
Abstract
Introduction
Osteocytes represent 95% of all bone cells. These cells are old osteoblasts that occupy the lacunar space and are surrounded by the bone matrix. They possess cytoplasmic dendrites that form a canalicular network for communication between osteocytes and the bone surface. They express some biomarkers (osteopontin, β3 integrin, CD44, dentin matrix protein 1, sclerostin, phosphate-regulating gene with homologies to endopeptidases on the X chromosome, matrix extracellular phosphoglycoprotein, or E11/gp38) and have a mechano-sensing role that is dependent upon the frequency, intensity, and duration of strain.
Discussion
The mechanical information transmitted into the cytoplasm also triggers a biological cascade, starting with NO and PGE2 and followed by Wnt/β catenin signaling. This information is transmitted to the bone surface through the canalicular network, particularly to the lining cells, and is able to trigger bone remodeling by directing the osteoblast activity and the osteoclastic resorption. Furthermore, the osteocyte death seems to play also an important role. The outcome of micro-cracks in the vicinity of osteocytes may interrupt the canalicular network and trigger cell apoptosis in the immediate surrounding environment. This apoptosis appears to transmit a message to the bone surface and activate remodeling. The osteocyte network also plays a recognized endocrine role, particularly concerning phosphate regulation and vitamin D metabolism. Both the suppression of estrogen following menopause and chronic use of systemic glucocorticoids induce osteocyte apoptosis. On the other hand, physical activity has a positive impact in the reduction of apoptosis. In addition, some osteocyte molecular elements like sclerostin, connexin 43, E11/gp38, and DKK1 are emerging as promising targets for the treatment of various osteo-articular pathologies.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Osteocytes constitute the main cellular component of mammalian bones. Osteocytes represent more than 95% of all the bone cells (20,000 to 80,000 cells/mm3 of bone tissue); there is approximately 20-fold more osteocytes than osteoblasts in a bone [1–3]. Osteocyte density has been evaluated at 31,900 and 93,200 cells/mm3 in bovine and murine bone, respectively [4].
In human bone, Frost estimated that the mean half life of osteocytes is 25 years [5]. Meanwhile, bone remodeling induces a bone tissue turnover of 4% to 10% per year [6], so the expected duration of the life of an osteocyte is not easy to determine and is not likely to be this long. The life duration of osteocytes is higher than that of osteoblasts, which is an estimated 3 months in human bone [6], and 10–20 days in mice-woven bone [7].
The role of osteocytes remained unknown for a long time and was probably underestimated. Over the last 15 years, many publications have aimed to highlight the role of this cell. Osteocytes are distinctive and isolated cells that are embedded within the bone matrix, whereas osteoclasts, osteoblasts, and lining cells are present at the bone surface. The specific location of these cells within the bone matrix may explain some astonishing metaphors in the literature, such as “choreography from the tomb” [8] and “buried alive” [3]. In another title, they are named “martyrs for the integrity of bone strength” due to the physiological role played by their apoptosis on bone remodeling regulation [9].
Morphology
Osteocytes have a dendritic morphology, while their cell body has fusiform shape in long bones or sometimes rounded in flat bones (Figs. 1 and 2) [10]. These cells are localized in an osteocytic lacuna and have been buried in the matrix [8]. They present some cytoplasmic “extensions”: the dendrites that extend into channels in the matrix called “the canaliculi”. Osteocytes communicate with each other and with the cells at the surface of the bone tissue via these dendrites [11]. The lacuno-canalicular system which represents only 1% of interstitial fluid volume constitutes a molecular exchange surface area which has been estimated at 400-fold higher than the Havers and Volkmann system and 133-fold higher than the trabecular bone system (1,200 m2 versus 3 and 9 m2, respectively for rat male [12]).

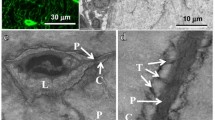
Bone tissue and osteocyte morphology. a Histological section of Wistar rat tibia after toluidine blue staining showing the fusiform shape of osteocytes. b Transmission electron microscopy of osteocyte in cortical bone tissue. O osteocyte, L lining cell, M mitochondria, N nucleus, C cytoplasm, D dendrites, arrow dendrite process dichotomy

Bone tissue anatomy, tissue and cellular architecture. a Multi-scale bone tissue anatomy: organ, osteon, osteocyte, and cytoplasmic processes. b Schematic representation of an osteocyte. Osteocytes are embedded into a mineralized bone matrix. c Confocal microscopy imaging of an osteocyte from Wistar rat tibia. d Osteocyte communication. Connexin-composed connexons form hemi-channels that control the passage of the E2 prostaglandin or ATP between the osteocyte cytoplasm and the extracellular compartment. These hemi-channels are also implied in the mechanism of action of bisphosphonates. Two separate hemi-channels from two adjacent osteocytes can contact each other to form a gap junction and can thereby exchange information. Gap junctions are thus involved in the communication between two osteocytes
Some studies have shown that according to the type of bone formed and the activity of the osteoblasts involved, the newly formed osteocytes can adopt a variable size and morphology in comparison with older osteocytes already embedded in the matrix [13]. Moreover, the morphology of embedded osteocytes is dependent on the bone type. Indeed, osteocytes found in trabecular bone are more rounded than osteocytes from cortical bone [14], with the latter adopting an elongated morphology [14]. In humans, osteocytes measure approximately 10 µm across the short axis and 20 µm along the long axis in long bones [4, 15].
The dimensions of the murine osteocyte lacunae are around 5 by 20 µm [16] with a gap present between the cell and the lacunar wall. Likewise, the cytoplasmic processes are approximately half the diameter of the canaliculi, with murine canaliculi ranging between 50 and 100 nm in diameter [16]. Osteocytes display polarity in terms of the distribution of their cell processes with the majority coming from the cell membrane facing the bone surface [11].
Origin and biomarkers
Osteoblasts are involved in bone matrix mineralization. At the end of the bone-forming phase, osteoblasts have one of three different fates: (1) they are embedded in the bone as osteocytes, (2) they are transformed into inactive surface osteoblasts called bone-lining cells, or (3) they undergo programmed cell death (apoptosis). An osteocyte is an old osteoblast buried in the matrix that it has itself produced; some of the pre-osteoblastic and osteoblastic characteristics remain detectable in these cells (osteopontin, integrin β3). During the process of bone formation, some osteoblasts are left behind in the upwardly advancing newly formed osteoid material and become entombed in the matrix as an “osteoid osteocyte”. During the process of “burial”, the future osteocyte maintains contact with the advancing osteoblasts at the surface by extending cellular processes [17], while the surrounding osteoid matrix becomes mineralized [17]. Aubin has suggested that only 10–20% of osteoblasts differentiate into osteocytes [17]. Molecular mechanisms regulating these processes are not fully understood and may depend on the location of bone formation, on the species, on the age and/or gender, and on the mode of ossification [3].
Cells at this early stage of osteoblast to osteocyte differentiation have been variously named as “large osteocytes”, “young osteocytes”, “osteoid osteocytes”, or “pre-osteocytes” [18]. These cells are larger than mature osteocytes and have numerous ribosomes, a well-developed endoplasmic reticulum and a wide Golgi complex, both involved in the synthesis of proteins and mucopolysaccharides [3]. The study of the different markers (membrane, nuclear, and cytoplasmic markers) allows for the profiling of osteocytes to determine the stage of cell maturation (young osteocyte versus mature osteocyte) [3]. Osteocyte differentiation is accompanied by the progressive reduction of several bone markers (alkaline phosphatase, bone sialoprotein, osteocalcin, collagen type I, Runx2), the preservation of some markers (osteopontin, β3 integrin, E11/gp38 antigen), and the appearance of new markers (CD44, dentin matrix protein 1 (DMP1), matrix extracellular phosphoglycoprotein (MEPE)) [3] (Table 1). Once the osteoid mineralizes, osteocyte ultrastructure undergoes further changes including a reduction in the endoplasmic reticulum and Golgi apparatus corresponding to a decrease in protein synthesis and secretion [19]. At this stage, many of the previously expressed bone markers are down regulated in the osteocyte (including alkaline phosphatase, bone sialoprotein, osteocalcin, collagen type I, Runx2) (Table 1) [19].
Currently, the molecular control of osteocytogenesis is largely unknown. Most of the in vitro osteocyte characteristics have been based on the use of the murine osteocyte-like cell line MLO-Y4 [20]. These cells, isolated from the long bones of transgenic mice, were characterized by long dendritic processes, osteopontin and connexin 43 expression, low levels of collagen type I, and alkaline phosphatase expression [20]. Osteocytogenesis is accompanied by increased expression of the genes for MEPE and DMP1, which have been associated with the osteocyte phenotype. In rodents, the restriction of mechanical stimulation (exercise) results in increased levels of the hypoxia-related hypoxia-induced transcription factor-1 alpha protein in osteocytes [21].
A recent study has determined a role for matrix metalloproteinase-2 in the regulation of osteocyte production and the generation of an appropriate canalicular system [22]. DMP1 is an extracellular matrix protein member of the SIBLING family [23]. A recent work has emphasized the relative osteocyte specificity of this molecule and implicated it in osteocyte function and signaling [24]. Toyosawa et al. noted an osteocyte specificity of DMP1 in rat bone located on cell processes and in the peri-cellular matrix [24]. It is possible that DMP1 is a target molecule for CBFA-1 and is absent in CBFA-1 knockout animals. DMP1 knockout is associated with a hypo-mineralized phenotype linked with elevated fibroblast growth factor 23 (FGF23) and defective osteocyte lacuno-canalicular network formation [25].
In 2000, two studies described a factor synthesized by osteoblasts and osteocytes, this factor being named either osteoblast/osteocyte factor-45 (OF45) or MEPE [26, 27]. The protein OF45/MEPE is another member of the SIBLING family that is known to influence mineralization directly in a BMP2 stimulated in vitro model of osteogenesis and to inhibit phosphate uptake by renal cell lines [28]. Targeted disruption of OF45/MEPE results in increased bone mass and a degree of resistance to age-related trabecular bone loss [29].
Mechano-reception and mechano-transduction
Long time regarded as simply a cell at the end of its lifetime, embedded in a mineral matrix, the osteocyte is now seen as the cell at the center of and the initiator of the bone remodeling process [30]. Indeed, osteocytes form an interconnected network of cells having the capacity to detect mechanical pressures and loads. Although it is widely accepted that the osteocyte is the cell responsible for sensing mechanical strain, there is debate within the field as to whether the osteocyte cell body or dendrites are primarily responsible for mechano-sensation [31, 32]. Nevertheless, this mechanical variation stimulates osteocytes to emit specific signals to cells present at the bone surface in response to this mechanical stimulus [33] (Fig. 3).

Mechano-transduction. The osteocyte mechano-transduction involves a stimulus (mechano-stimulator), a receptor apparatus (mechano-sensor), and a consequent signaling cascade (mechano-pathway)
Mechanical strains and weight bearing loading forces play a major role in the triggering of the bone remodeling process all along the adult life [34]. Indeed, the bone tissue is able to adapt continuously to these mechanical loads by adding bone matrix to improve resistance to increased loads or by resorbing bone in response to a decrease in use. The parameters locally influencing this balance between formation and bone resorption are now well characterized in vivo and include frequency, intensity, and duration of the mechanical stimulus [35, 36]. Thus, bone mass is influenced by the applied tension peak [35], whereas the bone formation rate is modulated by the stimulus frequency [36]. The existence of load and discharge cycles leads to a greater increase in the bone formation than when this same stimulus is applied only once [35, 36]. Lastly, bone quality and strength are improved if the mechanical stimulus is applied by short increments rather than over long periods [37]. The effects of these mechanical variations are well known and characterized macroscopically, and the most recent studies now seek to determine how mechanical modifications are detected at the cellular level and how this mechanical transduction is carried out.
Theoretical models and experimentations suggest that the lacuno-canalicular interstitial fluid flow varies with the extra-vascular pressure and with variations of mechanical loads applied to bone tissue and osteocytes [38]. Thus, mechanical forces applied to the bone induce interstitial fluid movements along canaliculi and osteocyte lacunae, and consequently, cause shear stress at the cellular level and deformations of the osteocyte plasma membrane [39, 40]. Theoretical models showed that shear stress applied to the osteocyte membrane during a physiological load was about 8–30 dyn/cm2 [38]. It would be a major breakthrough in this mechano-reception field if one were able to measure interstitial fluid flows and pressures along the lacuno-canalicular system. It has also been suggested that this mechanical information could be directly detected by ciliar or flagellar structures of the cellular membrane [41, 42]. These cilia systems are already known to be critical to mechano-reception in other organs, for example, in the inner ear. Moreover, as osteocytes seem sensitive to many other factors like hypoxia [43], it is highly possible that the osteocyte mechano-reception is carried out via several modalities [33].
Whatever the mechanism of this mechano-reception, osteocytes are able to respond to mechanical stimulation by modulating the expression and the secretion of many molecules [44], including insulin-like growth factors (IGF-I and IGF-II) [45–47], osteocalcin [45, 48], sclerostin [49, 50], c-fos [45, 46], the synthesis prostaglandin enzymes G/H [45, 51], prostanoids [52, 53], and nitric oxide (NO) [54–56]. Osteocyte mechano-reception may stimulate the Wnt/Lrp pathway as a negative regulator of sclerostin secretion [49], whereas the sclerostin itself is a negative regulator of the bone formation. Following the perception of the mechanical message and conversion into a chemical message, the osteocyte can propagate its message by two nonexclusive methods: firstly, by diffusion of produced molecules (for example, the paracrine effects of NO secretion), and secondly, by a method of local transmission through gap junctions. These junctions form a connection between the cytoplasms of two adjacent cells. Gap junctions are formed by molecules of the connexin family and Cx43 is the main connexin found in bone [20, 57]. These communicating junctions allow for the passage of molecules with a molecular weight of less than 1 kDa, such as prostaglandin PGE2 [53, 58]. Furthermore, Cx43-constituted hemi-channels were reported to be essential transducers of the anti-apoptotic effects of bisphosphonates on osteocytes [59]. For this reason, these communicating junctions are currently perceived as being critical to the osteocyte mechano-transmission.
Apoptosis, micro-cracks and bone remodeling
Osteocytes are cells that not only play a physiological role during their lifetime, but also achieve functions through their apoptosis (Fig. 4). Micro-cracks have a deleterious effect on the bone tissue if they are in excess, with possibilities of micro-fissures, micro-fractures, and fractures due to bone deficiency. However, the micro-cracks and the breaking of the lacunar network seem to play an important physiological role [60]. It has been observed that pro-apoptotic molecules are elevated in osteocytes found in the vicinity of the micro-crack, whereas anti-apoptotic molecules are expressed 1–2 mm from the micro-crack [60], suggesting that the apoptotic area may be restricted to the neighborhood of the micro-crack.

Osteocyte death—the sequence of events. a A micro-crack ( ) severes the canaliculi of several osteocyte dendritritic processes. b The micro-crack induces osteocyte apoptotic death (
) severes the canaliculi of several osteocyte dendritritic processes. b The micro-crack induces osteocyte apoptotic death ( ) and a biochemical signal is transmitted to the lining cells at the surface of the bone tissue. c Lining cells (
) and a biochemical signal is transmitted to the lining cells at the surface of the bone tissue. c Lining cells ( ) and osteocytes (
) and osteocytes ( ) both release local factors that attract cells in the circulation and cells in marrow into the remodeling compartment where osteoclastogenesis occurs. d Osteoclasts (
) both release local factors that attract cells in the circulation and cells in marrow into the remodeling compartment where osteoclastogenesis occurs. d Osteoclasts ( ) resorb the matrix and the micro-crack (lacunae resorption:
) resorb the matrix and the micro-crack (lacunae resorption:  ). e Reversal phase formation of the cement line, osteoblasts deposit newly formed osteoid matrix:
). e Reversal phase formation of the cement line, osteoblasts deposit newly formed osteoid matrix:  . f Newly reformed canaliculi and an osteocyte network
. f Newly reformed canaliculi and an osteocyte network
The message transmitted by osteocyte apoptosis travels through the canalicular network to the surface of the bone tissue and is sent on to the progenitor cells. The real nature of this message is not known. It may consist in fluid movement, biochemical signals, or electrical stimuli [33, 51]. This message leads to the initiation signals for remodeling, thereby, stimulating the bone resorption/formation cycle. It should be noted that this phenomenon could directly stimulate resorption without passing through the usual message pathways that involve the lining cells. This direct initiation could be linked to the molecular expression of (RANKL) Receptor Activator for Nuclear Factor κ B Ligand, also known as TNF-related activation-induced cytokine (TRANCE), osteoprotegerin ligand (OPGL), and ODF (osteoclast differentiation factor) by osteocytes and especially by the cytoplasmic dendritic processes in the canaliculi [61]. Furthermore, it has been demonstrated that osteocytes may be involved in a process of direct stimulation of osteoblasts. This modeling phenomenon does not require a preceding local resorption. This type of bone formation could exist near fractures or during growth [61, 62].
Until recently, no mechanism has been demonstrated which explains the death of osteocytes either in old age or in association with osteoporosis and osteoarthritis [60]. More recently, Noble reported that the sudden suppression of ovarian function in young women with endometriosis taking (GnRH) Gonadotropin-releasing hormone, also known as Luteinizing-hormone-releasing hormone (LHRH) and luliberin analogs is associated with high prevalence rates of detectable deoxyribonucleic acid breaks in osteocytes [15]. Animal experiments show that both modulation of mechanical loading and ovariectomy induce an increase in apoptosis of osteocytes and lead to micro-damages [63–66].
It is most likely that osteocyte death through apoptosis occurs at very high and at very low strain levels. The simplest explanation of the high rate of apoptosis of osteocytes seen after plastic deformation is that the micro-cracking of the matrix leads to mechanical damage of the osteocyte cell processes [60]. Noble et al. hypothesized that the death of osteocytes by apoptosis serves as a homing signal for osteoclasts and thus acts to encourage local bone resorption [67]. Alternatively, living osteocytes might act to inhibit osteoclastic resorption by expression of osteoprotegerin or other osteoclast inhibitors [29]. Osteocytes are also capable of generating the protein osteopontin, which contains a (RGD) is a standard amino acid abbreviation, meaning Arginine (Arg or R) – Glycine (Gly or G) – Aspartic Acid (Asp or D) integrin recognition site and appears able to induce osteoclast or osteoblast attachment to the bone matrix depending on the circumstances [68]. Osteocytes interact with the extracellular matrix through integrins [69, 70] and/or through dendritic processes in the canalicular wall [71]. Fluid movement through the canaliculi resulting from mechanical loading may induce a deformation of the extracellular matrix, generate tension in the tethering elements, or directly disturb the cell membrane [71, 72]. These changes may be transduced via integrin clustering into resultant survival signaling [73]. Moreover, dying osteocytes serve as initiators of targeted bone remodeling in response to physiologic or excessive strain to prevent the accumulation of micro-damages [6].
Osteocytes, by their apoptosis, are cells whose sacrifice acts to protect the integrity of bone strength [9]. Micro-cracks, apoptosis of the osteocytes, and the classical remodeling all lead to the resorption of the fragile matrix zone beyond the micro-crack. It is thought that the zones made up of old, highly mineralized bone are the most subject to micro-cracks. One theory of bone turnover is a finalist perspective, whereby, the remodeling leads to the disappearance of the micro-cracks and surrounding old mineralized bone and replaces this imperfect or “old” bone with newly formed bone that has properties that are more favorable.
A detectable space is present between the mineralized bone surface and the osteocyte surface. This space contains non-mineralized extracellular matrix enriched in non-collagenous proteins and proteoglycans. This matrix facilitates the formation of bone fluids and the regulation of osteocyte activity through soluble factors [74]. Numerous studies have suggested that osteocytes orchestrate bone remodeling, regulating osteoblast and osteoclast activities [44, 73]. While the exact process, which leads to the initiation of bone remodeling at a specific site is currently unknown, many data suggest that osteocytes play a crucial role [44].
In 1968, Baud showed that some osteocyte lacunae exhibit irregular borders and considered this shape as reminiscent of osteocyte osteolysis activity [18]. The hypothesis of osteocyte osteolysis recently got some support from studies in rodents [75, 76]. Few studies have attempted to assess whether or not osteocytes may also have matrix deposition and mineralization ability [76–78]. The parathormone could have an influence on osteocyte osteolysis and matrix synthesis [76].
Phosphate and vitamin D metabolism
Phosphate is very important at the cellular level, where it is a component of many metabolic cycles (for example, during the synthesis of adenosine triphosphate (ATP)) and in various essential cellular functions, such as muscular conduction or coagulation [79]. At the tissue level, phosphate ions are necessary for the bone matrix mineralization [80]. However, as extreme variations in the levels of circulating phosphate (hypophosphatemia and hyperphosphatemia) have harmful effects [79, 81], several pathways exist to control phosphatemia. These control loops make it possible to avoid large variations in phosphate production and/or degradation while allowing bone mineralization to continue.
Historically, the first discovery of these loops controlling phosphate involves the parathyroid hormone (PTH) and vitamin D axes [82, 83]. This loop also controls calcemia and phosphatemia. It is well known that hypocalcemia leads to the production of PTH, which in turn acts on osteoblasts to stimulate the release of calcium and phosphate from the skeletal reservoir [83] most likely via the upregulation of RANKL, the osteoblastic signal for osteoclastogenesis. It is unknown whether PTH acts directly on osteoclasts to increase the resorption of the mineral matrix [61] as PTH receptors have never been identified on osteoclasts. PTH also causes a reduction in the urinary calcium excretion from the distal tubule, an inhibition of the phosphate urinary reabsorption from the proximal tubule, and a stimulation of the production of di-hydroxylated vitamin D, the active form of vitamin D [82]. The renal production of active vitamin D, as a di-hydroxylated metabolite, leads to an increase in calcium and phosphate absorption by the small intestine enterocytes [82]. Finally, this PTH/activated vitamin D axis is able to counterbalance hypocalcemia while modulating phosphatemia [84].
More recently, a second pathway for the regulation of phosphate has been identified. FGF23, Klotho, phosphate-regulating gene with homologies to endopeptidases on the X chromosome (PHEX), and DMP1 have all been implicated in this second pathway [85]. Fibroblast growth factor 23 (FGF23) is a low molecular weight, fibroblastic growth factor of 32 kDa [86, 87]. It is produced and secreted mainly by osteocytes, but also by bone marrow venous pericytes, ventrolateral thalamic nodes, and lymphatic elements [88, 89]. The osteocyte basal expression of FGF23 is low and increases when the DMP1/PHEX pathway is inhibited [90]. FGF23 is produced in response to an increase in the levels of di-hydroxylated vitamin D [89], and it transmits a signal on binding to a dimeric trans-membrane receptor. Indeed, this receptor is composed by both an isotype of the FGF receptor (type-1c, -3c or -4) and by an essential cofactor which is represented by the Klotho glycoprotein [91, 92]. The gene coding for this Klotho protein was identified as one of the genes that controls aging as its loss or mutation leads to an accelerated senescence with the early appearance of ectopic calcifications, muscular and cutaneous atrophies, osteoporosis, arteriosclerosis, and pulmonary emphysema [89, 91, 92]. Moreover, the disruption of the Klotho-FGF23 axis may be a consequence of the expression of progerin that is involved in the development of progeria (Hutchinson–Gilford syndrome) [93]. Constitutively, the Klotho protein is expressed only in some organs (including the parathyroid gland, kidneys, testicles, ovaries, brain, pituitary gland, and choroids plexuses) [90, 94]. However, the normal biological action of the binding of FGF23 on its Klotho receptor is observed primarily at the parathyroid gland and kidneys. Osteocytes produce FGF23 in response to high rates of di-hydroxylated vitamin D and this production causes an inhibition of the renal reabsorption of phosphate from the distal tubule and a reduction in the di-hydroxylated vitamin D production [92, 95]. In addition, the binding of osteocyte FGF23 on parathyroid glandular cells will induce a suppression of PTH secretion leading to increased calciuria and phosphaturia [96]. In summary, FGF23 produced by osteocytes in response to increased rates of di-hydroxylated vitamin D will act on kidneys and parathyroid gland via Klotho and lead to a reduction of phosphatemia [97].
An excess of FGF23 leads to a hypophosphatemia and can lead to an osteomalacia. FGF23 is secreted in excess by some mesenchymal tumors [98, 99]. Constitutively, FGF23 is mildly expressed by osteocytes and its synthesis is tightly controlled by the combined and inhibiting action of the DMP1 and PHEX proteins [90, 100]. The dentin matrix protein 1 (DMP1) protein may be capable of interacting with the PHEX protein [85]. Phosphate-regulating gene with homologies to endopeptidases on the X chromosome (PHEX) is expressed by osteocytes and osteoblasts and is an endopeptidase of the plasma membrane's external surface [23, 101]. Biologically, both the native DMP1 protein and the PHEX enzyme cause an inhibition of FGF23 expression by acting negatively on the promoter sequences of this growth factor [25]. Therefore, increases in FGF23, in response to an increased di-hydroxylated vitamin D, could be inhibited by DMP1 or PHEX [85]. However, the intricate mechanism of this regulation currently remains unclear.
Effects of osteoporosis and physical activity on osteocytes
Osteoporosis is the consequence of a disturbance in the existing balance between osteoblastic bone formation and osteoclastic bone resorption [102]. This disease occurs mainly in post-menopausal women, and it is characterized by an increase in bone resorption without enough compensating formation of new bone [103, 104]. Osteoporosis is also defined by a loss of bone strength and a reduction of mass with a deterioration of bone quality, leading to an increased fracture risk [105].
It is presently unknown exactly how the suppression of estrogen can induce osteocyte apoptosis. If estrogen suppression has a direct effect on osteocytes, the two most probable possibilities are (1) the anti-oxidant effect of estrogen is critical for osteocytes that are vulnerable to anoxia or at least to hypoxia [43]; or (2) estrogens act directly via their osteocyte receptor [63]. Osteoporosis induced by chronic glucocorticoid treatment is often complicated by an area of local bone necrosis that is associated with the apoptosis of fracture-boarding osteocytes [106]. In this situation, the osteocyte apoptosis constitutes a cumulative and irrevocable effect that stops the lacuno-canalicular network and prevents the repair of micro-cracks, directly inducing bone fragility, and probably playing a major role in corticosteroid-induced osteoporosis and surrounding of fractures.
Recent work highlights the positive impact of physical activity on bone quality in post-menopausal women [107, 108]. However, the exact response of the osteocyte to physical activity or to the absence of mechanical loading is still largely unknown. Aguirre showed that mechanical stimulation of mouse osteocytes in culture attenuated apoptosis [109]. On the other hand, the reduction of mechanical loading in a tail-suspension murine model mimicking weightlessness increased the prevalence of osteocyte apoptosis, which was followed by osteoclast recruitment and associated bone resorption [109]. The same group also showed that mechanical stimulation of bone tissue activates extracellular regulated kinases pathways through integrin recruitment [73]. This serine–threonine kinase activated a kinase-induced decline in osteocyte apoptosis [73]. The global role of the osteocyte in mechano-sensing is generally accepted and the modeling processes usually include the triggering role of the osteocyte [4]. However, the molecular pathways involved in the mechano-sensing phenomenon remains debated [10, 44].
Therapeutic opening
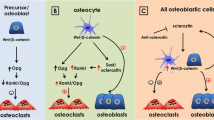
The osteocyte is now regarded as being both at the center of bone remodeling and as the initiator of the bone remodeling processes. The biology and functions of this cell are becoming the subject of more and more studies and are leading to a better understanding of the role and actions of osteocytes. In addition, molecular elements controlling osteocyte functions have now been identified and many have become possible therapeutic targets for osteo-articular pathology treatment. Among the most recent and most promising targets, the sclerostin protein that is encoded by the SOST gene, the connexin 43 which forms gap junctions, the E11/gp38 glycoprotein coded by the human E11 gene or murine gp38 gene, and the Wnt/β-catenin pathway controlled by the Dkk1 protein, have all been identified [110]. Sclerostin globally plays a negative role on bone formation [49], and this molecule is preferentially produced by the osteocytes and pre-osteocytes [3, 50].
The osteocyte SOST gene encodes the sclerostin protein [49, 111]. Its expression is controlled by the levels of PTH, and an increase in PTH secretion causes a reduction of SOST expression [112, 113]. However, this SOST regulation is also varied according to the bone localization. Indeed, an increase in PTH leads to the reduction of SOST only in bone epiphysis and diaphysis, while SOST expression rates remain unchanged in the metaphysis [62, 112]. It should be noted, however, that as sclerostin is expressed in mature osteocytes, particularly those close to cartilage zones that are not resorbed, it is very likely that sclerostin expression plays a role in the local targeting of bone remodeling [114–116]. For this reason, both the SOST gene and its sclerostin protein represent very promising therapeutic targets for the regulation of targeted bone remodeling [117, 118].
Connexin 43, which acts to form gap junctions between osteoblasts and osteocytes, is a current hot topic and is the subject of many studies. Osteocytes are able to contact adjacent cells by forming a connection using half-channels called “connexons” (Fig. 2). When two cells form a whole channel by the attachment of their separate connexons, a gap junction is formed and the cells are able to exchange information in the form of ions or small molecules [119]. Osteoblast and osteocyte connexons are formed by the juxtaposition of six connexin 43 proteins [120]. These connexin 43-composed connexons have the capacity to control the passage of E2 prostaglandin or ATP produced in response to mechanic stimulations [57]. Moreover, connexin 43 is deemed to be involved in the mechanism of action of bisphosphonates in osteoblasts and osteocytes [121–123]. Finally, it has recently been reported that connexin 43 could be regarded as a key molecule in inter-osteocyte communication [124, 125]. Connexin 43 may be involved in the regulation and the amplification of the osteocyte responses to mechanical stimulation and also in cell survival and apoptosis or in the differentiation from osteoblasts into osteocytes [126]. Thus, connexin 43 could be a potential therapeutic target.
The podoplanin protein (otherwise known as GP38, T1 alpha, or E11/gp38 protein) is coded by the E11 human or the gp38 murine gene and is selectively expressed by osteocytes in response to mechanical stimulation [127, 128]. This protein is also necessary for osteocyte dendrite elongation in response to shear stress [129]. However, as dendrite formation is an active process and not a passive mechanism, it is likely that the E11/gp38 protein is critical not only during dendritic formation, but also for osteocyte viability and function [129]. Indeed, its neutralization by antibodies led to the rapid degradation of dendritic podocytes, whereas its over-expression caused the formation of tubule-like membrane expansions [130, 131]. This molecule, therefore, seems to play an essential role in the osteocyte mechanical load perception capacities and may become a therapeutic target of interest.
The Wnt/β-catenin pathway is currently regarded as an important regulator of bone mass and bone cellular functions [44]. Indeed, this pathway is implicated in the differentiation and proliferation of osteoblasts as well as the synthesis of bone by osteoblasts, and it has been shown to play a role in the transmission of osteocyte signals that are generated by mechanical loads to the bone surface cells [132–135]. This pathway is particularly well controlled by several molecular inhibitors including the SOST gene and the Dkk1 gene and protein [136]. Indeed, mutations in SOST or Dkk1 in humans and in mice lead to an increase in bone mass [110, 117, 137–139]. Clinical trials aimed at the treatment of osteoporosis have shown that the Wnt/β-catenin pathway also induces anabolic effects within the clinical treatment setting [117, 140]. Thus, this Wnt/β-catenin pathway and/or its regulators (such as SOST or Dkk1) are also interesting therapeutic targets for new treatments of bone pathologies.
Summary and perspective
In conclusion, the osteocyte is no longer regarded as only an old osteoblast or as an inactive cell embedded in the bone matrix. Therefore, the functions of the viable osteocyte are now viewed as being equally or possibly more important than the functions of the dead or dying osteocyte. In fact, the osteocyte is currently considered as the mechano-sensing cell of the bone tissue. The osteocyte is thus perceived as being at the center of bone remodeling by coordinating both osteoblast activity and osteoclast resorption, but also as the initiator of the bone remodeling processes by locally sensing bone matrix deformation (i.e., cracked zones or overused areas). In that way, significant differences in osteocyte tridimensional morphologies and lacunae have been recently reported in human cortical bone from different pathologies with various bone mineral density (osteoarthritis, osteopenia, and osteopetrosis) [141], assuming that osteocytes may acquire phenotypical and adaptative differences according to various external mechanical strains. Furthermore, the osteocyte apoptosis has been recently reported as being insufficient for repair of micro-damage without physiological loading stimulation [142]. While manipulation of osteocyte responsiveness to mechanical loading offers potential for therapies aimed at preventing bone loss, the exact relationship between osteocyte morphology, bone architecture, mechano-responsiveness, and apoptosis is complex and needs further study.
Next to its mechano-sensibility, the osteocyte has also been shown to play a major role in phosphate homeostasis through PHEX, DMP1, and FGF23. Recent studies have furthermore shown that its sclerostin expression plays a critical role in the bone mass regulation and may be important in bone anabolic responses to PTH. At last, a recent study has demonstrated that the osteocyte marker DMP1 and SOST are regulated by muscle-related genes such as myogenin and Mef2 [143], supporting the idea that muscle-related gene network may play a role in the osteocyte cytoskeleton contractility and movements.
In summary, in the last few years, the perception of the osteocyte has changed from being viewed as an old inactive osteoblast to a highly active mechano-sensing and secretory cell that plays major roles in regulating osteoblast and osteoclast activity. Molecular pathways that control osteocyte functions might therefore become potential therapeutic targets for the treatment of bone diseases.
References
Marotti G (1996) The structure of bone tissues and the cellular control of their deposition. Ital J Anat Embryol = Arch Ital Anat Embryol 101:25–79
Parfitt AM (1990) Bone forming cells in clinical conditions. In: Hall BK (ed) Bone: a treatise the osteoblast and osteocyte. Telford Press, Caldwell, pp 351–429
Franz-Odendaal TA, Hall BK, Witten PE (2006) Buried alive: how osteoblasts become osteocytes. Dev Dyn 235:176–190
Mullender MG, van der Meer DD, Huiskes R, Lips P (1996) Osteocyte density changes in aging and osteoporosis. Bone 18:109–113
Frost HM (1966) Bone dynamics in metabolic bone disease. J Bone Jt Surg 48:1192–1203
Manolagas SC (2000) Birth and death of bone cells: basic regulatory mechanisms and implications for the pathogenesis and treatment of osteoporosis. Endocr Rev 21:115–137
McCulloch CA, Heersche JN (1988) Lifetime of the osteoblast in mouse periodontium. Anat Rec 222:128–135
Manolagas SC (2006) Choreography from the tomb: an emerging role of dying osteocytes in the purposeful, and perhaps not so purposeful, targeting of bone remodeling. BoneKEy-Osteovision 3:5–14
Seeman E (2006) Osteocytes—martyrs for integrity of bone strength. Osteoporos Int 17:1443–1448
Vatsa A, Breuls RG, Semeins CM, Salmon PL, Smit TH, Klein-Nulend J (2008) Osteocyte morphology in fibula and calvaria—is there a role for mechanosensing? Bone 43:452–458
Palumbo C, Palazzini S, Zaffe D, Marotti G (1990) Osteocyte differentiation in the tibia of newborn rabbit: an ultrastructural study of the formation of cytoplasmic processes. Acta Anat 137:350–358
Bronner F (1992) Bone and calcium homeostasis. Neurotoxicology 13:775–782
Marotti G, Muglia MA, Palumbo C (1994) Structure and function of lamellar bone. Clin Rheumatol 13(Suppl 1):63–68
Currey JD (2003) The many adaptations of bone. J Biomech 36:1487–1495
Noble BS (2008) The osteocyte lineage. Arch Biochem Biophys 473:106–111
Zhang P, Su M, Tanaka SM, Yokota H (2006) Knee loading stimulates cortical bone formation in murine femurs. BMC musculoskeletal disorders 7:73
Aubin JE, Turksen K (1996) Monoclonal antibodies as tools for studying the osteoblast lineage. Microsc Res Tech 33:128–140
Baud CA (1968) Submicroscopic structure and functional aspects of the osteocyte. Clin Orthop 56:227–236
Cameron DA, Paschall HA, Robinson RA (1967) Changes in the fine structure of bone cells after the administration of parathyroid extract. J Cell Biol 33:1–14
Kato Y, Windle JJ, Koop BA, Mundy GR, Bonewald LF (1997) Establishment of an osteocyte-like cell line, MLO-Y4. J Bone Miner Res 12:2014–2023
Gross TS, Akeno N, Clemens TL, Komarova S, Srinivasan S, Weimer DA, Mayorov S (2001) Selected contribution: osteocytes upregulate HIF-1alpha in response to acute disuse and oxygen deprivation. J Appl Physiol 90:2514–2519
Inoue K, Mikuni-Takagaki Y, Oikawa K, Itoh T, Inada M, Noguchi T, Park JS, Onodera T, Krane SM, Noda M, Itohara S (2006) A crucial role for matrix metalloproteinase 2 in osteocytic canalicular formation and bone metabolism. J Biol Chem 281:33814–33824
Fisher LW, Fedarko NS (2003) Six genes expressed in bones and teeth encode the current members of the SIBLING family of proteins. Connect Tissue Res 44(Suppl 1):33–40
Toyosawa S, Shintani S, Fujiwara T, Ooshima T, Sato A, Ijuhin N, Komori T (2001) Dentin matrix protein 1 is predominantly expressed in chicken and rat osteocytes but not in osteoblasts. J Bone Miner Res 16:2017–2026
Feng JQ, Ward LM, Liu S, Lu Y, Xie Y, Yuan B, Yu X, Rauch F, Davis SI, Zhang S, Rios H, Drezner MK, Quarles LD, Bonewald LF, White KE (2006) Loss of DMP1 causes rickets and osteomalacia and identifies a role for osteocytes in mineral metabolism. Nat Genet 38:1310–1315
Petersen DN, Tkalcevic GT, Mansolf AL, Rivera-Gonzalez R, Brown TA (2000) Identification of osteoblast/osteocyte factor 45 (OF45), a bone-specific cDNA encoding an RGD-containing protein that is highly expressed in osteoblasts and osteocytes. J Biol Chem 275:36172–36180
Rowe PS, de Zoysa PA, Dong R, Wang HR, White KE, Econs MJ, Oudet CL (2000) MEPE, a new gene expressed in bone marrow and tumors causing osteomalacia. Genomics 67:54–68
Rowe PS, Kumagai Y, Gutierrez G, Garrett IR, Blacher R, Rosen D, Cundy J, Navvab S, Chen D, Drezner MK, Quarles LD, Mundy GR (2004) MEPE has the properties of an osteoblastic phosphatonin and minhibin. Bone 34:303–319
Gowen LC, Petersen DN, Mansolf AL, Qi H, Stock JL, Tkalcevic GT, Simmons HA, Crawford DT, Chidsey-Frink KL, Ke HZ, McNeish JD, Brown TA (2003) Targeted disruption of the osteoblast/osteocyte factor 45 gene (OF45) results in increased bone formation and bone mass. J Biol Chem 278:1998–2007
Bonewald LF (2007) Osteocytes as dynamic multifunctional cells. Ann N Y Acad Sci 1116:281–290
Adachi T, Aonuma Y, Tanaka M, Hojo M, Takano-Yamamoto T, Kamioka H (2009) Calcium response in single osteocytes to locally applied mechanical stimulus: differences in cell process and cell body. J Biomech 42:1989–1995
Wang Y, McNamara LM, Schaffler MB, Weinbaum S (2008) Strain amplification and integrin based signaling in osteocytes. J Musculoskelet Neuronal Interact 8:332–334
Bonewald LF (2006) Mechanosensation and transduction in osteocytes. BoneKEy osteovision 3:7–15
Nicolella DP, Moravits DE, Gale AM, Bonewald LF, Lankford J (2006) Osteocyte lacunae tissue strain in cortical bone. J Biomech 39:1735–1743
Rubin CT (1984) Skeletal strain and the functional significance of bone architecture. Calcif Tissue Int 36(Suppl 1):S11–S18
Turner CH, Forwood MR, Otter MW (1994) Mechanotransduction in bone: do bone cells act as sensors of fluid flow? Faseb J 8:875–878
Robling AG, Hinant FM, Burr DB, Turner CH (2002) Improved bone structure and strength after long-term mechanical loading is greatest if loading is separated into short bouts. J Bone Miner Res 17:1545–1554
Weinbaum S, Cowin SC, Zeng Y (1994) A model for the excitation of osteocytes by mechanical loading-induced bone fluid shear stresses. J Biomech 27:339–360
Cowin SC (2002) Mechanosensation and fluid transport in living bone. J Musculoskelet Neuronal Interact 2:256–260
Han Y, Cowin SC, Schaffler MB, Weinbaum S (2004) Mechanotransduction and strain amplification in osteocyte cell processes. Proc Natl Acad Sci USA 101:16689–16694
Xiao Z, Zhang S, Mahlios J, Zhou G, Magenheimer BS, Guo D, Dallas SL, Maser R, Calvet JP, Bonewald L, Quarles LD (2006) Cilia-like structures and polycystin-1 in osteoblasts/osteocytes and associated abnormalities in skeletogenesis and Runx2 expression. J Biol Chem 281:30884–30895
Malone AM, Anderson CT, Tummala P, Kwon RY, Johnston TR, Stearns T, Jacobs CR (2007) Primary cilia mediate mechanosensing in bone cells by a calcium-independent mechanism. Proc Natl Acad Sci USA 104:13325–13330
Dodd JS, Raleigh JA, Gross TS (1999) Osteocyte hypoxia: a novel mechanotransduction pathway. Am J Physiol 277:C598–C602
Bonewald LF, Johnson ML (2008) Osteocytes, mechanosensing and Wnt signaling. Bone 42:606–615
Kawata A, Mikuni-Takagaki Y (1998) Mechanotransduction in stretched osteocytes–temporal expression of immediate early and other genes. Biochem Biophys Res Commun 246:404–408
Lean JM, Mackay AG, Chow JW, Chambers TJ (1996) Osteocytic expression of mRNA for c-fos and IGF-I: an immediate early gene response to an osteogenic stimulus. Am J Physiol 270:E937–E945
Skerry TM, Bitensky L, Chayen J, Lanyon LE (1989) Early strain-related changes in enzyme activity in osteocytes following bone loading in vivo. J Bone Miner Res 4:783–788
Mikuni-Takagaki Y, Suzuki Y, Kawase T, Saito S (1996) Distinct responses of different populations of bone cells to mechanical stress. Endocrinology 137:2028–2035
Robling AG, Niziolek PJ, Baldridge LA, Condon KW, Allen MR, Alam I, Mantila SM, Gluhak-Heinrich J, Bellido TM, Harris SE, Turner CH (2008) Mechanical stimulation of bone in vivo reduces osteocyte expression of Sost/sclerostin. J Biol Chem 283:5866–5875
Robling AG, Bellido T, Turner CH (2006) Mechanical stimulation in vivo reduces osteocyte expression of sclerostin. J Musculoskelet Neuronal Interact 6:354
Klein-Nulend J, Burger EH, Semeins CM, Raisz LG, Pilbeam CC (1997) Pulsating fluid flow stimulates prostaglandin release and inducible prostaglandin G/H synthase mRNA expression in primary mouse bone cells. J Bone Miner Res 12:45–51
Rawlinson SC, El-Haj AJ, Minter SL, Tavares IA, Bennett A, Lanyon LE (1991) Loading-related increases in prostaglandin production in cores of adult canine cancellous bone in vitro: a role for prostacyclin in adaptive bone remodeling? J Bone Miner Res 6:1345–1351
Vance J, Galley S, Liu DF, Donahue SW (2005) Mechanical stimulation of MC3T3 osteoblastic cells in a bone tissue-engineering bioreactor enhances prostaglandin E2 release. Tissue Eng 11:1832–1839
Basso N, Heersche JN (2006) Effects of hind limb unloading and reloading on nitric oxide synthase expression and apoptosis of osteocytes and chondrocytes. Bone 39:807–814
Klein-Nulend J, Semeins CM, Ajubi NE, Nijweide PJ, Burger EH (1995) Pulsating fluid flow increases nitric oxide (NO) synthesis by osteocytes but not periosteal fibroblasts—correlation with prostaglandin upregulation. Biochem Biophys Res Commun 217:640–648
Zaman G, Pitsillides AA, Rawlinson SC, Suswillo RF, Mosley JR, Cheng MZ, Platts LA, Hukkanen M, Polak JM, Lanyon LE (1999) Mechanical strain stimulates nitric oxide production by rapid activation of endothelial nitric oxide synthase in osteocytes. J Bone Miner Res 14:1123–1131
Cherian PP, Siller-Jackson AJ, Gu S, Wang X, Bonewald LF, Sprague E, Jiang JX (2005) Mechanical strain opens connexin 43 hemichannels in osteocytes: a novel mechanism for the release of prostaglandin. Mol Biol Cell 16:3100–3106
Siller-Jackson AJ, Burra S, Gu S, Xia X, Bonewald LF, Sprague E, Jiang JX (2008) Adaptation of connexin 43-hemichannel prostaglandin release to mechanical loading. J Biol Chem 283:26374–26382
Plotkin LI, Manolagas SC, Bellido T (2002) Transduction of cell survival signals by connexin-43 hemichannels. J Biol Chem 277:8648–8657
Noble BS, Reeve J (2000) Osteocyte function, osteocyte death and bone fracture resistance. Mol Cell Endocrinol 159:7–13
Ma YL, Cain RL, Halladay DL, Yang X, Zeng Q, Miles RR, Chandrasekhar S, Martin TJ, Onyia JE (2001) Catabolic effects of continuous human PTH (1–38) in vivo is associated with sustained stimulation of RANKL and inhibition of osteoprotegerin and gene-associated bone formation. Endocrinology 142:4047–4054
Silvestrini G, Ballanti P, Sebastiani M, Leopizzi M, Di Vito M, Bonucci E (2008) OPG and RANKL mRNA and protein expressions in the primary and secondary metaphyseal trabecular bone of PTH-treated rats are independent of that of SOST. J Mol Histol 39:237–242
Tomkinson A, Gevers EF, Wit JM, Reeve J, Noble BS (1998) The role of estrogen in the control of rat osteocyte apoptosis. J Bone Miner Res 13:1243–1250
Ikeda T, Yamaguchi A, Yokose S, Nagai Y, Yamato H, Nakamura T, Tsurukami H, Tanizawa T, Yoshiki S (1996) Changes in biological activity of bone cells in ovariectomized rats revealed by in situ hybridization. J Bone Miner Res 11:780–788
Cantatore FP, Loverro G, Ingrosso AM, Lacanna R, Sassanelli E, Selvaggi L, Carrozzo M (1995) Effect of oestrogen replacement on bone metabolism and cytokines in surgical menopause. Clin Rheumatol 14:157–160
Burr DB, Forwood MR, Fyhrie DP, Martin RB, Schaffler MB, Turner CH (1997) Bone microdamage and skeletal fragility in osteoporotic and stress fractures. J Bone Miner Res 12:6–15
Noble BS, Stevens H, Loveridge N, Reeve J (1997) Identification of apoptotic changes in osteocytes in normal and pathological human bone. Bone 20:273–282
Gerstenfeld LC (1999) Osteopontin in skeletal tissue homeostasis: an emerging picture of the autocrine/paracrine functions of the extracellular matrix. J Bone Miner Res 14:850–855
Gohel AR, Hand AR, Gronowicz GA (1995) Immunogold localization of beta 1-integrin in bone: effect of glucocorticoids and insulin-like growth factor I on integrins and osteocyte formation. J Histochem Cytochem 43:1085–1096
Aarden EM, Nijweide PJ, van der Plas A, Alblas MJ, Mackie EJ, Horton MA, Helfrich MH (1996) Adhesive properties of isolated chick osteocytes in vitro. Bone 18:305–313
You LD, Weinbaum S, Cowin SC, Schaffler MB (2004) Ultrastructure of the osteocyte process and its pericellular matrix. Anat Rec A Discov Mol Cell Evol Biol 278:505–513
Bakker A, Klein-Nulend J, Burger E (2004) Shear stress inhibits while disuse promotes osteocyte apoptosis. Biochem Biophys Res Commun 320:1163–1168
Plotkin LI, Mathov I, Aguirre JI, Parfitt AM, Manolagas SC, Bellido T (2005) Mechanical stimulation prevents osteocyte apoptosis: requirement of integrins, Src kinases, and ERKs. Am J Physiol Cell Physiol 289:C633–C643
Talmage DW, Talmage RV (2007) Calcium homeostasis: how bone solubility relates to all aspects of bone physiology. J Musculoskelet Neuronal Interact 7:108–112
Teti A, Zallone A (2009) Do osteocytes contribute to bone mineral homeostasis? Osteocytic osteolysis revisited. Bone 44:11–16
Tazawa K, Hoshi K, Kawamoto S, Tanaka M, Ejiri S, Ozawa H (2004) Osteocytic osteolysis observed in rats to which parathyroid hormone was continuously administered. J Bone Miner Metabol 22:524–529
Baylink DJ, Wergedal JE (1971) Bone formation by osteocytes. Am J Physiol 221:669–678
Jande SS, Belanger LF (1973) The life cycle of the osteocyte. Clin Orthop Rel Res 94:281–305
Amanzadeh J, Reilly RF Jr (2006) Hypophosphatemia: an evidence-based approach to its clinical consequences and management. Nat Clin Pract 2:136–148
Murshed M, Harmey D, Millan JL, McKee MD, Karsenty G (2005) Unique coexpression in osteoblasts of broadly expressed genes accounts for the spatial restriction of ECM mineralization to bone. Genes Dev 19:1093–1104
Block GA, Klassen PS, Lazarus JM, Ofsthun N, Lowrie EG, Chertow GM (2004) Mineral metabolism, mortality, and morbidity in maintenance hemodialysis. J Am Soc Nephrol 15:2208–2218
Rizzoli R, Fleisch H, Bonjour JP (1977) Role of 1, 25-dihydroxyvitamin D3 on intestinal phosphate absorption in rats with a normal vitamin D supply. J Clin Invest 60:639–647
Talmage RV, Doppelt SH, Fondren FB (1976) An interpretation of acute changes in plasma 45Ca following parathyroid hormone administration to thyroparathyroidectomized rats. Calcif Tissue Res 22:117–128
Shiraki M, Gee MV, Baum BJ, Roth GS (1986) Parathyroid hormone stimulates phosphate efflux through an apparently adenosine 3′, 5′-monophosphate-independent process in rat parotid cell aggregates. Endocrinology 118:2009–2015
Quarles LD (2008) Endocrine functions of bone in mineral metabolism regulation. J Clin Invest 118:3820–3828
Yamashita T, Yoshioka M, Itoh N (2000) Identification of a novel fibroblast growth factor, FGF-23, preferentially expressed in the ventrolateral thalamic nucleus of the brain. Biochem Biophys Res Commun 277:494–498
Shimada T, Mizutani S, Muto T, Yoneya T, Hino R, Takeda S, Takeuchi Y, Fujita T, Fukumoto S, Yamashita T (2001) Cloning and characterization of FGF23 as a causative factor of tumor-induced osteomalacia. Proc Natl Acad Sci USA 98:6500–6505
Itoh N, Ornitz DM (2004) Evolution of the Fgf and Fgfr gene families. Trends Genet 20:563–569
Tsujikawa H, Kurotaki Y, Fujimori T, Fukuda K, Nabeshima Y (2003) Klotho, a gene related to a syndrome resembling human premature aging, functions in a negative regulatory circuit of vitamin D endocrine system. Mol Endocrinol (Baltim Md) 17:2393–2403
Liu S, Zhou J, Tang W, Jiang X, Rowe DW, Quarles LD (2006) Pathogenic role of Fgf23 in Hyp mice. Am J Physiol 291:E38–E49
Kurosu H, Yamamoto M, Clark JD, Pastor JV, Nandi A, Gurnani P, McGuinness OP, Chikuda H, Yamaguchi M, Kawaguchi H, Shimomura I, Takayama Y, Herz J, Kahn CR, Rosenblatt KP, Kuro-o M (2005) Suppression of aging in mice by the hormone Klotho. Science (New York, NY) 309:1829–1833
Urakawa I, Yamazaki Y, Shimada T, Iijima K, Hasegawa H, Okawa K, Fujita T, Fukumoto S, Yamashita T (2006) Klotho converts canonical FGF receptor into a specific receptor for FGF23. Nature 444:770–774
Ortiz A (2008) Hutchinson–Gilford progeria syndrome. N Engl J Med 358:2410; author reply 2410-2411
Sitara D, Razzaque MS, Hesse M, Yoganathan S, Taguchi T, Erben RG, Juppner H, Lanske B (2004) Homozygous ablation of fibroblast growth factor-23 results in hyperphosphatemia and impaired skeletogenesis, and reverses hypophosphatemia in Phex-deficient mice. Matrix Biol 23:421–432
Kurosu H, Ogawa Y, Miyoshi M, Yamamoto M, Nandi A, Rosenblatt KP, Baum MG, Schiavi S, Hu MC, Moe OW, Kuro-o M (2006) Regulation of fibroblast growth factor-23 signaling by klotho. J Biol Chem 281:6120–6123
Ben-Dov IZ, Galitzer H, Lavi-Moshayoff V, Goetz R, Kuro-o M, Mohammadi M, Sirkis R, Naveh-Many T, Silver J (2007) The parathyroid is a target organ for FGF23 in rats. J Clin Invest 117:4003–4008
Nabeshima Y (2008) The discovery of alpha-Klotho and FGF23 unveiled new insight into calcium and phosphate homeostasis. Cell Mol Life Sci 65:3218–3230
Hannan FM, Athanasou NA, Teh J, Gibbons CL, Shine B, Thakker RV (2008) Oncogenic hypophosphataemic osteomalacia: biomarker roles of fibroblast growth factor 23, 1, 25-dihydroxyvitamin D3 and lymphatic vessel endothelial hyaluronan receptor 1. Eur J Endocrinol 158:265–271
Koriyama N, Nishimoto K, Kodama T, Nakazaki M, Kurono Y, Yoshida H, Tei C (2006) Oncogenic osteomalacia in a case with a maxillary sinus mesenchymal tumor. Am J Med Sci 332:142–147
Liu S, Zhou J, Tang W, Menard R, Feng JQ, Quarles LD (2008) Pathogenic role of Fgf23 in Dmp1-null mice. Am J Physiol 295:E254–E261
Bai X, Miao D, Panda D, Grady S, McKee MD, Goltzman D, Karaplis AC (2002) Partial rescue of the Hyp phenotype by osteoblast-targeted PHEX (phosphate-regulating gene with homologies to endopeptidases on the X chromosome) expression. Mol Endocrinol (Baltim Md) 16:2913–2925
Marie P, Debiais F, Cohen-Solal M, de Vernejoul MC (2000) New factors controlling bone remodeling. Joint Bone Spine 67:150–156
Egermann M, Schneider E, Evans CH, Baltzer AW (2005) The potential of gene therapy for fracture healing in osteoporosis. Osteoporos Int 16(Suppl 2):S120–S128
Fromigue O, Modrowski D, Marie PJ (2004) Growth factors and bone formation in osteoporosis: roles for fibroblast growth factor and transforming growth factor beta. Curr Pharm Des 10:2593–2603
Lane JM, Gardner MJ, Lin JT, van der Meulen MC, Myers E (2003) The aging spine: new technologies and therapeutics for the osteoporotic spine. Eur Spine J 12(Suppl 2):S147–S154
Weinstein RS, Nicholas RW, Manolagas SC (2000) Apoptosis of osteocytes in glucocorticoid-induced osteonecrosis of the hip. J Clin Endocrinol Metab 85:2907–2912
Tolomio S, Ermolao A, Travain G, Zaccaria M (2008) Short-term adapted physical activity program improves bone quality in osteopenic/osteoporotic postmenopausal women. J Phys Activ Health 5:844–853
Kitagawa J, Nakahara Y (2008) Associations of daily walking steps with calcaneal ultrasound parameters and a bone resorption marker in elderly Japanese women. J Physiol Anthropol 27:295–300
Aguirre JI, Plotkin LI, Stewart SA, Weinstein RS, Parfitt AM, Manolagas SC, Bellido T (2006) Osteocyte apoptosis is induced by weightlessness in mice and precedes osteoclast recruitment and bone loss. J Bone Miner Res 21:605–615
Morvan F, Boulukos K, Clement-Lacroix P, Roman Roman S, Suc-Royer I, Vayssiere B, Ammann P, Martin P, Pinho S, Pognonec P, Mollat P, Niehrs C, Baron R, Rawadi G (2006) Deletion of a single allele of the Dkk1 gene leads to an increase in bone formation and bone mass. J Bone Miner Res 21:934–945
Winkler DG, Sutherland MK, Geoghegan JC, Yu C, Hayes T, Skonier JE, Shpektor D, Jonas M, Kovacevich BR, Staehling-Hampton K, Appleby M, Brunkow ME, Latham JA (2003) Osteocyte control of bone formation via sclerostin, a novel BMP antagonist. EMBO J 22:6267–6276
Silvestrini G, Ballanti P, Leopizzi M, Sebastiani M, Berni S, Di Vito M, Bonucci E (2007) Effects of intermittent parathyroid hormone (PTH) administration on SOST mRNA and protein in rat bone. J Mol Histol 38:261–269
Bellido T (2006) Downregulation of SOST/sclerostin by PTH: a novel mechanism of hormonal control of bone formation mediated by osteocytes. J Musculoskelet Neuronal Interact 6:358–359
Keller H, Kneissel M (2005) SOST is a target gene for PTH in bone. Bone 37:148–158
van Bezooijen RL, ten Dijke P, Papapoulos SE, Lowik CW (2005) SOST/sclerostin, an osteocyte-derived negative regulator of bone formation. Cytokine Growth Factor Rev 16:319–327
Poole KE, van Bezooijen RL, Loveridge N, Hamersma H, Papapoulos SE, Lowik CW, Reeve J (2005) Sclerostin is a delayed secreted product of osteocytes that inhibits bone formation. Faseb J 19:1842–1844
Li X, Ominsky MS, Niu QT, Sun N, Daugherty B, D'Agostin D, Kurahara C, Gao Y, Cao J, Gong J, Asuncion F, Barrero M, Warmington K, Dwyer D, Stolina M, Morony S, Sarosi I, Kostenuik PJ, Lacey DL, Simonet WS, Ke HZ, Paszty C (2008) Targeted deletion of the sclerostin gene in mice results in increased bone formation and bone strength. J Bone Miner Res 23:860–869
Li X, Ominsky MS, Warmington KS, Morony S, Gong J, Cao J, Gao Y, Shalhoub V, Tipton B, Haldankar R, Chen Q, Winters A, Boone T, Geng Z, Niu QT, Ke HZ, Kostenuik PJ, Simonet WS, Lacey DL, Paszty C (2009) Sclerostin antibody treatment increases bone formation, bone mass, and bone strength in a rat model of postmenopausal osteoporosis. J Bone Miner Res 24:578–588
Unger VM, Kumar NM, Gilula NB, Yeager M (1999) Three-dimensional structure of a recombinant gap junction membrane channel. Science (New York, NY) 283:1176–1180
Goodenough DA, Goliger JA, Paul DL (1996) Connexins, connexons, and intercellular communication. Annu Rev Biochem 65:475–502
Plotkin LI, Weinstein RS, Parfitt AM, Roberson PK, Manolagas SC, Bellido T (1999) Prevention of osteocyte and osteoblast apoptosis by bisphosphonates and calcitonin. J Clin Invest 104:1363–1374
Follet H, Li J, Phipps RJ, Hui S, Condon K, Burr DB (2007) Risedronate and alendronate suppress osteocyte apoptosis following cyclic fatigue loading. Bone 40:1172–1177
Plotkin LI, Lezcano V, Thostenson J, Weinstein RS, Manolagas SC, Bellido T (2008) Connexin 43 is required for the anti-apoptotic effect of bisphosphonates on osteocytes and osteoblasts in vivo. J Bone Miner Res 23:1712–1721
Stains JP, Civitelli R (2005) Gap junctions in skeletal development and function. Biochim Biophys Acta 1719:69–81
Stains JP, Civitelli R (2005) Gap junctions regulate extracellular signal-regulated kinase signaling to affect gene transcription. Mol Biol Cell 16:64–72
Civitelli R (2008) Connexin 43 modulation of osteoblast/osteocyte apoptosis: a potential therapeutic target? J Bone Miner Res 23:1709–1711
Nose K, Saito H, Kuroki T (1990) Isolation of a gene sequence induced later by tumor-promoting 12-O-tetradecanoylphorbol-13-acetate in mouse osteoblastic cells (MC3T3-E1) and expressed constitutively in ras-transformed cells. Cell Growth Differ 1:511–518
Wetterwald A, Hoffstetter W, Cecchini MG, Lanske B, Wagner C, Fleisch H, Atkinson M (1996) Characterization and cloning of the E11 antigen, a marker expressed by rat osteoblasts and osteocytes. Bone 18:125–132
Zhang K, Barragan-Adjemian C, Ye L, Kotha S, Dallas M, Lu Y, Zhao S, Harris M, Harris SE, Feng JQ, Bonewald LF (2006) E11/gp38 selective expression in osteocytes: regulation by mechanical strain and role in dendrite elongation. Mol Cell Biol 26:4539–4552
Schulze E, Witt M, Kasper M, Lowik CW, Funk RH (1999) Immunohistochemical investigations on the differentiation marker protein E11 in rat calvaria, calvaria cell culture and the osteoblastic cell line ROS 17/2.8. Histochem Cell Biol 111:61–69
Sprague L, Wetterwald A, Heinzman U, Atkinson MJ (1996) Phenotypic changes following over-expression of sense or antisense E11 cDNA in ROS 17/2.8 cells. J Bone Miner Res 11:S132
Heino TJ, Hentunen TA, Vaananen HK (2004) Conditioned medium from osteocytes stimulates the proliferation of bone marrow mesenchymal stem cells and their differentiation into osteoblasts. Exp Cell Res 294:458–468
Hartmann C (2006) A Wnt canon orchestrating osteoblastogenesis. Trends Cell Biol 16:151–158
Bodine PV, Komm BS (2006) Wnt signaling and osteoblastogenesis. Rev Endocr Metab Disord 7:33–39
Westendorf JJ, Kahler RA, Schroeder TM (2004) Wnt signaling in osteoblasts and bone diseases. Gene 341:19–39
Brott BK, Sokol SY (2002) Regulation of Wnt/LRP signaling by distinct domains of Dickkopf proteins. Mol Cell Biol 22:6100–6110
Balemans W, Patel N, Ebeling M, Van Hul E, Wuyts W, Lacza C, Dioszegi M, Dikkers FG, Hildering P, Willems PJ, Verheij JB, Lindpaintner K, Vickery B, Foernzler D, Van Hul W (2002) Identification of a 52 kb deletion downstream of the SOST gene in patients with van Buchem disease. J Med Genet 39:91–97
Staehling-Hampton K, Proll S, Paeper BW, Zhao L, Charmley P, Brown A, Gardner JC, Galas D, Schatzman RC, Beighton P, Papapoulos S, Hamersma H, Brunkow ME (2002) A 52-kb deletion in the SOST-MEOX1 intergenic region on 17q12–q21 is associated with van Buchem disease in the Dutch population. Am J Med Genet 110:144–152
Balemans W, Ebeling M, Patel N, Van Hul E, Olson P, Dioszegi M, Lacza C, Wuyts W, Van Den Ende J, Willems P, Paes-Alves AF, Hill S, Bueno M, Ramos FJ, Tacconi P, Dikkers FG, Stratakis C, Lindpaintner K, Vickery B, Foernzler D, Van Hul W (2001) Increased bone density in sclerosteosis is due to the deficiency of a novel secreted protein (SOST). Hum Mol Genet 10:537–543
Li X, Ominsky MS, Warmington KS, Morony S, Gong J, Cao J, Gao Y, Shalhoub V, Tipton B, Haldankar R, Chen Q, Winters A, Boone T, Geng Z, Niu QT, Ke HZ, Kostenuik PJ, Simonet WS, Lacey DL, Paszty C (2008) Sclerostin antibody treatment increases bone formation, bone mass and bone strength in a rat model of postmenopausal osteoporosis. J Bone Miner Res 24(4):578–588
van Hove RP, Nolte PA, Vatsa A, Semeins CM, Salmon PL, Smit TH, Klein-Nulend J (2009) Osteocyte morphology in human tibiae of different bone pathologies with different bone mineral density—is there a role for mechanosensing? Bone 45:321–329
Waldorff EI, Christenson KB, Cooney LA, Goldstein SA (2009) Microdamage repair and remodeling requires mechanical loading. J Bone Miner Res. Oct 12. [Epub ahead of print]. PMID: 19821772. doi:10.1359/jbmr.091016
Dean AK, Harris SE, Kalajzic I, Ruan J (2009) A systems biology approach to the identification and analysis of transcriptional regulatory networks in osteocytes. BMC bioinformatics 10(Suppl 9):S5
Acknowledgments
The authors want to thank Eric Dolléans for technical help during sample preparation and cutting. Confocal microscopy was performed in collaboration with David Gosset and Chantal Pichon from the Centre de Biophysique Moléculaire, CNRS UPR4301, University of Orléans and Inserm, Orléans, France. Transmission electron microscopy was achieved in collaboration with Rustem Uzbekov, Pierre-Yves Sizaret, and Brigitte Arbeille from the Laboratoire de Biologie Cellulaire, Faculté de Médecine, Tours, France.
Author information
Authors and Affiliations
Corresponding author
Additional information
Rochefort and Pallu contributed equally to this paper.
Rights and permissions
About this article
Cite this article
Rochefort, G.Y., Pallu, S. & Benhamou, C.L. Osteocyte: the unrecognized side of bone tissue. Osteoporos Int 21, 1457–1469 (2010). https://doi.org/10.1007/s00198-010-1194-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00198-010-1194-5