPX4 drives ferroptosis. Autophagy. 2023;19(7):1982–96.
Yang Y, Lin Y, Wang M, Yuan K, Wang Q, Mu P, et al. Targeting ferroptosis suppresses osteocyte glucolipotoxicity and alleviates diabetic osteoporosis. Bone Research. 2022;10(1):26.
Yang Y, Wang P, Guo J, Ma T, Hu Y, Huang L, et al. Zinc Overload Induces Damage to H9c2 Cardiomyocyte Through Mitochondrial Dysfunction and ROS-Mediated Mitophagy. Cardiovasc Toxicol. 2023;23(11–12):388–405.
Zalckvar E, Berissi H, Mizrachy L, Idelchuk Y, Koren I, Eisenstein M, et al. DAP-kinase-mediated phosphorylation on the BH3 domain of beclin 1 promotes dissociation of beclin 1 from Bcl-XL and induction of autophagy. EMBO Rep. 2009;10(3):285–92.
Zhang X, Yang Q. Association between serum copper levels and lung cancer risk: A meta-analysis. J Int Med Res. 2018;46(12):4863–73.
Zhang Y, Li B, Chen C, Gao Z. Hepatic distribution of iron, copper, zinc and cadmium-containing proteins in normal and iron overload mice. Biometals : an International Journal on the Role of Metal Ions in Biology, Biochemistry, and Medicine. 2009;22(2):251–9.
Zhang J, Tripathi DN, Jing J, Alexander A, Kim J, Powell RT, et al. ATM functions at the peroxisome to induce pexophagy in response to ROS. Nat Cell Biol. 2015;17(10):1259–69.
Zhang J, Qiao P, Yao G, Zhao H, Wu Y, Wu S. Ionizing Radiation Exacerbates the Bone Loss Induced by Iron Overload in Mice. Biol Trace Elem Res. 2020;196(2):502–11.
Zhang B, Pan C, Feng C, Yan C, Yu Y, Chen Z, et al. Role of mitochondrial reactive oxygen species in homeostasis regulation. Redox Report : Communications in Free Radical Research. 2022;27(1):45–52.
Zhang S, Qiu Y, Feng Y, Zhang Y, Li Y, Wang B, et al. Calpain-2 Facilitates Autophagic/Lysosomal Defects and Apoptosis in ARPE-19 Cells and Rats Induced by Exosomes from RPE Cells under NaIO(3) Stimulation. Oxid Med Cell Longev. 2023;2023:3310621.
Zhou X, Lin P, Yamazaki D, Park KH, Komazaki S, Chen SR, et al. Trimeric intracellular cation channels and sarcoplasmic/endoplasmic reticulum calcium homeostasis. Circ Res. 2014;114(4):706–16.
Zhou X, Hao W, Shi H, Hou Y, Xu Q. Calcium homeostasis disruption - a bridge connecting cadmium-induced apoptosis, autophagy and tumorigenesis. Oncology Research and Treatment. 2015;38(6):311–5.
Zhou Y, Liao J, Mei Z, Liu X, Ge J. Insight into Crosstalk between Ferroptosis and Necroptosis: Novel Therapeutics in Ischemic Stroke. Oxid Med Cell Longev. 2021;2021:9991001.
Zhou H, Zhou YL, Mao JA, Tang LF, Xu J, Wang ZX, et al. NCOA4-mediated ferritinophagy is involved in ionizing radiation-induced ferroptosis of intestinal epithelial cells. Redox Biol. 2022a;55:102413.
Zhou Y, Zhao X, Zhang L, Xia Q, Peng Y, Zhang H, et al. Iron overload inhibits cell proliferation and promotes autophagy via PARP1/SIRT1 signaling in endometriosis and adenomyosis. Toxicology. 2022b;465:153050.
Acknowledgements
Not applicable.
Funding
Open access funding provided by Wenzhou Medical University. This work was jointly supported by Zhejiang Provincial University Student Sci&Tech Innovation Activity Plan and New Seedling Talent Plan (NO.2023R413079).
Author information
Authors and Affiliations
Contributions
LY was in charge of writing of manuscript. Material preparation, documentation and analysis are performed by Fen fen Gao, Ruo ting Ge and Rui Liu. MSM was in charge of the revision of manuscript. LXD conceived and reviewed the manuscript and was responsible for the overall content as guarantor. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethical Approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Lai, Y., Gao, F.f., Ge, R.t. et al. Metal ions overloading and cell death. Cell Biol Toxicol 40, 72 (2024). https://doi.org/10.1007/s10565-024-09910-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s10565-024-09910-4