
Abstract
Although musculoskeletal diseases such as osteoporosis are diagnosed and treatment outcome is evaluated based mainly on routine clinical outcomes of bone mineral density (BMD) by DXA and biochemical markers, it is recognized that these two indicators, as valuable as they have proven to be in the everyday clinical practice, do not fully account for manifested bone strength. Thus, the term bone quality was introduced, to complement considerations based on bone turnover rates and BMD. Bone quality is an “umbrella” term that incorporates the structural and material/compositional characteristics of bone tissue. Vibrational spectroscopic techniques such as Fourier transform infrared microspectroscopy (FTIRM) and imaging (FTIRI), and Raman spectroscopy, are suitable analytical tools for the determination of bone quality as they provide simultaneous, quantitative, and qualitative information on all main bone tissue components (mineral, organic matrix, tissue water), in a spatially resolved manner. Moreover, the results of such analyses may be readily combined with the outcomes of other techniques such as histology/histomorphometry, small angle X-ray scattering, quantitative backscattered electron imaging, and nanoindentation.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Why bone quality?
The main clinical tool in the effort to estimate/predict bone strength and potential fracture risk is the evaluation of clinical risk factors together with bone mineral density (BMD) measured by dual X-ray absorptiometry (DXA). Although BMD is an important contributing factor to bone strength, it is not the sole factor that determines bone fragility [1]. Clinical data show a substantial overlap in BMD values between populations that do and do not develop fractures [2–4]. Based on BMD, it has been suggested that for a certain bone mass, an individual’s fracture risk increases with age [5] while the 1-year relative risk associated with a given change in BMD has been shown to be independent of age [6]. Many studies have also revealed that mechanical variables directly related to fracture risk are either independent [7] or partly accounted for, by bone mass itself [8, 9]. The combination of BMD outcomes with biomarkers corresponding to bone turnover rates has improved the clinical situation, but even the combination of these two clinically utilized outcomes does not fully account for the observed bone strength. As a result, bone quality was introduced as an important determinant of bone strength [10]. Bone quality’s pivotal role becomes evident when one considers several clinical paradigms such as the finding that a prior fracture is a strong predictor of future ones independently of BMD suggesting intrinsic skeletal defects [11], or the clinical realization that BMD gains are disproportionate to the fracture risk reduction evident in osteoporotic patients receiving antiresorptives treatment [12]. Finally, conditions such as premenopausal idiopathic osteoporosis (IOP) where widely employed clinical tools such as DXA and biochemical markers fail to either account for the demonstrated bone strength further emphasize the importance of bone quality [13–15]. Bone quality is a broad term encompassing the structural and material properties of bone [16]. Although structural properties are equally important with the material properties of bone in the determination of its mechanical characteristics in health and disease, they are beyond the scope of the present review, and the reader is encouraged to consider review articles that are focusing on the role of bone architecture as well as methods to assess it [17–21].
Need for microscopic imaging techniques when assessing matrix and mineral composition/quality and bone strength
When assessing matrix and mineral quality, one has to account for tissue heterogeneity at the microscopic level. In healthy humans, bone surfaces may be undergoing formation or resorption, or they may be relatively inactive, and these bone turnover processes occur throughout life in both cortical and trabecular bone [22]. Bone remodeling is essential to the ability of bone to repair damaged tissue and successfully adapt to variable loading conditions. In humans, it occurs on both cortical (periosteal and endosteal surfaces, Haversian canals) and trabecular compartments, with trabecular rates higher than cortical ones throughout life [22]. The rate of cortical bone turnover, which is due to rapid growth, modeling and remodeling reaches values of about 50% per year in the mid-shaft of the femur during the first 2 years of life and eventually declines to a rate of 2–5% per year in the elderly, where maintaining the skeleton is of utmost importance. Rates of remodeling in trabecular bone are higher throughout life [22–24]. As a matter of fact, mineral and matrix composition/quality in healthy humans has been shown to vary as a function of both patient age as well as tissue age within the same patient [25]. Since organic matrix and mineral chemistry are dependent on turnover rates but also factors other than bone turnover [26, 27], it becomes evident then that microscopic imaging techniques are appropriate since they are capable of accounting for both types of age: patient and tissue age [16, 28]. This attribute is critical as published data have shown the correlation between tissue age, bone tissue composition/quality, and mechanical properties [25, 29, 30].
Healthy bone is characterized by adequate stiffness and strength combined with sufficient toughness [31]. Bone’s mineral component is responsible for its stiffness, while the organic matrix diminishes its inherent brittleness. Although bone stiffness is an average (albeit not linear) of all microanatomical values, strength and toughness depend on minuscule material defects [31]. For fracture resistance, the most important attribute of bone is its aptitude to absorb impact-generated energy without reaching complete failure, i.e., its toughness [31], which is inversely related to its stiffness [31]. Bone material toughness depends on two interrelated factors: the quality of its building blocks (mineralized collagen fibers) at multiple organizational scales and the interface quality between these blocks (one example would be the cement line) [16, 31]. The study of both toughness determining factors, and especially the quality of interfaces, is therefore optimized when microscopic analyses are utilized.
As a result of all these considerations, analysis of matrix and mineral properties as a function of specific microanatomical location (based on selection criteria such as tissue age, or mineralization extent determined by either microradiography or quantitative backscattered electron imaging [32]) is desirable as it affords the discrimination between bone turnover-dependent and independent effects. Techniques such as μCT, small angle X-ray scattering (SAXS), microradiography, quantitative backscattered electron imaging (qBEI), scanning acoustic microscopy, atomic force microscopy, Raman microspectroscopy, and Fourier transform infrared microspectroscopy (FTIRM) and imaging (FTIRI) are particularly suitable for studies aiming at exploring bone quality [16, 28, 32–36]. In the present review, we will focus on FTIRM, FTIRI, and Raman microspectroscopic imaging techniques.
Fourier transform infrared and Raman spectroscopy
The mutual principle underlying both techniques is the transition between vibrational energy states of molecules; infrared spectra arise directly from absorption of energy in the infrared range, while Raman spectra from the scattering of visible or ultraviolet photons that have either gained or lost part of their energy upon interaction with the vibrating molecular bonds. Each molecule has its own unique vibrational characteristics. Additionally, the neighboring molecular environment influences the vibrational features, thus offering additional information on the “molecular neighborhood.” The position, intensity, integrated area, and width of a vibrational band can be used for monitoring a particular functional group or regions of a particular chemical species [28]. Since information on both the molecule and its neighborhood is readily available, most vibrational spectroscopic bands are composite ones, consisting of underlying peaks specific to a specific molecule-molecular neighborhood state. Thus, resolution of these broad bands to their underlying peaks offers the most detailed molecular picture possible. This is achieved through the utilization of second derivative spectroscopy (to decipher the exact number and position of these underlying peaks in a mathematically objective manner) coupled with curve-fitting routines (to determine their relative contribution) [37–40].
Vibrations are for the most part only either infrared or Raman active; thus, the two techniques offer complimentary information regarding the analyzed sample [41]. In the 1970s and 1980s, spectra-structure correlations were developed for proteins, nucleic acids, and lipid assemblies as well as molecules that are vital for the biological functions of the macromolecular moieties [28]. It was simultaneously realized that the proper spatial organization of the structural components in both cells and tissues is crucial for their optimum function, deviation from which results in pathological states [28, 42–44]. The earliest attempt to address this insight occurred in 1949 [45], when a reflecting microscope was coupled with an IR spectrometer to acquire utilizable spectra of nerve fibers and small crystals. On the other hand, the challenging signal-to-noise ratio of common Raman scattering delayed the introduction of Raman microscopic and imaging applications until 1974 [46]. Ever since, wide-ranging technical advances have resulted in a broader application of vibrational microscopy and imaging [28]. Recent new technological gains have greatly enhanced the afforded spatial resolution and rapid data acquisition. A contemporary infrared setup based on a scattering-type scanning near-field optical microscope equipped with a coherent-continuum infrared light source allows the acquisition of infrared spectra with an AFM-comparable spatial resolution (20 nm) [47]. Similar results are achievable through a second setup utilizing a pulsed, tunable infrared laser which excites molecular absorption in a sample in attenuated total reflection (ATR) mode, on a ZnSe prism [48]. Tip enhanced Raman scattering (TERS) combines surface enhanced Raman spectroscopy (SERS) with atomic force microscopy (AFM) analysis allowing for analyses with spatial resolution in the nanometer scale [49]. The development of stimulated Raman scattering microscopy permits the acquisition of images at specific Raman-shifted frequencies at video rates [50]. Finally, spatially offset Raman spectroscopy (SORS) is being actively developed so as to offer a method for noninvasive in vivo Raman measurements with custom-designed fiber optic probes placed on the skin of a subject [51].
Unique advantage of vibrational spectroscopic techniques
Bone is a composite material consisting of mineral, organic matrix, and water, with a highly complex and amazing hierarchical structure [16]. All three components contribute, both individually and in concert, to the determination of bone mechanical properties such as stiffness (essential for preventing bone bending under the load of body; often measured by the elastic modulus (or Young’s modulus) [31]), strength (maximum stress the bone material can sustain prior to failure), and toughness (absorption of impact energy without reaching complete failure; roughly measured by the energy to failure) [31]. Since bone is a heterogeneous material, its overall stiffness is a nonlinear average of all the local stiffness values [31]. On the other hand, bone strength and toughness may not be calculated as an average of bone constituents’ local values, as they depend on the nucleation and progression of cracks; thus, they are dependent on miniscule material defects (pores, microdamage, interfaces, fiber organization) [31]. Interestingly, toughness and stiffness are inversely related [31]. Although plenty of analytical techniques offer a wealth of information on individual bone components, to the best of our knowledge, thermogravimetric and vibrational spectroscopic analyses are the only two methods that provide simultaneous information on all three bone components. The former requires tissue homogenate and is destructive (no further analyses are feasible on the same tissue), while the latter offers both qualitative and quantitative information in a nondestructive, spatially resolved manner, thus allowing their combinatorial use with other techniques, enabling the gaining of a wealth of information on the same bone surface.
Tissue requirements
In principle, both FTIR [52] and Raman [42, 53] techniques can analyze fresh bone tissue provided there is an adequately flat surface. On the other hand, reflection FTIR is hampered by challenges such as subtle changes in the raw spectral bands, the significance of which remains to be determined, whereas blood in the fresh bone tissue may result in uncorrectable fluorescence interference with the bone characteristic Raman bands, rendering the spectra unusable. The overwhelming majority of tissue available for spectroscopic analysis (especially human tissue) has undergone tissue dehydration through a series of acetones or alcohols, followed by embedding in a plastic medium, most often poly methyl methacrylate (PMMA). The choice of fixative solution as well as embedding material is of utmost importance for subsequent spectroscopic analysis as the various media have different, profound effects on bone tissue [54, 55]. The most commonly used (due to the fact that the least amount of artifacts is introduced) protocol involves fixation of tissue in alcohol and embedding in PMMA resin. Thin tissue sections (2–5 μm) are required for FTIR analysis, whereas Raman can analyze both bone blocks with a polished surface as well as sections that are more than 5 μm thick. Details of spectral acquisition and processing have been published elsewhere [56, 57].
Parameters assessed by vibrational spectroscopic analysis and their importance
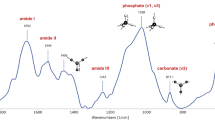
Below, we show typical examples of FTIR and Raman spectra of bone tissue (Fig. 1).

Typical FTIR (top) and Raman (bottom) spectra, with various bands of utility for the analysis of bone tissue appropriately labeled
(i) Mineral to matrix ratio
This parameter has been validated against ash weight measurements, as well as mineral content by quantitative backscatter electron imaging [58–60].
The mineral to matrix ratio is the most widely reported parameter of vibrational spectroscopic analysis and is easily determined in experiments utilizing either infrared or Raman methods [28]. Its physical principle is that in vibrational spectroscopy, the integrated area of a band is directly proportional to the concentration of the specific molecular moiety giving rise to the specific band. Most commonly, the ratio of the integrated areas of the v1,v3PO4 (stretching and bending vibrations) to amide I (mainly from the peptide bond C=O stretching vibration with minor contributions from the out-of-phase CN stretching vibration, the CCN deformation, and the NH in-plane bend) or II (out-of-phase combination of the NH in-plane bend and the CC and NC stretching vibration with smaller contributions from the CO in-plane bend and the CC and NC stretching vibrations) bands in FTIR, and either v1 or v2PO4 to either amide I or amide III band in Raman, provides what is commonly reported as the mineral/matrix ratio. This is a form of bone density that, unlike other measures such as BMD by DXA, or tissue mineral content (TMC) or BMD or degree of mineralization of bone (DMB) by μCT, directly measures and accounts for the amount of the organic matrix in the sample volume analyzed. It strongly correlates with ash weight measurements by thermogravimetric analysis [58] and with mineralization data by qBEI in secondary mineralized bone packets [59]. The fact that this ratio encompasses the amount of organic matrix is of great importance and utility, as the mechanical properties of bone are determined by the quantity and quality of both its mineral and the matrix (primarily type I collagen fibrils) constituents [61]. The importance of the organic matrix amount in the determination of bone strength has been shown in published reports which demonstrate that bone displays a region of elastic (recoverable) deformation, followed by a region of plastic (permanent) deformation. Utilizing tensile testing of gradually decalcified bovine bone tissue, Burstein and Reilly suggested that the mineral offers mainly compressive, while the collagen tensile strength [61], though it is more likely that the interaction between the two is responsible for the unique mechanical characteristics of bone. The spectroscopically determined mineral/matrix ratio is directly proportional to bending stiffness and failure moment and a superior predictor of bone-bending stiffness compared to BMD alone [62]. At actively forming trabecular surfaces of healthy females, this ratio increases as a function of tissue age and decreases as a function of patient age at constant tissue age [25]. It is unaltered in juvenile osteoporosis [63], decreased in osteoporosis [64] compared to premenopausal healthy controls, and modified by various osteoporosis therapies [65–69]. Unlike outcomes based on either DXA, μCT, or qBEI, in premenopausal patients diagnosed with IOP, this ratio at actively forming trabecular surfaces was altered between IOP patients that sustained fragility fractures vs. those who did not [14].
An interesting option is inherent with Raman analysis, as this ratio can be calculated based on either the ratio of the integrated areas of the v1 to amide I or v2PO4 to amide III band; the former is tissue organization independent, while the latter is dependent on both the amount of mineral and organic matrix present in the sample analyzed, as well as collagen fiber and mineral crystallite orientation. All four peaks are present in the Raman spectra of bone tissue; thus, they offer the option to combine quantitative data with tissue organization considerations, without the need of extra analyses [70, 71]. Such information is also available through FTIR analysis, but since a polarizer needs to be introduced in the beam path, the required time for analysis is tripled as several spectra of the same pixels need to be acquired to be able to incorporate tissue orientation considerations [72].
(ii) Mineral maturity/crystallinity
This parameter has been validated against X-ray diffraction outcomes [37, 73–75].
Bone mineral consists of poorly crystalline, highly substituted, apatite crystals. Pure apatites consist of calcium (Ca2+), phosphate (PO4 3−), and hydroxyl (OH−) ions. Biological apatites are characterized by various crystal lattice substitutions such as magnesium (Mg2+), potassium (K+), and/or sodium (Na+) for Ca2+, acidic phosphate (HPO4 2−) for PO4, and carbonate (CO3 2−), which can actually substitute for either OH− (type A), or PO4 3− (type B), or be loosely bound on the crystal surface (labile [76]). Additionally, crystal vacancies may be present in the crystallites. The type and extent of substitutions influences the crystal solubility as well as the size and shape. In the literature, the chemical makeup of these crystals (and how close it is to that of pure, highly crystalline hydroxyapatite) is usually referred to as crystal maturity, while the size and shape as crystallinity. Interestingly, it is changes in the chemical makeup that drive changes in size and shape; thus, the term mineral maturity/crystallinity is often used.
Vibrational spectroscopic techniques such as FTIR and Raman [28] are particularly useful in establishing the chemical makeup of the poorly crystalline apatitic crystals in bone, i.e., the presence of impurities, and based on comparison to X-ray line broadening analysis, on their shape and size [75, 77]. Bone mineral maturity/crystallinity is significantly dependent on both subject and tissue age within the same subject. Healthy bone consists of crystallites whose size and shape fall within a certain range. Values outside this range have been encountered in clinical cases with prevalent fragility fractures such as osteoporosis and fluorosis [16, 64, 78, 79]. In FTIR, the information is attainable through the resolution and quantification of the underlying peaks in the v1,v3PO4 band, using second derivative spectroscopic and curve-fitting analysis [37, 39]. In Raman spectroscopic analysis, this bone quality index is assessed from either the full width at half height of the v1PO4 band or the wavelength at maxima of the same band [75].
(iii) Carbonate content
This parameter has been validated against chemically determined carbonate content in synthetic analogs of bone mineral, as well as bone tissue [76, 80, 81].
As stated in the previous subsection, bone mineral crystallites are poorly crystalline, highly substituted apatites. One of the most abundant substitutions is carbonate, which may be in the form of type A, type B, or labile [76], as outlined previously [82]; the type and extent of the substitution influences the crystals’ solubility and size and shape. In healthy bone, the average carbonate content is about 6 wt%, although it is known to vary according to subject and tissue age, as well as anatomical compartment (trabecular vs. cortical) [81]. The extent of carbonate substitution has been reported to be altered in bone susceptible to fragility fractures in both animal models and humans [83–92].
Determination of relative carbonate content in bone tissue is readily available in both FTIR and Raman analyses. In FTIR, two bands are evident that are due to carbonate: one ∼850–890 cm−1 and one ∼1400–1500 cm−1. Both bands are composite ones, overlapping with either acidic phosphate peaks (the former) or noncollagenous protein peaks (the latter); thus, they require further resolution to their respective underlying peaks using second derivative spectroscopy followed by curve-fitting routines so as to gain information on the extent as well as the type of carbonate substitution [37, 81]. In Raman spectroscopy, the most frequently used spectral band to report type B carbonate content is ∼1070 cm−1, while for type A, a weak band ∼1103 cm−1 is used [42]. Both of these bands are composite ones as well, overlapping with phosphate, lipid, and PMMA peaks; thus, as is the case with FTIR, further processing is required so as to resolve the carbonate-related peak.
Using homogenized human iliac crest biopsy tissue, it has been shown that carbonate content slightly increases with increasing subject age, even in the absence of accompanying changes in crystallinity after the age of 30 [93]. Unfortunately, since homogenized tissue was used, no correlation to tissue age within the same subject was feasible. Later studies employing vibrational spectroscopic techniques and attempting to establish the correlation of carbonate content with tissue age have been conflicting, with some reports alluding to a direct relationship between the two, while others showing the opposite [37, 81, 94]. This apparent conflict is most likely due to the fact that in most studies, no strict tissue age control was applied (for example using fluorescent double labels to determine the exact tissue age), or the broad raw bands were used in the calculations unjustifiably ignoring the spectral contribution of other moieties to these unresolved bands. Moreover, although most frequently the carbonate content based on vibrational spectroscopic analyses is reported as the carbonate to phosphate ratio, a few reports have introduced the ratio of carbonate to organic matrix [44]. As the normalization is to the organic matrix content, this ratio is obviously not reflecting the extent of carbonate substitution in the mineral crystallites, but is rather another, crude way of expressing mineral to matrix ratio. Nevertheless, since altered carbonate content has been reported in bone tissue susceptible to fragility fractures, future studies are needed so as to explain and consolidate the glaring discrepancies that exist in the published literature concerning this bone mineral quality metric.
(iv) Relative tissue water content/nanoporosity
The direct method in fresh bone tissue has been validated against gravimetric analysis results [95].
Recently published reports have reintroduced to the spotlight the importance of bone tissue water content in the determination of its mechanical behavior [96, 97]. Vibrational spectroscopic techniques and in particular Raman analysis can provide information on tissue hydration. Unal et al. recently identified four Raman peaks (∼3220, 3325, 3453, and 3584 cm−1) that correlated with water loss determined by thermogravimetric analysis in a series of experiments involving the gradual dehydration of fresh, demineralized bovine femur bone [95]. The advantage of this approach is that fresh tissue is utilized, and water is directly measured through quantification of its hydroxyl groups. On the other hand, potential limitations include potential spectral interference from apatite when undemineralized bone is analyzed and the fact that such analysis requires a custom-made, noncommercially available instrument. Moreover, although fresh bone tissue may be secured during experiments involving animal models, it should be kept in mind that most of the human bone tissue available for spectroscopic analysis has already been dehydrated and embedded in plastic (most often PMMA), as part of standard clinical practice.
There is also an indirect way of estimating tissue hydration during Raman analysis of fixed, embedded, mineralized bone tissue. Since PMMA will infiltrate bone tissue in spaces that were occupied by tissue water prior to fixation, it follows that its relative content may be used as a tissue water surrogate, or nanoporosity within the mineralized tissue [25]. It may be expressed as PMMA/amide III based on the integrated area of the spectral slice ∼494–509 cm−1 (PMMA)/amide III. In healthy females, this parameter is greatly dependent on tissue age, inversely correlates with healthy subject age, has the lowest values in subjects aged 20–45 years [25], and eventually increases in patients diagnosed with postmenopausal osteoporosis. This spectroscopic parameter has been recently shown to be altered in children diagnosed with mild forms of osteogenesis imperfecta [98], in agreement with thermogravimetric analysis results in a mouse animal model [60].
(v) Collagen cross-links
This parameter has been validated against biochemically characterized collagen cross-linked peptides, as well as biochemically analyzed model tissues [26, 38, 99, 100]. It has also been correlated with mechanical properties at the nano- and whole bone organ levels [26, 101].
The organic matrix of bone consists of collagen and a series of noncollagenous proteins and lipids. Some 85–90% of the total bone protein consists of collagen fibers [102]. Type I collagen, the principal component of the organic matrix of bone, as well as other connective tissues, is a large fibrous protein with a highly repetitive amino acid sequence [103] {Gly (glycine) – X – Y}n (often X is proline and Y is hydroxyproline), allowing three polypeptide chains (called α chains; type I collagen is composed of two α1 and one α2 chains) to fold into a unique triple helical structure, consisting of three domains: the –NH2 terminal nontriple helical, the triple helical, and the –COOH terminal nontriple helical domains. The single uninterrupted triple helical domain represents more than 95% of the molecule [103]. The most distinctive feature of mineralizing type I collagen in mineralized tissues is its cross-linking chemistry and molecular packing structure [103]. At present, seven major collagen cross-links have been established as naturally occurring intermolecular cross-links. They are (1) dehydrodihydroxylysinonorleucine (deH-DHLNL), (2) dehydrohydroxylysinonorleucine (deH-HLNL), (3) dehydrohistidinohydroxymerodesmosine (deH-HHMD), (4) pyridinoline (Pyr), (5) deoxypyridinoline (d-Pyr; lysyl analog of Pyr), (6) pyrrole, and (7) histidinohydroxylysinonorleucine (HHL). The first three are NaBH4-reducible (their reduced forms are referred to as DHLNL, HLNL, and HHMD, respectively) and the rest are nonreducible compounds [103]. Cross-linking is initiated with the conversion of the specific peptidyl Hyl, mainly at the C domain of the collagen molecules soon after the molecule is synthesized. These aldehydes are located at the boundaries of the “hole” zones and react with the juxtaposed Hyl on neighboring molecules to form bifunctional cross-links (deH-DHLNL) prior to mineralization. These cross-links are capable of tautomerizing between an iminium and a keto form. When two such cross-links are able to contact each other, they can condense to form a complex cross-link, pyridinoline. As a minor form, the lysyl analog of pyridinoline (deoxypyridinoline or lysylpyridinoline) is present in some tissues. It comprises two Hyls and one Lys and appears to be present mainly in mineralized tissues. The ratio of pyridinoline to deoxypyridinoline cross-links in human bone is 3 or 4 to 1, and their content appears to reach the maximum concentration by ∼10–15 years of age and remain in the same range throughout adult life [103]. Because cross-link condensation reactions, except for the initial oxidation step, are spontaneous, turnover rate is an important factor (but not the sole one) in regulating cross-link maturation [103].
The intermolecular cross-linking affords the fibrillar matrices with mechanical properties such as tensile strength and viscoelasticity. There have been suggestions that not only is the matrix content in osteoporotic bone decreased relative to age- and sex-matched controls [104] but that biochemical alterations in the collagen molecules also exist [105, 106]. The decreased mechanical strength and the change in the thermal stability can be directly related to changes in posttranslational modifications, i.e., lysine hydroxylation and the intermolecular cross-link profile. The increased hydroxylation and change in cross-linking are consistent with increased turnover of the collagen, possibly in an attempt to initiate a repair mechanism which, in fact, leads to an acceleration in the increase in fragility of bone. Increased collagen hydroxylation in osteoporotic as opposed to osteoarthritic femoral heads has also been reported [107]. Hip fracture patients are frequently vitamin B6 deficient suggesting that B6 deficiency may be a risk factor for osteoporotic fractures [108] (Vitamin B6 is an essential co-factor for lysyl oxidase, the enzyme needed to make reactive aldehydes on collagen [109, 110]). The importance of collagen intermolecular cross-links to the mechanical performance of bone is very apparent in lathyrism, where it has been shown in animal models that alterations in enzymatic collagen cross-links, even when restricted to microanatomical locations, are sufficient to influence bone strength [99, 101].
Vibrational spectroscopy has been used previously for analyzing three-dimensional structures of collagen and other proteins. There are many vibrational bands characteristic of peptide groups and side chains from which information on protein structures may be obtained. Among these, the amide I band is especially sensitive to secondary structures [111]. In such studies, information on protein structures is extracted from resolution of broad envelopes consisting of component bands arising from the amide I modes of various secondary structures. This may be achieved through employment of resolution enhancement techniques such as such as second derivative spectroscopy, deconvolution, and differential spectroscopy. We would strongly recommend the use of second derivative spectroscopy as it is mathematically objective (no initial assumptions are required to be made by the operator). This technique allows enhanced identification of small and overlapping absorption peaks that are not resolved in the original spectrum, albeit at a loss in signal-to-noise ratio [112–114]. Using such techniques, spectral parameters have been developed that correlate with biochemically determined enzymatic collagen cross-links (specifically pyridinoline, deoxypyridinoline, and divalent) for FTIR [26, 38, 99], and by extrapolation for Raman spectroscopy [42, 101], and which enable the description of them in a spatially resolved manner, a unique feature of spectroscopic analysis of mineralized tissues. Although spectroscopic theory allows in principle this extrapolation from FTIR to Raman, the considerably lower signal-to-noise ratio of Raman compared to FTIR in the spectral region of amide I should be kept in mind when minor underlying peaks shown in the FTIR amide I spectra to correlate with deoxypyridinoline and divalent enzymatic collagen cross-links are utilized in Raman spectra without prior proper validation. On the other hand, we have recently validated the use of the pyridinoline-correlating subpeak in Raman amide I spectral regions [100].
Based on such analyses, it has been shown that in healthy females, the relative Pyd content of trabeculae is significantly dependent on tissue age while exhibiting a Gaussian distribution as a function of subject age at constant tissue age [25]. Moreover, it was the most discriminant bone quality index discerning between changes due to aging and postmenopausal osteoporosis [25]. The spectroscopically monitored collagen cross-links (and their spatial distribution) may be an underestimated, yet major contributor to bone strength and/or toughness. In cases where bone resistance to fracture cannot be explained by either BMD or architecture, or predicted by finite element analysis, the ratio of pyridinoline to divalent collagen cross-links correlates with fracture incidence [13–15, 68, 115]. Its potential clinical value may be seen in the case of IOP, a rather rare condition in premenopausal women, in which fragility fractures and/or low BMD occur in otherwise healthy women with normal gonadal function [14]. The IOP-diagnosed population includes women with abnormally low BMD and no fragility fractures, as well as women with normal BMD and fragility fractures. Although different techniques (histomorphometry, qCT, DXA, qBEI, biochemical to determine advance glycation endproducts, finite element analysis) are able to discriminate healthy controls and IOP women, to date, only vibrational spectroscopic techniques, and in particular the pyridinoline/divalent collagen cross-link ratio, are capable of further discrimination within the IOP group based on fracture incidence [13–15].
(vi) Relative proteoglycan content
This parameter has been validated against a series of standard proteoglycans, as well as model tissues [116–121].
Proteoglycans are noncollagenous constituents of the extracellular matrix, present in both cartilage and bone among many other tissues. They are characterized by the presence of one or more glycosaminoglycan polymers attached to a protein core via an O-linkage through a serine residue [122]. In bone, they fulfill multiple roles involving the organic matrix assembly, the modulation of both organic matrix mineralization and remodeling rates [123–126], as well as preventing mineralization of the perilacunar matrix around the osteocyte lacunae, and the canaliculi in compact lamellar bone, safeguarding the unhindered interstitial fluid movement [127]. They are subject to subtle posttranslational modifications, some of which are subject and tissue age dependent [128], in size, sulfation, and/or charge density, which are crucial for their specific role [123]. Finally, an important chemical property of proteoglycans is their capacity to swell by binding large amounts of water (through their GAG chains) and fill in spaces [129].
While information on proteoglycans is available with both FTIR [130] and Raman [116] spectroscopic analyses, only Raman (spectral band ∼1375 cm−1; CH3 symmetric deformation of glycosaminoglycan groups) can readily provide this in mineralized tissues [116] as the marker band for proteoglycans in FTIR ∼1060 cm−1 (indicative of sugar rings) is overlapped by the v3PO4 band. To date, vibrational spectroscopic analysis is not discriminant enough between different proteoglycan species, but the Raman spectral signature of proteoglycans arises from the glycosaminoglycan (GAG) chains [116], and in bone, chondroitin 4-sulfate accounts for ∼90% of the total GAG content, while being predominantly encountered in biglycan and decorin [131].
In healthy humans, at actively forming trabecular surfaces, the relative glycosaminoglycan content of bone is strongly dependent on tissue age and shows a weaker correlation with subject age [25]. It exhibits a Gaussian distribution when considered as a function of subject age, and its values are significantly lower in diseases with a prevalence of fragility fractures such as postmenopausal osteoporosis and osteogenesis imperfecta [25, 98].
(vii) Relative lipid content
This parameter has been validated against a series of chemically characterized lipids [132, 133].
Lipids have been identified in the published literature as nucleators of collagen fiber mineralization, revealed to be present in a layer just immediately behind the first deposited bone mineral [134, 135]. They are also part of the matrix vesicle membranes, implicated in endochondral and bone mineralization [136–148]. Although information on their relative content is available during both FTIR [149] and Raman analysis [135] of bone tissue, Raman is better suited as in the relevant spectra a band ∼1300 cm−1 (arising from methylene twisting vibrations) is present, which although relatively weak, is not overlapped by the spectral signature of any other bone constituent [135]. The ratio of the integrated area of this band normalized to the integrated area of the amide III band may be used to describe the relative lipid content in mineralized tissues [28]. Establishing the relative lipid content in bone tissue may be very important for another, yet often overlooked reason. Oxidized lipids serve as a substratum for advanced glycation endproducts (AGEs) formation and accumulation [150]. Since increased accumulation of AGEs has been related with elevated fracture risk [151], it becomes evident that including considerations pertaining to lipid content and their spatial distribution is desirable when attempting to understand factors contributing to the determination of bone strength. In healthy females, in the area of trabecular bone, it has been shown to be dependent on tissue age while exhibiting a Gaussian behavior as a function of subject age, at constant tissue age [25].
(viii) Other parameters
Using vibrational spectroscopic analysis, it is feasible to obtain information on several other bone quality indices.
Some single-detector infrared instruments (mostly using synchrotron radiation) provide access to the v4PO4 spectral region (500–650 cm−1). This spectral region offers the advantage of a much more straightforward (is easier to discern the underlying constituent peaks of the raw band) determination of mineral maturity/crystallinity, calculated either as a peak splitting function calculated as a ratio of the areas above and below the double peak of the phosphate anti-symmetric bending frequency (v4) at approximately 550 to 650 cm−1 [73] or through curve-fitting routines based on second derivative spectral resolution [74], and acid phosphate content (∼530–560 cm−1) [74, 152].
The peak width at half height of the pixel distribution of any of the previously described bone quality indices, often referred to as “heterogeneity” [44], has been reported in FTIR imaging and shown to decrease with bisphosphonate treatment [153] and in bones from patients with fractures as compared to nonfractured controls [154]. This may be an important piece of information in the puzzle of factors leading to bone fragility.
Acid phosphate substitution into the apatite crystal lattice can be measured with FTIR imaging [39] and has been shown to be inversely proportional to crystallinity, thus is expected to be an important determinant of mineralized tissues’ mechanical properties and their response to treatment [64, 155–157]. Using uni- and multivariate analyses of FTIR data from model compounds and spectral images from mineralized tissues, Spevak et al. were able to define two peak height ratios in the v1,v3 phosphate spectral band (namely the 1112/1096 cm−1 and the 1127/1096 cm−1) that provide information on the acid phosphate substitution within these samples. Both of these ratios correlated significantly with the previously published mineral maturity/crystallinity spectral index [37, 158], based on the ration of the 1030 cm−1 and the 1020 cm−1 subpeaks of the v1,v3 phosphate band. Moreover, since acid phosphate substitution is known to be associated with new mineral deposition [155, 159], Spevak et al. suggested that it may be feasible to use images of acid phosphate substitution in place of tetracycline labeling to locate the most recent site of bone deposition [39], although this suggestion has not been validated to date and is unclear how it may be utilized in cases where the acid phosphate content is altered due to either disease and/or treatment.
Nonenzymatic collagen cross-links (AGEs) are evaluable by Raman spectroscopic analysis. AGEs are a diverse group of compounds, formed through the nonenzymatic glycation or glycoxidation of proteins, lipids, and nucleic acids [160] with the best-studied being carboxymethyl-lysine (CML) and pentosidine (PEN) [161–164]. Using Raman spectroscopic analysis of tissues, the relative content of CML and PEN may be monitored based on the integrated area ratio of bands at 1150 (CML) cm−1 or 1495 (PEN) cm−1/1450 cm−1 (methylene side chains (CH2)) [165–170]. In mineralized tissues, AGEs formation and accumulation has been shown to result in more brittle bone that is less capable to deform prior to fracturing, and thought to be a pivotal culprit for the increased fracture incidence encountered in diabetes [151, 170]. In animal models, the spectroscopically determined AGEs have been shown to correlate with inflammation and affiliated bone loss, as well as propagation toughness (a measure of the ability of the bone matrix to resist crack propagation and avoid catastrophic failure) [169, 170]. This measurement is better performed using fresh, nondehydrated or embedded bone tissue, as the fixation and embedding processes usually result in loss of lipids, which may result in subsequent reduction of the AGEs spectroscopic signal below instrumental detection limit [170].
Examples
Due to its excellent spatial resolution (∼1 μm), Raman analysis is not particularly well suited for large imaging analyses, but rather smaller, more anatomically focused one. An example is provided in Fig. 2, in the cortical (endosteal surface) bone compartment of an iliac crest biopsy from a 65-year-old postmenopausal osteoporosis patient who had been treated with teriparatide for 2 years. Raman microspectroscopic instruments can be readily coupled to a fluorescent microscope, thus enabling the selection of areas for analysis based on the presence of double fluorescent labels. Since the protocol for the administration of the double labels is precise, the analyses may be performed as a function of tissue age. (a) Picture through the coupled fluorescent microscope of a Raman instrument, with the tetracycline double labels evident. The green box indicates the area of Raman imaging. Interestingly, the integrated area of the whole, uncorrected Raman spectra accurately shows the presence of fluorescence labels due to the sensitivity of the Raman detectors to fluorescence (b; top image). The tissue water content (b; middle image) and mineral/matrix (b; bottom image) can be readily calculated and, as seen, are inversely related. When an anatomically equivalent bone surface (cortical, endosteal) surface is analyzed (c) that is not undergoing active bone formation (no fluorescent double labels present), Raman imaging analysis shows no presence of fluorescent labels (c; top image), low tissue water content (c; middle image), and high mineral/matrix values (c; bottom image) (Figs. 3 and 4).

a–c An example of information that may be gained through Raman imaging of bone tissue

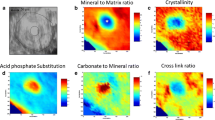
FTIR imaging is well suited for imaging both small and large areas with a spatial resolution of ∼6.3 μm. The advent of faster detectors as well as the evolution of the associated software allows the analysis of a whole iliac crest biopsy surface within 6–8 h depending on spectral resolution used. An example is given in this figure, in which the whole cortex of a thin (∼4 μm) section from the same iliac crest biopsy used in Fig. 2 is analyzed. a Photomicrograph of the cortex. b The spatial distribution of the mineral content (blue = no mineral, red = maximum mineral content). c The spatial distribution of the organic matrix content (blue = 0, red = maximum mineral content). d The spatial distribution of PMMA (blue = 0, red = maximum)

An example of further information available through FTIR imaging analysis, in the area of trabecular bone in a thin section from the iliac crest biopsy used in Figs. 2 and 3. a A photomicrograph of the trabecula analyzed. b The calculated spatial distribution of the organic matrix content, showing the lamellar structure. c The calculated spatial distribution of the mineral maturity/crystallinity. d The calculated spatial distribution of the pyridinoline/divalent enzymatic collagen cross-link ratio
Potential pitfalls
Much like every other analytical technique, potential pitfalls are associated with vibrational spectroscopic analysis, that the user and/or reader ought to be aware of when selecting specific methods and interpreting the results.
Lasers used in Raman spectroscopy are inherently polarized; thus, certain spectral bands are dependent exclusively on the concentration of the chemical species giving rise to them, while others are dependent on both concentration and tissue organization. The best example of the latter is the most commonly reported mineral/matrix ratio calculated from the v1PO4 and amide I bands, as opposed to the ratio calculated from the v2PO4 and amide III bands [70].
Spectral bands are usually composite ones. Their constituent underlying peaks are unique to specific chemical/biochemical environments. As a result, valuable information may be gained by resolving the raw bands into their constituent peaks. This can be achieved through the utilization of spectral resolution techniques coupled with curve-fitting routines. When engaging in such activity, one should always rely on spectroscopically sound resolution methods such as deconvolution (though minimal initial parameter input by the operator is required) and second derivative spectroscopy (mathematically objective, thus strongly recommended) [38]. Moreover, the calculation and further processing of average spectra (from individual ones acquired over a large tissue area) should be avoided, as this results in smoother average spectra of superior S/N, at the expense of biological variability (which is then underestimated). We also strongly advise against choosing a set number of peaks at specific wavelengths to curve-fitting series of spectra acquired in different tissues and/or different anatomical areas within the same mineralized tissue surface/block/section, especially if organic matrix is being analyzed. Organic matrix composition may be different even between two neighboring 1-μm3 voxels [171]; as vibrational spectroscopic analysis is sensitive to protein secondary structure (thus, its results are greatly dependent on tissue composition heterogeneity), fitting all spectra with a set number of peaks at predetermined wavelengths will result in mathematical solutions that may not necessarily be biologically relevant [38].
As mentioned earlier, the peak width at half height of the pixel distribution of the previously described bone quality indices (during vibrational spectroscopic imaging analysis) is often used as a surrogate of bone tissue heterogeneity for the specific parameter considered [44, 153, 154]. Although tissue heterogeneity is accepted as a major determinant of bone mechanical properties, the reader should always keep in mind that to date, spectroscopically derived heterogeneity is based on analysis of a few, randomly selected areas of the available bone tissue, and no thorough validation exists thus far as to the validity of such an approach (whether the values of these randomly selected areas are adequately representing the heterogeneity of the whole bone tissue available for analysis). Moreover, they treat the distributions as Gaussian ones, even in cases where it is evident that the distribution of values is not normal.
Finally, both FTIR and Raman microspectroscopic imaging methods report on tissue properties that may be dependent on bone turnover (thus changes in mean tissue age are driving potential changes in monitored tissue properties), and/or factors independent of bone turnover [172, 173]. As such, we would advise against random selection of anatomical areas for analysis. Either the whole bone surface should be considered for analysis (a time-consuming endeavor), or microanatomical areas should be chosen for analysis based on robust criteria, such as tissue age or mineralization extent among others.
Conclusions/outlook
Vibrational spectroscopic techniques offer a wealth of unique information on bone material properties and structure. Yet, at the time of this review, they are not amenable for routine use in the clinic, as they require a bone tissue biopsy to be excised from the patient, something that is not a routine diagnostic procedure in the clinical practice. On the other hand, they have proven to be invaluable analytical tools in instances where pathophysiologic mechanisms are investigated, especially when the routinely used clinical outcomes cannot explain or predict fracture incidence, or when the effect of various therapies on bone material properties is investigated (especially involving clinical trials). They are also particularly suitable for studies involving animal models. Attempts are currently underway to further develop these techniques so as to circumvent the need for a biopsy [174, 175], though it should be kept in mind that only the periosteal bone surface would be available for analysis.
Both FTIR and Raman outcomes are thought to be important contributors in the determination of bone’s mechanical characteristics. Thus, it is of great interest to correlate the outcomes of the spectroscopic analysis with those of mechanical tests at the nano-, micro-, as well as the whole organ levels. This is already actively pursued between nanoindentation and vibrational spectroscopic analysis [176–214], although not all of the publications report on analysis of the same area by both techniques. On the other hand, the published reports to date show an excellent correlation between spectroscopically determined bone quality indices, nanomechanical, and whole bone mechanical properties, especially when tissue age and mineralization extent were accounted for when selecting areas of analysis [26, 101]. Although few reports already exist on the subject [83, 84, 170, 188, 215–218], great potential is also envisioned for the correlation of spectroscopically determined bone quality indices with outcomes of microindentation, as the latter is actively being developed to become an everyday clinical tool in the effort to identify patients at increased fracture risk based on mechanical rather than mineral density considerations.
Recently, reports have been published entailing the coupling of an infrared spectrometer to an AFM microscope, allowing the study of bone material properties with a lateral spatial resolution of 20–100 nm [219, 220]. This is a considerable improvement over the 6.3- or 0.6-μm spatial resolution capability afforded by FTIR imaging and Raman analysis, respectively, thus promising novel bone compositional information at the nanometer level. This should become readily achievable once a couple of concerns are addressed regarding the actual spot of analysis (to date, a range rather than a precise value is provided), the partial spectral shift evident in the published spectra (the extent of which seems to be instrument dependent), and whether there are heat-related alterations induced on the analyzed areas.
Great promise also holds for the use of these techniques in combination with other imaging modalities such as time-lapse micro-CT [34] (thus correlating material and structural properties), scanning acoustic microscopy (correlating with mechanical outcomes) [35], and backscattered electron imaging (to obtain the distribution of mineral) [32], offering both structural and material outcomes on the very same bone surface.
References
Marshall D, Johnell O, Wedel H (1996) Meta-analysis of how well measures of bone mineral density predict occurrence of osteoporotic fractures. BMJ 312:1254–1259
Cummings SR (1985) Are patients with hip fractures more osteoporotic? Review of the evidence. Am J Med 78:487–494
McCreade RB, Goldstein AS (2000) Biomechanics of fracture: is bone mineral density sufficient to assess risk? J Bone Miner Res 15:2305–2308
Schnitzler CM (1993) Bone quality: a determinant for certain risk factors for bone fragility. Cacif Tissue Int 53:S27–S31
Hui S, Slemenda CW, Johnston CC (1988) Age and bone mass as predictors of fracture in a prospective study. J Clin Invest 81:1804–1809
Siris ES, Brenneman SK, Miller PD, Barrett-Connor E, Chen YT, Sherwood LM, Abbott TA (2004) Predictive value of low BMD for 1-year fracture outcomes is similar for postmenopausal women ages 50–64 and 65 and older: results from the National Osteoporosis Risk Assessment (NORA). J Bone Miner Res 19:1215–1220
Jepsen KJ, Schaffler MB (2001) Bone mass does not adequately predict variations in bone fragility: a genetic approach. Trans Orthop Res Soc 47th Annual Meeting 114
Kanis JA, Melton LJI, Christiansen C, Johnston CJ, Haltaev N (1994) Perspective: the diagnosis of osteoporosis. J Bone Miner Res 9:1137–1142
Kann P, Graeben S, Beyer J (1994) Age-dependence of bone material quality shown by the measurement of frequency of resonance in the ulna. Calcif Tissue Int 54:96–100
Watts NB (2002) Bone quality: getting closer to a definition. J Bone Miner Res 17:1148–1150
Klotzbuecher CM, Ross PD, Landsman PB, Abbott TA 3rd, Berger M (2000) Patients with prior fractures have an increased risk of future fractures: a summary of the literature and statistical synthesis. J Bone Miner Res 15:721–739
Watts NB, Cooper C, Lindsay R, Eastell R, Manhart MD, Barton IP, van Staa TP, Adachi JD (2004) Relationship between changes in bone mineral density and vertebral fracture risk associated with risedronate: greater increases in bone mineral density do not relate to greater decreases in fracture risk. J Clin Densitom 7:255–261
Malluche HH, Porter DS, Mawad H, Monier-Faugere MC, Pienkowski D (2013) Low-energy fractures without low T-scores characteristic of osteoporosis: a possible bone matrix disorder. J Bone Joint Surg Am 95:e1391–e1396
Misof BM, Gamsjaeger S, Cohen A et al (2012) Bone material properties in premenopausal women with idiopathic osteoporosis. J Bone Miner Res 27:2551–2561
Paschalis EP, Shane E, Lyritis G, Skarantavos G, Mendelsohn R, Boskey AL (2004) Bone fragility and collagen cross-links. J Bone Miner Res 19:2000–2004
Fratzl P, Gupta HS, Paschalis EP, Roschger P (2004) Structure and mechanical quality of the collagen-mineral composite in bone. J Mater Chem 14:2115–2123
Griffith JF, Genant HK (2008) Bone mass and architecture determination: state of the art. Best Pract Res Clin Endocrinol Metab 22:737–764
Ma S, Boughton O, Karunaratne A, Jin A, Cobb J, Hansen U, Abel R (2016) Synchrotron imaging assessment of bone quality. Clin Rev Bone Miner Metab 14:150–160
Oei L, Koromani F, Rivadeneira F, Zillikens MC, Oei EH (2016) Quantitative imaging methods in osteoporosis. Quant Imaging Med Surg 6:680–698
Peyrin F, Dong P, Pacureanu A, Langer M (2014) Micro- and nano-CT for the study of bone ultrastructure. Curr Osteoporos Rep 12:465–474
Engelke K, Libanati C, Fuerst T, Zysset P, Genant HK (2013) Advanced CT based in vivo methods for the assessment of bone density, structure, and strength. Curr Osteoporos Rep 11:246–255
Einhorn TA (1996) The bone organ system: form and function. In: Marcus R, Feldman D, Kelsey J (eds) Osteoporosis. Academic, New York
Bullough P (1990) The tissue diagnosis of metabolic bone disease. The Orthop Clinics of No America 21:65–79
Bullough P (1992) Atlas of orthopaedic pathology. Gower Medical Publishing, New York
Paschalis EP, Fratzl P, Gamsjaeger S et al (2016) Aging versus postmenopausal osteoporosis: bone composition and maturation kinetics at actively-forming trabecular surfaces of female subjects aged 1 to 84 years. J Bone Miner Res 31:347–357
Paschalis EP, Tatakis DN, Robins S et al (2011) Lathyrism-induced alterations in collagen cross-links influence the mechanical properties of bone material without affecting the mineral. Bone 49:1232–1241
McLean RR, Jacques PF, Selhub J, Tucker KL, Samelson EJ, Broe KE, Hannan MT, Cupples LA, Kiel DP (2004) Homocysteine as a predictive factor for hip fracture in older persons. N Engl J Med 350:2042–2049
Gamsjaeger S, Mendelsohn R, Boskey A, Gourion-Arsiquaud S, Klaushofer K, Paschalis E (2014) Vibrational spectroscopic imaging for the evaluation of matrix and mineral chemistry. Curr Osteoporos Rep 12:454–464
Burket JC, Brooks DJ, MacLeay JM, Baker SP, Boskey AL, van der Meulen MC (2013) Variations in nanomechanical properties and tissue composition within trabeculae from an ovine model of osteoporosis and treatment. Bone 52:326–336
Donnelly E, Boskey AL, Baker SP, van der Meulen MC (2010) Effects of tissue age on bone tissue material composition and nanomechanical properties in the rat cortex. J Biomed Mater Res A 92:1048–1056
Wagermaier W, Klaushofer K, Fratzl P (2015) Fragility of bone material controlled by internal interfaces. Calcif Tissue Int 97:201–212
Roschger P, Paschalis EP, Fratzl P, Klaushofer K (2008) Bone mineralization density distribution in health and disease. Bone 42:456–466
Montagner F, Kaftandjian V, Farlay D, Brau D, Boivin G, Follet H (2015) Validation of a novel microradiography device for characterization of bone mineralization. J Xray Sci Technol 23:201–211
Tower RJ, Campbell GM, Muller M, Gluer CC, Tiwari S (2015) Utilizing time-lapse micro-CT-correlated bisphosphonate binding kinetics and soft tissue-derived input functions to differentiate site-specific changes in bone metabolism in vivo. Bone 74:171–181
Raum K, Grimal Q, Varga P, Barkmann R, Gluer CC, Laugier P (2014) Ultrasound to assess bone quality. Curr Osteoporos Rep 12:154–162
Kerschnitzki M, Kollmannsberger P, Burghammer M, Duda GN, Weinkamer R, Wagermaier W, Fratzl P (2013) Architecture of the osteocyte network correlates with bone material quality. J Bone Miner Res 28:1837–1845
Paschalis EP, DiCarlo E, Betts F, Sherman P, Mendelsohn R, Boskey AL (1996) FTIR microspectroscopic analysis of human osteonal bone. Calcif Tissue Int 59:480–487
Paschalis EP, Gamsjaeger S, Tatakis DN, Hassler N, Robins SP, Klaushofer K (2015) Fourier transform infrared spectroscopic characterization of mineralizing type I collagen enzymatic trivalent cross-links. Calcif Tissue Int 96:18–29
Spevak L, Flach CR, Hunter T, Mendelsohn R, Boskey A (2013) Fourier transform infrared spectroscopic imaging parameters describing acid phosphate substitution in biologic hydroxyapatite. Calcif Tissue Int 92:418–428
Jackson M, Mantsch H (1995) The use and misuse of FTIR spectroscopy in the determination of protein structure. Crit Rev Biochem Mol Biol 30:95–120
Socrates G (2001) Infrared and Raman characteristic group frequencies: tables and charts. Wiley, Chichester
Morris MD, Mandair GS (2011) Raman assessment of bone quality. Clin Orthop Relat Res 469:2160–2169
Gong B, Mandair GS, Wehrli FW, Morris MD (2014) Novel assessment tools for osteoporosis diagnosis and treatment. Curr Osteoporos Rep 12:357–365
Boskey AL (2013) Bone composition: relationship to bone fragility and antiosteoporotic drug effects. Bonekey Rep 2:447
Barer R, Cole ARH, Thompson HW (1949) Infrared spectroscopy with the reflecting microscope in physics, chemistry, and biology. Nature 163:198–201
Rosasco GJ, Etz E (1974) Investigation of the Raman spectra of individual micron sized particles. Proceeding Abstracts, 4th International Conference on Raman Spectroscopy. Brunswick, Maine
Huth F, Govyadinov A, Amarie SW, Nuansing W, Keilmann F, Hillenbrand R (2012) Working principle of nano-FTIR-nano-FTIR absorption spectroscopy of molecular fingerprints at 20 nm spatial resolution. Nano Lett 12:3973–3978
Marcott C, Lo M, Hu Q, Kjoller K, Boskey A, Noda I (2014) Using 2D correlation analysis to enhance spectral information available from highly spatially resolved AFM-IR spectra. J Mol Struct 1069:284–289
Pettinger B (2010) Single-molecule surface- and tip-enhanced Raman spectroscopy. Molecular Physics: An International Journal at the Interface Between Chemistry and Physics 108:2039–2059
Saar BG, Freudiger CW, Stanley CM, Holtom GR, Xie XS (2010) Video-rate molecular imaging in vivo with stimulated Raman scattering. Science 330:1368–1370
Draper ER, Morris MD, Camacho NP, Matousek P, Towrie M, Parker AW, Goodship AE (2005) Novel assessment of bone using time-resolved transcutaneous Raman spectroscopy. J Bone Miner Res 20:1968–1972
Acerbo AS, Carr GL, Judex S, Miller LM (2012) Imaging the material properties of bone specimens using reflection-based infrared microspectroscopy. Anal Chem 84:3607–3613
Esmonde-White K, Esmonde-White F (2014) Raman spectroscopy in biomineralization. In: DiMasi E, Gower L (eds) Biomineralization sourcebook. CRC, Boca Raton, pp 59–72
Aparicio S, Doty S, Camacho N, Paschalis E, Spevak L, Mendelsohn R, Boskey A (2002) Optimal methods for processing mineralized tissues for Fourier transform infrared microspectroscopy. Calcif Tissue Int 70:422–429
Yeni YN, Yerramshetty J, Akkus O, Pechey C, Les CM (2006) Effect of fixation and embedding on Raman spectroscopic analysis of bone tissue. Calcif Tissue Int 78:363–371
Paschalis E (2012) Fourier transform infrared imaging of bone. In: Helfrich M, Ralston S (eds) Bone research protocols, second edn. Springer, New York, pp 517–525
Goodyear S, Aspden R (2012) Raman microscopy of bone. In: Helfrich M, Ralston S (eds) Bone research protocols, second edn. Springer, New York, pp 527–534
Boskey AL, Pleshko N, Doty SB, Mendelsohn R (1992) Applications of Fourier transform infrared (FT-IR) microscopy to the study of mineralization in bone and cartilage. Cells and Materials 2:209–220
Roschger A, Gamsjaeger S, Hofstetter B et al (2014) Relationship between the v(2)PO(4)/amide III ratio assessed by Raman spectroscopy and the calcium content measured by quantitative backscattered electron microscopy in healthy human osteonal bone. J Biomed Opt 19:065002
Rodriguez-Florez N, Garcia-Tunon E, Mukadam Q, Saiz E, Oldknow KJ, Farquharson C, Millan JL, Boyde A, Shefelbine SJ (2015) An investigation of the mineral in ductile and brittle cortical mouse bone. J Bone Miner Res 30:786–795
Reilly DT, Burstein AH (1974) The mechanical properties of cortical bone. JBJS 56A:1001–1022
Donnelly E, Chen DX, Boskey AL, Baker SP, van der Meulen MC (2010) Contribution of mineral to bone structural behavior and tissue mechanical properties. Calcif Tissue Int 87:450–460
Garcia I, Chiodo V, Ma Y, Boskey A (2016) Evidence of altered matrix composition in iliac crest biopsies from patients with idiopathic juvenile osteoporosis. Connect Tissue Res 57:28–37
Boskey AL, DiCarlo E, Paschalis E, West P, Mendelsohn R (2005) Comparison of mineral quality and quantity in iliac crest biopsies from high- and low-turnover osteoporosis: an FT-IR microspectroscopic investigation. Osteoporos Int 16:2031–2038
Durchschlag E, Paschalis EP, Zoehrer R, Roschger P, Fratzl P, Recker R, Phipps R, Klaushofer K (2006) Bone material properties in trabecular bone from human iliac crest biopsies after 3- and 5-year treatment with risedronate. J Bone Miner Res 21:1581–1590
Gamsjaeger S, Buchinger B, Zoehrer R, Phipps R, Klaushofer K, Paschalis EP (2011) Effects of one year daily teriparatide treatment on trabecular bone material properties in postmenopausal osteoporotic women previously treated with alendronate or risedronate. Bone 49:1160–1165
Gamsjaeger S, Hofstetter B, Zwettler E, Recker R, Gasser JA, Eriksen EF, Klaushofer K, Paschalis EP (2013) Effects of 3 years treatment with once-yearly zoledronic acid on the kinetics of bone matrix maturation in osteoporotic patients. Osteoporos Int 24:339–347
Paschalis EP, Glass EV, Donley DW, Eriksen EF (2005) Bone mineral and collagen quality in iliac crest biopsies of patients given teriparatide: new results from the fracture prevention trial. J Clin Endocrinol Metab 90:4644–4649
Paschalis EP, Boskey AL, Kassem M, Eriksen EF (2003) Effect of hormone replacement therapy on bone quality in early postmenopausal women. J Bone Miner Res 18:955–959
Gamsjaeger S, Masic A, Roschger P, Kazanci M, Dunlop JW, Klaushofer K, Paschalis EP, Fratzl P (2010) Cortical bone composition and orientation as a function of animal and tissue age in mice by Raman spectroscopy. Bone 47:392–399
Kazanci M, Roschger P, Paschalis EP, Klaushofer K, Fratzl P (2006) Bone osteonal tissues by Raman spectral mapping: orientation-composition. J Struct Biol 156:489–496
Gadaleta SJ, Landis WJ, Boskey AL, Mendelsohn R (1996) Polarized FT-IR microscopy of calcified turkey leg tendon. Connect Tissue Res 34:203–211
Termine JD, Posner AS (1966) Infra-red determinaion of the percentage of crystallinity in apatitic calcium phosphates. Nature 211:268–270
Miller LM, Vairavamurthy V, Chance MR, Mendelsohn R, Paschalis EP, Betts F, Boskey AL (2001) In situ analysis of mineral content and crystallinity in bone using infrared micro-spectroscopy of the nu(4) PO(4)(3-) vibration. Biochim Biophys Acta 1527:11–19
Kazanci M, Fratzl P, Klaushofer K, Paschalis EP (2006) Complementary information on in vitro conversion of amorphous (precursor) calcium phosphate to hydroxyapatite from Raman microspectroscopy and wide-angle X-ray scattering. Calcif Tissue Int 79:354–359
Rey C, Renugopalakrishnan V, Shimizu M, Collins B, Glimcher MJ (1991) A resolution-enhanced Fourier transform infrared spectroscopic study of the environment of the CO3(2-) ion in the mineral phase of enamel during its formation and maturation. Calcif Tissue Int 49:259–268
Gadaleta SJ, Paschalis EP, Betts F, Mendelsohn R, Boskey AL (1996) Fourier transform infrared spectroscopy of the solution-mediated conversion of amorphous calcium phosphate to hydroxyapatite: new correlations between X-ray diffraction and infrared data. Calcif Tissue Int 58:9–16
Fratzl P, Roschger P, Eschberger J, Abendroth B, Klaushofer K (1994) Abnormal bone mineralization after fluoride treatment in osteoporosis: a small-angle X-ray-scattering study. J Bone Miner Res 9:1541–1549
Paschalis EP, Betts F, DiCarlo E, Mendelsohn R, Boskey AL (1997) FTIR microspectroscopic analysis of human iliac crest biopsies from untreated osteoporotic bone. Calcif Tissue Int 61:487–492
Penel G, Leroy G, Rey C, Bres E (1998) MicroRaman spectral study of the PO4 and CO3 vibrational modes in synthetic and biological apatites. Calcif Tissue Int 63:475–481
Ou-Yang H, Paschalis EP, Mayo WE, Boskey AL, Mendelsohn R (2001) Infrared microscopic imaging of bone: spatial distribution of CO3(2-). J Bone Miner Res 16:893–900
LeGeros RZ (2002) Properties of osteoconductive biomaterials: calcium phosphates. Clin Orthop Relat Res 81–98
Bacchetta J, Farlay D, Abelin-Genevois K, Lebourg L, Cochat P, Boivin G (2015) Bone impairment in oxalosis: an ultrastructural bone analysis. Bone 81:161–167
Duboeuf F, Burt-Pichat B, Farlay D, Suy P, Truy E, Boivin G (2015) Bone quality and biomechanical function: a lesson from human ossicles. Bone 73:105–110
Iwasaki Y, Kazama JJ, Yamato H, Fukagawa M (2011) Changes in chemical composition of cortical bone associated with bone fragility in rat model with chronic kidney disease. Bone 48:1260–1267
Naruse K, Uchida K, Suto M, Miyagawa K, Kawata A, Urabe K, Takaso M, Itoman M, Mikuni-Takagaki Y (2016) Alendronate does not prevent long bone fragility in an inactive rat model. J Bone Miner Metab 34:615–626
Bozkurt O, Bilgin MD, Evis Z, Pleshko N, Severcan F (2016) Early alterations in bone characteristics of type I diabetic rat femur: a Fourier transform infrared (FT-IR) imaging study. Appl Spectrosc 70:2005–2015
Oest ME, Gong B, Esmonde-White K, Mann KA, Zimmerman ND, Damron TA, Morris MD (2016) Parathyroid hormone attenuates radiation-induced increases in collagen crosslink ratio at periosteal surfaces of mouse tibia. Bone 86:91–97
Macica CM, King HE, Wang M, McEachon CL, Skinner CW, Tommasini SM (2016) Novel anatomic adaptation of cortical bone to meet increased mineral demands of reproduction. Bone 85:59–69
Masci M, Wang M, Imbert L et al (2016) Bone mineral properties in growing Col1a2(+/G610C) mice, an animal model of osteogenesis imperfecta. Bone 87:120–129
Boskey AL, Donnelly E, Boskey E, Spevak L, Ma Y, Zhang W, Lappe J, Recker RR (2016) Examining the relationships between bone tissue composition, compositional heterogeneity, and fragility fracture: a matched case-controlled FTIRI study. J Bone Miner Res 31:1070–1081
Tamminen IS, Yli-Kyyny T, Isaksson H, Turunen MJ, Tong X, Jurvelin JS, Kroger H (2013) Incidence and bone biopsy findings of atypical femoral fractures. J Bone Miner Metab 31:585–594
Hanschin RG, Stern WB (1995) X-ray diffraction studies on the lattice perfection of human bone apatite (Crista iliaca). Bone 16:355S–363S
Boskey AL, Coleman R (2010) Aging and bone. J Dent Res 89:1333–1348
Unal M, Yang S, Akkus O (2014) Molecular spectroscopic identification of the water compartments in bone. Bone 67:228–236
Granke M, Does MD, Nyman JS (2015) The role of water compartments in the material properties of cortical bone. Calcif Tissue Int 97:292–307
Unal M, Akkus O (2015) Raman spectral classification of mineral- and collagen-bound water’s associations to elastic and post-yield mechanical properties of cortical bone. Bone 81:315–326
Paschalis EP, Gamsjaeger S, Fratzl-Zelman N et al (2016) Evidence for a role for nanoporosity and pyridinoline content in human mild osteogenesis imperfecta. J Bone Miner Res 31:1050–1059
Paschalis EP, Verdelis K, Doty SB, Boskey AL, Mendelsohn R, Yamauchi M (2001) Spectroscopic characterization of collagen cross-links in bone. J Bone Miner Res 16:1821–1828
Gamsjaeger S, Robins S, Tatakis D, Klaushofer K, Paschalis E (2017) Identification of pyridinoline trivalent collagen cross-links by Raman microspectroscopy. Calcif Tissue Int. doi:10.1007/s00223-016-0232-5
McNerny EM, Gong B, Morris MD, Kohn DH (2015) Bone fracture toughness and strength correlate with collagen cross-link maturity in a dose-controlled lathyrism mouse model. J Bone Miner Res 30:455–464
Termine JD, Robey PG (1996) Bone matrix proteins and the mineralization process. In Favus MJ (ed) Primer on the metabolic bone diseases and disorders of mineral metabolism, 3rd edition, An Official Publication of the American Society for Bone and Mineral Research. Lippincott-Raven
Robins SP (2007) Biochemistry and functional significance of collagen cross-linking. Biochem Soc Trans 35:849–852
Dodds RA, Ferris BD (1987) Changes in orientation of non-collagenous bone matrix in osteoporosis. In: Christiansen C, Johansen JS, Riis BJ (eds) Osteoporosis 1987. Osteopress APS, Copenhagen, pp 309–312
Bailey AJ, Wotton SF, Sims TJ, Thompson PW (1992) Post-translational modifications in the collagen of human osteoporotic femoral head. Biochem Biophys Res Comm 185:801–805
Bailey AJ, Wotton SF, Sims TJ, Thompson PW (1993) Biochemical changes in the collagen of human osteoporotic bone matrix. Connect Tissue Res 29:119–132
Kowitz J, Knippel M, Schuhr T, Mach J (1997) Alteration in the extent of collagen I hydroxylation, isolated from femoral heads of women with a femoral neck fracture caused by osteoporosis. Calcif Tissue Int 60:501–505
Reynolds TM, Marshall PD, Brain AM (1992) Hip fracture patients may be vitamin B6-deficient. Controlled study of serum pyridoxal-5′-phosphate. Acta Orthop Scand 63:635–638
Benke BJ, Fleshood HL, Pitot HC (1972) Osteoporotic bone disease in the pyridoxine-deficient rat. Biochem Med 6:526–535
Masse PG, Rimnac CM, Yamauchi M, Coburn PS, Rucker BR, Howell SD, Boskey AL (1996) Pyridoxine deficiency affects biomechanical properties of chick tibial bone. Bone 18:567–574
Torii H, Tasumi M (1996) Theoretical analyses of the amide I infrared bands of globular proteins. In: Mantsch HH, Chapman D (eds) Infrared spectroscopy of biomolecules. Wiley-Liss, Chichester, pp 1–18
Dong A, Huang P, Caughey WS (1990) Protein secondary structures in water from second-derivative amide I infrared spectra. Biochemistry 29:3303–3308
Susi H, Byler DM (1983) Protein structure by Fourier transform infrared spectroscopy: second derivative spectra. Biochem Biophys Res Commun 115:391–397
Kennedy DF, Crisma M, Toniolo C, Chapman D (1991) Studies of peptides forming 3(10)- and alpha-helices and beta-bend ribbon structures in organic solution and in model biomembranes by Fourier transform infrared spectroscopy. Biochemistry 30:6541–6548
Blank RD, Baldini TH, Kaufman M, Bailey S, Gupta R, Yershov Y, Boskey AL, Coppersmith SN, Demant P, Paschalis EP (2003) Spectroscopically determined collagen Pyr/deH-DHLNL cross-link ratio and crystallinity indices differ markedly in recombinant congenic mice with divergent calculated bone tissue strength. Connect Tissue Res 44:134–142
Gamsjaeger S, Klaushofer K, Paschalis E (2014) Raman analysis of proteoglycans simultaneously in bone and cartilage. J Raman Spectrosc 45:794–800
Rieppo L, Saarakkala S, Narhi T, Helminen HJ, Jurvelin JS, Rieppo J (2012) Application of second derivative spectroscopy for increasing molecular specificity of Fourier transform infrared spectroscopic imaging of articular cartilage. Osteoarthr Cartil 20:451–459
Rieppo L, Saarakkala S, Narhi T, Holopainen J, Lammi M, Helminen HJ, Jurvelin JS, Rieppo J (2010) Quantitative analysis of spatial proteoglycan content in articular cartilage with Fourier transform infrared imaging spectroscopy: critical evaluation of analysis methods and specificity of the parameters. Microsc Res Tech 73:503–512
Rieppo L, Narhi T, Helminen HJ, Jurvelin JS, Saarakkala S, Rieppo J (2013) Infrared spectroscopic analysis of human and bovine articular cartilage proteoglycans using carbohydrate peak or its second derivative. J Biomed Opt 18:097006
Oinas J, Rieppo L, Finnila MA, Valkealahti M, Lehenkari P, Saarakkala S (2016) Imaging of osteoarthritic human articular cartilage using Fourier transform infrared microspectroscopy combined with multivariate and univariate analysis. Sci Rep 6:30008
Boskey A, Pleshko Camacho N (2007) FT-IR imaging of native and tissue-engineered bone and cartilage. Biomaterials 28:2465–2478
Scott JE (1988) Proteoglycan-fibrillar collagen interactions. Biochem J 252:313–323
Gualeni B, de Vernejoul MC, Marty-Morieux C, De Leonardis F, Franchi M, Monti L, Forlino A, Houillier P, Rossi A, Geoffroy V (2013) Alteration of proteoglycan sulfation affects bone growth and remodeling. Bone 54:83–91
Bi Y, Nielsen KL, Kilts TM, Yoon A, Karsdal AM, Wimer HF, Greenfield EM, Heegaard AM, Young MF (2006) Biglycan deficiency increases osteoclast differentiation and activity due to defective osteoblasts. Bone 38:778–786
Xu T, Bianco P, Fisher LW et al (1998) Targeted disruption of the biglycan gene leads to an osteoporosis-like phenotype in mice. Nat Genet 20:78–82
Mochida Y, Duarte WR, Tanzawa H, Paschalis EP, Yamauchi M (2003) Decorin modulates matrix mineralization in vitro. Biochem Biophys Res Commun 305:6–9
Thompson WR, Modla S, Grindel BJ, Czymmek KJ, Kirn-Safran CB, Wang L, Duncan RL, Farach-Carson MC (2011) Perlecan/Hspg2 deficiency alters the pericellular space of the lacunocanalicular system surrounding osteocytic processes in cortical bone. J Bone Miner Res 26:618–629
Grzesik WJ, Frazier CR, Shapiro JR, Sponseller PD, Robey PG, Fedarko NS (2002) Age-related changes in human bone proteoglycan structure. J Biol Chem 277:43638–43647
Wang Q, Yang YY, Niu HJ, Zhang WJ, Feng QJ, Chen WF (2013) An ultrasound study of altered hydration behaviour of proteoglycan-degraded articular cartilage. Impact of osteogenesis imperfecta. BMC Musculoskelet Disord 14:289
Rieppo L, Rieppo J, Jurvelin JS, Saarakkala S (2012) Fourier transform infrared spectroscopic imaging and multivariate regression for prediction of proteoglycan content of articular cartilage. PLoS One 7:e32344
Smith AJ, Singhrao SK, Newman GR, Waddington RJ, Embery G (1997) A biochemical and immuno-electron microscopical analysis of chondroitin sulphate-rich proteoglycans in human alveolar bone. Histochem J 29:1–9
Czamara K, Majzner K, Pacia MZ, Kochan K, Kaczor A, Baranska M (2015) Raman spectroscopy of lipids: a review. J Raman Spectrosc 46:4–20
Penel G, Delfosse C, Descamps M, Leroy G (2005) Composition of bone and apatitic biomaterials as revealed by intravital Raman microspectroscopy. Bone 36:893–901
Goldberg M, Boskey AL (1996) Lipids and biomineralizations. Prog Histochem Cytochem 31:1–187
During A, Penel G, Hardouin P (2015) Understanding the local actions of lipids in bone physiology. Prog Lipid Res 59:126–146
Akiva A, Malkinson G, Masic A, Kerschnitzki M, Bennet M, Fratzl P, Addadi L, Weiner S, Yaniv K (2015) On the pathway of mineral deposition in larval zebrafish caudal fin bone. Bone 75:192–200
Akisaka T, Gay CV (1985) The plasma membrane and matrix vesicles of mouse growth plate chondrocytes during differentiation as revealed in freeze-fracture replicas. Am J Anat 173:269–286
Wuthier RE (1976) Lipids of matrix vesicles. Fed Proc 35:117–121
Hsu HH, Anderson HC (1977) A simple and defined method to study calcification by isolated matrix vesicles. Effect of ATP and vesicle phosphatase. Biochim Biophys Acta 500:162–172
Schraer H, Gay CV (1977) Matrix vesicles in newly synthesizing bone observed after ultracryotomy and ultramicroincineration. Calcif Tissue Res 23:185–188
Hsu HH, Anderson HC (1978) Calcification of isolated matrix vesicles and reconstituted vesicles from fetal bovine cartilage. Proc Natl Acad Sci U S A 75:3805–3808
Schmidt JR, Kliemt S, Preissler C, Moeller S, von Bergen M, Hempel U, Kalkhof S (2016) Osteoblast-released matrix vesicles, regulation of activity and composition by sulfated and non-sulfated glycosaminoglycans. Mol Cell Proteomics 15:558–572
Mebarek S, Abousalham A, Magne D, Do le D, Bandorowicz-Pikula J, Pikula S, Buchet R (2013) Phospholipases of mineralization competent cells and matrix vesicles: roles in physiological and pathological mineralizations. Int J Mol Sci 14:5036–5129
Mahamid J, Addadi L, Weiner S (2011) Crystallization pathways in bone. Cells Tissues Organs 194:92–97
Aido M, Kerschnitzki M, Hoerth R, Burghammer M, Montero C, Checa S, Fratzl P, Duda GN, Willie BM, Wagermaier W (2014) Relationship between nanoscale mineral properties and calcein labeling in mineralizing bone surfaces. Connect Tissue Res 55(Suppl 1):15–17
Akiva A, Kerschnitzki M, Pinkas I, Wagermaier W, Yaniv K, Fratzl P, Addadi L, Weiner S (2016) Mineral formation in the larval zebrafish tail bone occurs via an acidic disordered calcium phosphate phase. J Am Chem Soc 138:14481–14487
Kerschnitzki M, Akiva A, Ben Shoham A, Asscher Y, Wagermaier W, Fratzl P, Addadi L, Weiner S (2016) Bone mineralization pathways during the rapid growth of embryonic chicken long bones. J Struct Biol 195:82–92
Kerschnitzki M, Akiva A, Ben Shoham A et al (2016) Transport of membrane-bound mineral particles in blood vessels during chicken embryonic bone development. Bone 83:65–72
Gazi E, Gardner P, Lockyer NP, Hart CA, Brown MD, Clarke NW (2007) Direct evidence of lipid translocation between adipocytes and prostate cancer cells with imaging FTIR microspectroscopy. J Lipid Res 48:1846–1856
Ramasamy R, Vannucci SJ, Yan SS, Herold K, Yan SF, Schmidt AM (2005) Advanced glycation end products and RAGE: a common thread in aging, diabetes, neurodegeneration, and inflammation. Glycobiology 15:16R–28R
Tang SY, Zeenath U, Vashishth D (2007) Effects of non-enzymatic glycation on cancellous bone fragility. Bone 40:1144–1151
Miller LM, Tibrewala J, Carlson CS (2000) Examination of bone chemical composition in osteoporosis using fluorescence-assisted synchrotron infrared microspectroscopy. Cell Mol Biol 46:1035–1044
Boskey AL, Spevak L, Weinstein RS (2009) Spectroscopic markers of bone quality in alendronate-treated postmenopausal women. Osteoporos Int 20:793–800
Gourion-Arsiquaud S, Lukashova L, Power J, Loveridge N, Reeve J, Boskey AL (2013) Fourier transform infrared imaging of femoral neck bone: reduced heterogeneity of mineral-to-matrix and carbonate-to-phosphate and more variable crystallinity in treatment-naive fracture cases compared with fracture-free controls. J Bone Miner Res 28:150–161
Courtland HW, Nasser P, Goldstone AB, Spevak L, Boskey AL, Jepsen KJ (2008) Fourier transform infrared imaging microspectroscopy and tissue-level mechanical testing reveal intraspecies variation in mouse bone mineral and matrix composition. Calcif Tissue Int 83:342–353
Huang RY, Miller LM, Carlson CS, Chance MR (2002) Characterization of bone mineral composition in the proximal tibia of cynomolgus monkeys: effect of ovariectomy and nandrolone decanoate treatment. Bone 30:492–497
Ruppel ME, Burr DB, Miller LM (2006) Chemical makeup of microdamaged bone differs from undamaged bone. Bone 39:318–324
Paschalis EP, Betts F, DiCarlo E, Mendelsohn R, Boskey AL (1997) FTIR microspectroscopic analysis of normal human cortical and trabecular bone. Calcif Tissue Int 61:480–486
Rey C, Shimizu M, Collins B, Glimcher MJ (1991) Resolution-enhanced Fourier transform infrared spectroscopy study of the environment of phosphate ion in the early deposits of a solid phase of calcium phosphate in bone and enamel and their evolution with age: 2. Investigations in the nu3PO4 domain. Calcif Tissue Int 49:383–388
Frye EB, Degenhardt TP, Thorpe SR, Baynes JW (1998) Role of the Maillard reaction in aging of tissue proteins. Advanced glycation end product-dependent increase in imidazolium cross-links in human lens proteins. J Biol Chem 273:18714–18719
Monnier VM, Sell DR, Genuth S (2005) Glycation products as markers and predictors of the progression of diabetic complications. Ann N Y Acad Sci 1043:567–581
Thornalley PJ, Langborg A, Minhas HS (1999) Formation of glyoxal, methylglyoxal and 3-deoxyglucosone in the glycation of proteins by glucose. Biochem J 344(Pt 1):109–116
Makita Z, Radoff S, Rayfield EJ, Yang Z, Skolnik E, Delaney V, Friedman EA, Cerami A, Vlassara H (1991) Advanced glycosylation end products in patients with diabetic nephropathy. N Engl J Med 325:836–842
Brownlee M, Cerami A, Vlassara H (1988) Advanced glycosylation end products in tissue and the biochemical basis of diabetic complications. N Engl J Med 318:1315–1321
Beattie JR, Pawlak AM, Boulton ME, Zhang J, Monnier VM, McGarvey JJ, Stitt AW (2010) Multiplex analysis of age-related protein and lipid modifications in human Bruch’s membrane. FASEB J 24:4816–4824
Beattie JR, Pawlak AM, McGarvey JJ, Stitt AW (2011) Sclera as a surrogate marker for determining AGE-modifications in Bruch’s membrane using a Raman spectroscopy-based index of aging. Invest Ophthalmol Vis Sci 52:1593–1598
Glenn JV, Beattie JR, Barrett L, Frizzell N, Thorpe SR, Boulton ME, McGarvey JJ, Stitt AW (2007) Confocal Raman microscopy can quantify advanced glycation end product (AGE) modifications in Bruch’s membrane leading to accurate, nondestructive prediction of ocular aging. FASEB J 21:3542–3552
Pawlak A, Renwick Beattie J, Gelnn J, Stitt A, McGarvey J (2008) Raman spectroscopy of advanced glycation end products (AGEs), possible markers for progressive retinal dysfunction. J Raman Spectrosc 39:1635–1642
Gamsjaeger S, Srivastava AK, Wergedal JE, Zwerina J, Klaushofer K, Paschalis EP, Tatakis DN (2014) Altered bone material properties in HLA-B27 rats include reduced mineral to matrix ratio and altered collagen cross-links. J Bone Miner Res 29(11):2382–2391
Rubin MR, Paschalis EP, Poundarik A, Sroga GE, McMahon DJ, Gamsjaeger S, Klaushofer K, Vashishth D (2016) Advanced glycation endproducts and bone material properties in type 1 diabetic mice. PLoS One 11:e0154700
Ingram RT, Clarke BL, Fisher LW, Fitzpatrick LA (1993) Distribution of noncollagenous proteins in the matrix of adult human bone: evidence of anatomic and functional heterogeneity. J Bone Miner Res 8:1019–1029
Dempster DW, Zhou H, Recker RR et al (2016) A longitudinal study of skeletal histomorphometry at 6 and 24 months across four bone envelopes in postmenopausal women with osteoporosis receiving teriparatide or zoledronic acid in the SHOTZ trial. J Bone Miner Res 31:1429–1439
Paschalis EP, Gamsjaeger S, Hassler N, Klaushofer K, Burr D (2017) Ovarian hormone depletion affects cortical bone quality differently on different skeletal envelopes. Bone 95:55–64
Schulmerich MV, Cole JH, Kreider JM, Esmonde-White F, Dooley KA, Goldstein SA, Morris MD (2009) Transcutaneous Raman spectroscopy of murine bone in vivo. Appl Spectrosc 63:286–295
Okagbare PI, Begun D, Tecklenburg M, Awonusi A, Goldstein SA, Morris MD (2012) Noninvasive Raman spectroscopy of rat tibiae: approach to in vivo assessment of bone quality. J Biomed Opt 17:90502–90501
Aruwajoye OO, Aswath PB, Kim HK (2016) Material properties of bone in the femoral head treated with ibandronate and BMP-2 following ischemic osteonecrosis. J Orthop Res
Balooch M, Habelitz S, Kinney JH, Marshall SJ, Marshall GW (2008) Mechanical properties of mineralized collagen fibrils as influenced by demineralization. J Struct Biol 162:404–410
Bi X, Patil CA, Lynch CC, Pharr GM, Mahadevan-Jansen A, Nyman JS (2011) Raman and mechanical properties correlate at whole bone- and tissue-levels in a genetic mouse model. J Biomech 44:297–303
Burket J, Gourion-Arsiquaud S, Havill LM, Baker SP, Boskey AL, van der Meulen MC (2011) Microstructure and nanomechanical properties in osteons relate to tissue and animal age. J Biomech 44:277–284
Chauhan S, Manoj K, Rastogi S, Khan SA, Prasad A (2017) Biomechanical investigation of the effect of extracorporeal irradiation on resected human bone. J Mech Behav Biomed Mater 65:791–800
Edwards JR, Nyman JS, Lwin ST et al (2010) Inhibition of TGF-beta signaling by 1D11 antibody treatment increases bone mass and quality in vivo. J Bone Miner Res 25:2419–2426
Ganeko K, Masaki C, Shibata Y, Mukaibo T, Kondo Y, Nakamoto T, Miyazaki T, Hosokawa R (2015) Bone aging by advanced glycation end products: a multiscale mechanical analysis. J Dent Res 94:1684–1690
Hammond MA, Wallace JM (2015) Exercise prevents beta-aminopropionitrile-induced morphological changes to type I collagen in murine bone. BoneKEy reports 4:645
Hofmann T, Heyroth F, Meinhard H, Franzel W, Raum K (2006) Assessment of composition and anisotropic elastic properties of secondary osteon lamellae. J Biomech 39:2282–2294
Huesa C, Yadav MC, Finnila MA, Goodyear SR, Robins SP, Tanner KE, Aspden RM, Millan JL, Farquharson C (2011) PHOSPHO1 is essential for mechanically competent mineralization and the avoidance of spontaneous fractures. Bone 48:1066–1074
Imbert L, Auregan JC, Pernelle K, Hoc T (2014) Mechanical and mineral properties of osteogenesis imperfecta human bones at the tissue level. Bone 65:18–24
Katsamenis OL, Chong HM, Andriotis OG, Thurner PJ (2013) Load-bearing in cortical bone microstructure: selective stiffening and heterogeneous strain distribution at the lamellar level. J Mech Behav Biomed Mater 17:152–165
Kavukcuoglu NB, Denhardt DT, Guzelsu N, Mann AB (2007) Osteopontin deficiency and aging on nanomechanics of mouse bone. J Biomed Mater Res A 83:136–144
Kavukcuoglu NB, Patterson-Buckendahl P, Mann AB (2009) Effect of osteocalcin deficiency on the nanomechanics and chemistry of mouse bones. J Mech Behav Biomed Mater 2:348–354
Kim G, Boskey AL, Baker SP, van der Meulen MC (2012) Improved prediction of rat cortical bone mechanical behavior using composite beam theory to integrate tissue level properties. J Biomech 45:2784–2790
Kim G, Cole JH, Boskey AL, Baker SP, van der Meulen MC (2014) Reduced tissue-level stiffness and mineralization in osteoporotic cancellous bone. Calcif Tissue Int 95:125–131
Makowski AJ, Uppuganti S, Wadeer SA, Whitehead JM, Rowland BJ, Granke M, Mahadevan-Jansen A, Yang X, Nyman JS (2014) The loss of activating transcription factor 4 (ATF4) reduces bone toughness and fracture toughness. Bone 62:1–9
Maruyama N, Shibata Y, Mochizuki A, Yamada A, Maki K, Inoue T, Kamijo R, Miyazaki T (2015) Bone micro-fragility caused by the mimetic aging processes in alpha-klotho deficient mice: in situ nanoindentation assessment of dilatational bands. Biomaterials 47:62–71
Nyman JS, Lynch CC, Perrien DS, Thiolloy S, O’Quinn EC, Patil CA, Bi X, Pharr GM, Mahadevan-Jansen A, Mundy GR (2011) Differential effects between the loss of MMP-2 and MMP-9 on structural and tissue-level properties of bone. J Bone Miner Res 26:1252–1260
Ojanen X, Isaksson H, Toyras J, Turunen MJ, Malo MK, Halvari A, Jurvelin JS (2015) Relationships between tissue composition and viscoelastic properties in human trabecular bone. J Biomech 48:269–275
Pathak S, Vachhani SJ, Jepsen KJ, Goldman HM, Kalidindi SR (2012) Assessment of lamellar level properties in mouse bone utilizing a novel spherical nanoindentation data analysis method. J Mech Behav Biomed Mater 13:102–117
Raghavan M, Sahar ND, Kohn DH, Morris MD (2012) Age-specific profiles of tissue-level composition and mechanical properties in murine cortical bone. Bone 50:942–953
Sinder BP, Lloyd WR, Salemi JD, Marini JC, Caird MS, Morris MD, Kozloff KM (2016) Effect of anti-sclerostin therapy and osteogenesis imperfecta on tissue-level properties in growing and adult mice while controlling for tissue age. Bone 84:222–229
Szabo ME, Zekonyte J, Katsamenis OL, Taylor M, Thurner PJ (2011) Similar damage initiation but different failure behavior in trabecular and cortical bone tissue. J Mech Behav Biomed Mater 4:1787–1796
Yu DG, Nie SB, Liu FX, Wu CL, Tian B, Wang WG, Wang XQ, Zhu ZA, Mao YQ (2015) Dynamic alterations in microarchitecture, mineralization and mechanical property of subchondral bone in rat medial meniscal tear model of osteoarthritis. Chin Med J 128:2879–2886
Zhang Y, McNerny EG, Terajima M et al (2016) Loss of BMP signaling through BMPR1A in osteoblasts leads to greater collagen cross-link maturation and material-level mechanical properties in mouse femoral trabecular compartments. Bone 88:74–84
Aguado E, Mabilleau G, Goyenvalle E, Chappard D (2017) Hypodynamia alters bone quality and trabecular microarchitecture. Calcif Tissue Int 100(4):332–340
Busa B, Miller LM, Rubin CT, Qin YX, Judex S (2005) Rapid establishment of chemical and mechanical properties during lamellar bone formation. Calcif Tissue Int 77:386–394
Feng L, Jasiuk I (2011) Multi-scale characterization of swine femoral cortical bone. J Biomech 44:313–320
Green DE, Adler BJ, Chan ME, Lennon JJ, Acerbo AS, Miller LM, Rubin CT (2013) Altered composition of bone as triggered by irradiation facilitates the rapid erosion of the matrix by both cellular and physicochemical processes. PLoS One 8:e64952
Isaksson H, Malkiewicz M, Nowak R, Helminen HJ, Jurvelin JS (2010) Rabbit cortical bone tissue increases its elastic stiffness but becomes less viscoelastic with age. Bone 47:1030–1038
Judex S, Boyd S, Qin YX, Miller L, Muller R, Rubin C (2003) Combining high-resolution micro-computed tomography with material composition to define the quality of bone tissue. Curr Osteoporos Rep 1:11–19
Katti KS, Gu C, Katti DR (2016) Anisotropic properties of human cortical bone with osteogenesis imperfecta. Biomech Model Mechanobiol 15:155–167
Lopez Franco GE, Huang A, Pleshko Camacho N, Stone DS, Blank RD (2006) Increased Young’s modulus and hardness of Col1a2oim dentin. J Dent Res 85:1032–1036
Malluche HH, Porter DS, Monier-Faugere MC, Mawad H, Pienkowski D (2012) Differences in bone quality in low- and high-turnover renal osteodystrophy. J Am Soc Nephrol 23:525–532
Matsumoto T, Itamochi S, Hashimoto Y (2016) Effect of concurrent use of whole-body vibration and parathyroid hormone on bone structure and material properties of ovariectomized mice. Calcif Tissue Int 98:520–529
Mieczkowska A, Irwin N, Flatt PR, Chappard D, Mabilleau G (2013) Glucose-dependent insulinotropic polypeptide (GIP) receptor deletion leads to reduced bone strength and quality. Bone 56:337–342
Miller LM, Little W, Schirmer A, Sheik F, Busa B, Judex S (2007) Accretion of bone quantity and quality in the developing mouse skeleton. J Bone Miner Res 22:1037–1045
Wen XX, Wang FQ, Xu C, Wu ZX, Zhang Y, Feng YF, Yan YB, Lei W (2015) Time related changes of mineral and collagen and their roles in cortical bone mechanics of ovariectomized rabbits. PLoS One 10:e0127973
Torres-del-Pliego E, Vilaplana L, Guerri-Fernandez R, Diez-Perez A (2013) Measuring bone quality. Curr Rheumatol Rep 15:373
Kaya S, Basta-Pljakic J, Seref-Ferlengez Z et al (2016) Lactation-induced changes in the volume of osteocyte lacunar-canalicular space alter mechanical properties in cortical bone tissue. J Bone Miner Res. doi:10.1002/jbmr.3044
Mirzaali MJ, Schwiedrzik JJ, Thaiwichai S, Best JP, Michler J, Zysset PK, Wolfram U (2016) Mechanical properties of cortical bone and their relationships with age, gender, composition and microindentation properties in the elderly. Bone 93:196–211
Tomanik M, Nikodem A, Filipiak J (2016) Microhardness of human cancellous bone tissue in progressive hip osteoarthritis. J Mech Behav Biomed Mater 64:86–93
Gourion-Arsiquaud S, Marcott C, Hu Q, Boskey AL (2014) Studying variations in bone composition at nano-scale resolution: a preliminary report. Calcif Tissue Int 95:413–418
Amarie S, Zaslansky P, Kajihara Y, Griesshaber E, Schmahl WW, Keilmann F (2012) Nano-FTIR chemical mapping of minerals in biological materials. Beilstein J Nanotechnol 3:312–323
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflicts of interest
None.
Rights and permissions
About this article

Cite this article
Paschalis, E.P., Gamsjaeger, S. & Klaushofer, K. Vibrational spectroscopic techniques to assess bone quality. Osteoporos Int 28, 2275–2291 (2017). https://doi.org/10.1007/s00198-017-4019-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00198-017-4019-y