
Abstract
Osteoporosis and atherosclerosis, two multifactorial and degenerative entities, are major public health problems. These diseases accompany the aging process and share common risk factors. Furthermore, several common pathophysiological factors have been suggested. These include similar molecular pathways involving bone and vascular mineralization, estrogen deficiency, parathyroid hormone, homocysteine, lipid oxidation products, inflammatory process, as well as vitamin D and K. Moreover, the use of statins, biphosphonates, beta-blockers and experimental dual-purpose therapies based on the biological linkage of the above entities may simultaneously benefit bone loss and vascular disease. This review considers a potential link between osteoporosis and atherosclerosis beyond aging. These common factors may lead to appropriate treatment strategies.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Osteoporosis and atherosclerosis are major public health problems that often coexist in both genders worldwide, particularly in the elderly. Some, but not all, studies reported a relationship dependent on age [1–6]. Several studies also suggested a causal relationship between atherosclerosis and osteoporosis [7–22, 24–26], so that the presence of the one is a predictor of the other (Table 1).
According to the National Institutes of Health (NIH) consensus conference in 2000, osteoporosis is defined as “a skeletal disorder characterized by compromised bone strength predisposing a person to an increased risk of fracture” [23]. Previous studies have shown an association between osteoporosis and cardiovascular mortality [7–11], aortic and coronary calcification [12–16], carotid atherosclerosis [17–19], peripheral arterial disease [20] and stroke [21, 22] in both sexes, especially in women.
Epidemiological data
It has been reported [7] that low bone mineral content in postmenopausal women was associated with increased mortality in later life, especially from cardiovascular disease (CVD) (about twofold increased risk of dying from CVD). The relative risk (RR) was higher in the early postmenopausal period, when each decrease of one standard deviation (SD) (0.4 g/cm2) in bone mass was associated with a 43% increase in mortality. A prospective study in women over 65 years old [8], reported that the same decrease in bone mineral density (BMD) of the femoral neck was followed by a 1.3-fold increase in mortality from coronary artery disease (CAD), after adjustment for risk factors, such as hypertension, diabetes mellitus (DM), smoking, advanced age and low physical activity. Another large prospective study in women over 65 years old showed that diminished BMD at the proximal radius was strongly associated with deaths from stroke [RR: 1.74; 95% confidence interval (CI) 1.12–2.70] and each SD decrease in BMD was associated with a 1.19-fold increase in mortality (95% CI 1.04–1.36), adjusted for age and duration of follow-up [9]. Furthermore, in a retrospective analysis low BMD appeared to independently predict significant CAD in women, with a higher odds ratio than the above risk factors [odds ratio (OR) 5.6, 95% CI 2.6 to 12.0, p < 0.0001] [10]. The reverse relationship has also been reported. In other words, there was a substantially increased risk of hip fracture in women with CVD [11]. This increase was attributed to the similar biological and molecular pathways that link these two conditions and will be discussed later in this review [11].
Regarding aortic calcification (a surrogate marker of the atherosclerotic burden), some studies reported an inverse relationship with BMD [12, 13]. After adjusting for age and potential confounders, measures for aortic calcification (by computed tomography) predicted 26.1% of the variance in BMD (p < 0.0001) [13]. In the same study, the investigators reported that postmenopausal women aged ≤ 60 years with aortic calcification had more than double the rate of bone loss (3.4 vs. 1.4% yearly) compared with those without calcified tissues. The prevalence of arterial calcium in the aorta (but not in carotid and coronary arteries, after adjustment for common risk factors) was also greater in both sexes with lower BMD in another study [14]. Except for arterial calcium, the stiffness of the aorta, assessed by means of augmentation index and central aortic systolic and pulse pressures, was greater in osteoporotic postmenopausal women than control subjects [15]. Finally, coronary calcium evaluated by electron beam tomography of the heart was significantly higher in women with osteoporosis than in those without [16].
A study including 2,733 women (aged 55–74 years; follow-up for 6 years) evaluated the potential link between osteoporosis and carotid calcification assessed by ultrasound. The prevalence of carotid echogenic plaques was significantly related to low BMD and the age-adjusted RR of fracture was higher among women with echogenic plaques than among those without: 1.7 (95% CI 1.0–2.7) [18]. The investigators of the San Antonio Family Osteoporosis Study also demonstrated a correlation between decreased BMD and increased carotid artery intimal medial thickness (IMT), after adjusting for known environmental factors. In particular, they observed a negative association between IMT and BMD in both Mexican American men and women over 60 years of age [19]. Moreover, in the Rotterdam Study, a prospective cohort study of individuals aged ≥ 55 and over, women with a low femoral neck BMD had a significantly increased risk of peripheral arterial disease (OR 1.49, 95% CI 1.16–1.91). However, this association was not found in men (OR 1.14, 95% CI 0.84–1.53) [20]. Finally, low BMD in the femoral neck has also been associated with a high risk of stroke in women [21, 22]. It was suggested that each SD (0.13 g/cm2) decrease in BMD in female but not male subjects was related to a 1.9-fold increased risk of stroke [21].
In general, only few studies showed a correlation between atherosclerosis and low bone mass in men. Two large epidemiological studies in older men have shown that low BMD is associated with increased severity of calcified carotid plaques [24] and CVD [25]. In another study, BMD was a strong predictor of total mortality in men as well as in women, but the number of fatal strokes was too low to evaluate the relationship between BMD and stroke mortality [26].
Mechanisms potentially underlying the association between osteoporosis and atherosclerosis
There are several possible links between osteoporosis and CVD independently of the aging process. Apart from the fact that both diseases share common risk factors, such as hypertension [27], DM [28], smoking [29], alcohol abuse [30] and low level of physical activity [30], they have common pathogenetic pathways (Table 2).
Vascular calcification-bone mineralization processes
Vascular calcification is an ongoing process and it may occur without (medial calcification) or within (neo-intimal calcification) atherosclerotic plaques [31]. This process appears to share some common characteristics with bone mineralization. In particular, hydroxyapatite, the basic component of the mineral phase of bone, is also present in calcium deposits in atherosclerotic lesions [32]. Furthermore, cells with osteoblastic or osteoclastic potential have been observed in the arterial wall, as well as bone matrix proteins, including gamma carboxyglutamate (GLa) proteins, osteopontin and bone morphogenetic proteins (BMP), especially BMP-2 [33].
GLa proteins comprise a part of a family of mineral binding proteins, which includes osteocalcin, several coagulation factors (factors VII and IX) and anti-coagulation factors (proteins C and S). Gla residues bind and incorporate calcium into hydroxyapatite crystals [31]. Mice that are deficient in GLa proteins demonstrate extensive vascular calcification, abnormal cartilage calcification and osteoporosis, which suggest that dysregulation of these proteins may result in abnormal mineralization of both bones and arteries [34].
Osteopontin (OP) is a major non-collagenous bone matrix glycoprotein, which binds to integrins, especially the αvβ3 one. Integrins comprise essential receptors for osteoclast migration to resorption sites [31]. Apart from its role in osteoclast attachment to bone, OP has also been associated with arterial calcification in patients with advanced atherosclerosis. In CAD, OP seems to be localized in calcified atherosclerotic lesions, in association with high serum levels. OP may also induce endothelial dysfunction, by decreased formation of nitric oxide, a regulator of cardiovascular homeostasis [35].
BMPs comprise a group of growth factors characterized by their ability to induce the formation of bone and cartilage. Seven such proteins have been discovered, 6 of which (BMP-2 to BMP-7) belong to the transforming growth factor (TGF)-β superfamily. TGF-β plays a pivotal role in the regulation of osteoblast differentiation and proliferation [36]. Their effects are demonstrated by intracellular mediators, including stimulatory and inhibitory Smad proteins. The most important, Smad6, an inhibitor of BMP signalling, plays a crucial role in the development of cardiovascular system. Indeed, Smad6-deficient mice exhibit cartilaginous metaplasia and ossification of the aorta, indicating the role of this protein in vascular calcification [37]. BMP-2 expression in the arterial wall, activated by several factors, such as TNF-a, oxidized lipids and hyperglycemia, seems to be a feature of atherosclerotic calcification [38].
Data from several studies evaluated the role of osteoprotegerin (OPG), another marker of bone remodeling, in the common pathogenesis of atherosclerosis and osteoporosis. OPG is a member of tumor necrosis factor (TNF) receptor family, which regulates osteoclastogenesis by inhibiting receptor activator of nuclear factor-kB ligand (RANKL)-mediated osteoclastic bone resorption in vitro and in vivo. OPG is secreted mainly by osteoblast lineage cells [39]. OPG is also produced by cells of the cardiovascular system, including coronary artery smooth muscle cells and endothelial cells [40]. OPG-deficient mice tend to exhibit both osteoporosis with multiple fractures and calcification of the aorta and renal arteries, suggesting that OPG can influence vascular calcification [41]. In one study, serum levels of OPG were about 30% greater in women with DM than in euglycaemic ones [42]. Similar data were reported by another study of 522 men, which showed that OPG levels were higher in patients with DM, hypertension and advanced CAD. Increased OPG levels may therefore represent an insufficient compensatory mechanism to prevent further vascular damage [43]. This raises interest for a therapeutic potential of exogenous administration of OPG to patients with osteoporosis and CAD. In general, activation of nuclear factor kB (NFkB) (except for the RANK/RANKL/OPG system) appears to be one of the most important events that link atherosclerosis to osteoporosis. It can be induced by several stimuli, including cytokines (mainly TNF-a), viruses, LDL, oxidants and immune stimuli. Induction of NFkB exerts proatherogenic effects in the vessel wall [44].
Our attempt to enlighten the above linkage should include the pivotal role of Wnts, a family of 19 secreted signalling glycoproteins, in the regulation of osteoblastogenesis and bone formation. They bind to receptor complexes, including LDL receptor-related proteins (LRP)-5 and LRP-6, as well as frizzled proteins [45]. This complex initiates an intracellular cascade of events leading to the stabilization of β-catenin and its subsequent translocation into the nucleus, where associated with the transcription factors Tcf/Lef, it triggers gene expression involving bone development [46]. Wnt ligands have also been implicated in the regulation of the pathologic calcification in the vasculature, as well as the osteoblastic transdifferentiation of vascular smooth cells in vitro, although the exact mechanisms explaining this process have not been elucidated [47]. Furthermore, emerging data suggest an antagonism of Wnt signalling by oxidative stress with increasing age, which may be a common molecular mechanism contributing to the development not only of osteoporosis, but also several pathologies such as atherosclerosis, insulin resistance and hyperlipidemia [48]. Evaluating the role of Wnt/LRP on a genetic basis, one should mention that dual gene mutations of LRP-5 and its ligand, apolipoprotein E (especially the e4 allele), in mice are associated with hypercholesterolemia, advanced atherosclerosis and low bone mass [35]. Recently, a missense mutation in LRP6 was identified in a family with autosomal dominant early CAD, features of the metabolic syndrome and osteoporosis [49]. All these data provide an attractive field for further investigation of the role of LRP-5/6 and Wnt signalling in a biological linkage of atherosclerosis and osteoporosis.
Another family of transcription factors includes core binding factor a1 (Cbfa1) and runt-related transcriptional factor-2 (Runx2), which induce osteoblastic differentiation at the early stage and inhibit it at the late stage. They promote the expression of bone matrix protein genes and the mineralization in immature mesenchymal and osteoblastic cells in vitro. Runx2 also forms a complex with Tcf/Lef, comprising an important regulator of bone formation [50]. Moreover, Runx2 expression has been identified in atherosclerotic human vascular tissue specimens, but not in normal vessels, indicating a potential role in vascular calcification. It appears to participate in the induction of vascular smooth cell calcification by oxidative stress [51].
As mentioned above, BMP-2 is expressed in calcified atherosclerotic lesions. It is necessary for the osteogenic differentiation of mesenchymal cells, including vascular smooth cells. Its effect is usually exerted in synergy with Msx2, a homeodomain transcription factor that controls osteoblastic differentiation and mineralization in the developing skull [38]. Genetic evidence suggests that the Msx2 gene is a direct gene target of BMP-2. Indeed, Msx2-deficient mice exhibit a generalized skeletal osteoblast deficiency and a low turnover osteoporosis syndrome [52]. In a similar way, Msx2 induces osteogenic versus adipogenic differentiation of aortic myofibroblasts. This action is performed via up-regulation of another transcription factor, osterix (Osx), which is also stimulated by BMP-2. Osx directs osteoblast-specific differentiation, up-regulates ALP expression and is necessary for mineralization. It was also shown that Msx2 can act independently of Runx2, by Wnt/LRP5 signalling pathway [53].
Estrogens and homocysteine
It is well documented that women after the menopause demonstrate an accelerated bone loss, suggesting that estrogen deficiency plays a pivotal role in this process [54]. The beneficial effects of estrogens on the cardiovascular system are also well established [55]. Furthermore, bone and coronary arteries are target organs for estrogens. Estrogen receptors have been detected on osteoblasts, osteoclasts and coronary artery smooth muscle cells [56]. They are also expressed in osteoblasts as well as in chondrocytes in men, whose BMD is positively correlated with estrogen concentrations [57].
Except for the direct effects of estrogens on bone loss and atherosclerosis, their deficiency affects these two progressive diseases indirectly. A decrease in estrogen levels is associated with an increase in serum parathyroid hormone (PTH) [58]. Increased PTH secretion results in accelerated bone loss and soft tissue calcium deposition, including vascular and myocardial calcification [59]. Furthermore, estrogens may be inversely related to serum levels of homocysteine [60] and lipids, especially oxidized low density lipoprotein (LDL) [61].
Homocysteine is a possible risk factor for atherosclerosis [62]. Homocysteinuria, a genetically inherited disease caused by a deficiency of cystathionine β synthetase or a mutant form of methylenetetrahydrofolate reductase (MTHFR), is characterized by elevated plasma homocysteine concentrations. Its clinical manifestations, apart from skeletal disorders and osteoporosis, include a tendency towards premature atherosclerosis and thromboembolism. Homocysteine seems to interfere with the formation of collagen cross-links, prevents insolubilization of fibrils, inhibits lysyl oxidase and may delay the synthesis of more complex cross-links in collagen. There is also evidence that postmenopausal women with heterozygous mutation in MTHFR and therefore hyperhomocysteinemia demonstrate a decrease in BMD [63]. This supports the hypothesis that homocysteine participates in the interaction between estrogen and bone metabolism. Moreover, a prospective study demonstrated a 10.9% decrease in plasma homocysteine levels in postmenopausal women continuously treated with micronised 17β-estradiol combined with cyclic dydrogesterone compared with baseline pre-hormone replacement levels [64].
Lipid oxidation
Lipid oxidation products such as minimally oxidized LDL (MM-LDL) promote arterial calcification and its accumulation in the subendothelial space of skeletal bone arteries inhibits bone formation. MM-LDL acts through activating peroxisome proliferator-activated receptors α and γ (PPARα and PPARγ). It promotes bone loss by directing progenitor marrow stromal cells to undergo adipogenic instead of osteogenic differentiation, as indicated by a reduced expression of bone alkaline phosphatase (b-ALP) and osteocalcin, two markers of bone formation [65]. The accumulation of oxidized lipids in tissue mimics chronic infection and thereby stimulates an immune response that promotes the hardening of soft tissue (to wall off infectious agents) and the softening of hard tissue (to dissolve a substrate for growth of infectious agents) [66]. Moreover, the enzyme 12/15 lipoxygenase (12/15LO), which is responsible for the oxidative modification of LDL [67], plays a functional role in the modulation of oxidative stress and is active in bone and arterial wall [68]. The importance of this enzyme rises from the fact that its inhibition in animals seems to enhance bone mass and diminish generation of atherosclerotic lesions [68, 69].
Other markers of oxidative stress involve the isoprostanes, lipid oxidation products derived from arachidonic acid. The two most extensively studied members of this family are 8-isoprostaglandin F2a (isoPGF2a) and 8-isoprostaglandin E2a (isoPGE2a) [70]. Isoprostanes are present in atherosclerotic plaques, have vasoconstrictor effects, inducing the release of endothelin-1 in endothelium and modulate the aggregation of platelets [71]. They provide a common biological linkage towards the association between atherosclerosis and osteoporosis, as they seem to inhibit osteoblastic differentiation of preosteoblasts and enhance osteoclastic differentiation and activity. This was evidenced using tartrate-resistant acid phosphatase (TRAP) activity, a marker of bone resorption, which was increased after treatment of marrow-derived preosteoclasts with isoPGE2a [71]. Another study evaluating the role of isoPGF2a, showed increased levels in hypercholesterolaemic subjects, combined with lower bone mass and higher levels of b-ALP and osteocalcin than in normocholesterolaemic ones [72]. All these findings lend support to the “lipid hypothesis of osteoporosis.
Inflammatory process
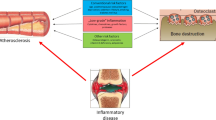
Atherosclerosis is thought to include an ongoing inflammatory process. Markers of inflammation such as interleukin-6 (IL-6) and C-reactive protein (CRP) have been associated with all-cause and cardiovascular mortality in both sexes [73]. On the other hand, IL-6 is known to stimulate osteoclasts to increase the rate of bone resorption [74]. As far as CRP is concerned, the association between serum high sensitivity CRP (hs-CRP) levels and BMD in pre-and postmenopausal women was stronger in subjects with lower BMD, after adjustment for age, body mass index (BMI) and menopausal status, combined with higher serum total ALP activity (all, P for trend <0.001) [75]. Similar results were obtained from another study which showed that the increase in the levels of hs-CRP in pre-and post-menopausal women was followed by an increase in serum NTX, a marker of bone resorption, as mentioned above [76]. Thus, a systemic inflammatory process may be a common mechanism for the development of low bone mass and atherosclerosis.
Vitamins D and K
The role of vitamin D in bone metabolism through calcium absorption is well established [77]. Genetic studies provide further support for a role of vitamin D in the pathogenesis of both osteoporosis and atherosclerosis. Vitamin D receptors (VDR) are present in endothelial [78] and smooth muscle cells [79] of the arterial wall. Some studies suggest that polymorphisms of VDR may contribute to the common risk between osteoporosis and atherosclerosis [80]. In particular, the BSmI polymorphism has been associated with slowed calcium absorption and lower BMD [81]. On the other hand, the frequency of BsmI B allele has been reported to be higher in patients with angiographically documented CAD [82] and DM [83], although one study found no relation at all [84].
As mentioned above, GLa proteins are involved in the regulation of the calcification processes in bone and vascular tissue. GLa is formed by vitamin K, which acts as a coenzyme for glutamate carboxylase, converting glutamate to γ-carboxyglutamate. As a result, reduced availability of vitamin K leads to functionally defective GLa proteins, with low affinity for the hydroxyapatite bone matrix and therefore low BMD [85]. Indeed, administration of vitamin K seems to retard femoral neck bone loss in postmenopausal women [86]. On the other hand, impaired vitamin K status has been associated with the presence of atherosclerotic calcification [87], providing evidence for its role in vascular mineralization.
Therapeutic implications of the association between atherosclerosis and osteoporosis
Statins
The beneficial actions of statins in patients with atherosclerosis and CVD are well established [88–90]. Statins appear to also exert beneficial effects on bone metabolism [91, 92]. Given the potential significance of hyperlipidemia in the pathogenesis of osteoporosis, these effects of statins might be attributed to their lipid-lowering action. However, the beneficial effects of statins on bone metabolism also appear to be lipid-lowering independent. Statins act by inhibiting the enzyme 3-hydroxy-3-methylglutaryl-CoA (HMG-CoA) reductase, the rate-limiting enzyme involved in endogenous cholesterol biosynthesis, which catalyzes the reduction of HMG-CoA to mevalonic acid. Apart from reducing cholesterol biosynthesis, the inhibition of mevalonate also leads to a reduction in the synthesis of important intermediates, such as the isoprenoids farnesyl pyrophosphate and geranylgeranyl pyrophosphate [93]. These intermediates are involved in the posttranslational prenylation of several proteins (e.g., Ras, Rho and Rac) that modulate a variety of cellular processes including cellular signalling, differentiation and proliferation [94]. Prenylated signalling proteins are essential for osteoclast function, consistent with some observations that statins inhibit osteoclast activity in vitro [95]. Apart from inhibition of osteoclast activity there is evidence that statins also induce bone formation, perhaps through an increase in BMP-2 [35]. In a prospective study, simvastatin therapy was associated with an increase in serum osteocalcin concentration [96], while another study demonstrated an increase in the levels of vitamin D after administration of atorvastatin [97].
Moreover, a substantial increase in bone formation and trabecular bone volume has been reported in female rats after 5 weeks of administration of simvastatin [98]. An analysis of four large prospective studies suggests that statins may prevent osteoporotic fractures. Each study found a strong trend towards fewer hip fractures among women who reported the use of statins, even after adjusting for a number of other factors [99]. In a large retrospective analysis, exposure to statins was associated with a decreased fracture risk even after short duration (a few weeks to a few months). In contrast, there was little evidence that fibrates or other lipid-lowering drugs alter bone fracture risk [91]. However, non-confirmatory data have arisen from a large randomized case-controlled trial, evaluating the effect of pravastatin use on the fracture rate, although this study involved a non osteoporotic population [100]. Pravastatin seems to be ineffective at increasing BMP-2 as it binds less strongly to plasma proteins than do other statins, such as lovastatin, simvastatin or fluvastatin [101].
A meta-analysis of 19 studies assessed the effect of statins on BMD and the risk of fractures. Twelve studies concluded that statins exert a beneficial effect on BMD, 6 studies that they had no effect and one study reported a deleterious effect. Furthermore, there was a different impact on bone mass between statins in regard to their lipophilic or hydrophilic properties. In particular, lipophilic statins (lovastatin, simvastatin) showed a significant effect on total hip (TH) and femoral neck (FN) BMD, whereas hydrophilic ones (atorvastatin, pravastatin, fluvastatin) had no effect on BMD. TH and FN BMD was significantly higher (by about 0.20 SD) among statin users than among controls, although lumbar spine BMD was only marginally increased, suggesting that these drugs act mainly on cortical bone. The discrepancies observed between all these studies might be attributed to several reasons, including low and variable uptake of statins into bones, low systemic bioavailability (important hepatic first-pass effect), different sensitivities of osteoblasts and osteoclasts to statins, variable risk of osteoporosis between patients, insufficient duration of exposure (statins may need to be taken for more than 1 year) and drug dosages (the doses showing benefit in animal studies were tenfold higher than those used in dyslipidemia [102].
Biphosphonates
The decrease in production of mevalonate achieved by statins seems to also be an important biochemical pathway in the action of biphosphonates which are widely used in the treatment of osteoporosis, via inhibition of osteoclastic activity. Biphosphonates can inhibit the development of atherosclerosis in animal experiments and this action seems to be independent of lowering serum calcium levels [103]. The protective effect of biphosphonates has been attributed to their direct action on the vessel wall by sensitizing macrophages to undergo apoptosis, preventing foam cell formation by inhibiting the uptake of LDL and affecting cell replication [104]. Furthermore, etidronate, a biphosphonate, was suggested to have an inhibitory effect on atherosclerosis in patients with low bone mass [105]. Why biphosphonates, though acting on the same biochemical pathway as statins, only have anti-resorptive effects (whereas statins directly stimulate bone formation) is yet unexplained. There might be different sensitivities of osteoblasts and osteoclasts to statins and biphosphonates [102]. However, all these promising preliminary findings need to be confirmed in large double-blind randomized controlled trials.
Antihypertensive drugs
Beta-blockers appear to induce bone formation and/or inhibit bone resorption in animals as well as reduce the risk of fracture in humans [106]. Their action seems to be based on the regulation of bone remodelling by the sympathetic nervous system. Sympathetic nerve fibres have been detected in bone tissue and functional adrenergic receptors are expressed on osteoblasts and osteoclasts [107]. Adrenergic stimulation has been reported to cause both anabolic and catabolic effects on bone, mediated by α- and β-adrenergic receptors, respectively. The β-adrenergic stimulation of bone resorption might be mediated by directly activated osteoclasts [108]. A large prospective population-based study associated the use of β-blockers with a reduced risk of fractures in middle-aged and older subjects from the general population [109]. However, these agents cannot be recommended as preventive therapy for fractures until randomized controlled trials establish their efficacy.
Although there is no general consensus that hypertension should be considered an established risk factor for osteoporosis, we will discuss the role of some antihypertensive drugs (apart from β-blockers) on bone metabolism. High blood pressure can induce abnormalities in calcium metabolism and increase bone mineral loss in women [29]. Some studies demonstrated an effect of thiazide diuretics in preventing osteoporosis, perhaps by reducing the urinary calcium excretion [110, 111]. In both of these randomized controlled trials the daily administration of hydrochlorothiazide (50 mg) prevented postmenopausal bone loss and its benefit seemed to be sustained for at least the first 4 years of treatment [111]. The bone-protective effect of thiazides cannot only be attributed to the increased renal calcium reabsorption via inhibition of Na+-Cl- cotransporter (NCC) at the kidney distal tubule, but also to a direct effect on bone. It seems that osteoblasts also express NCC and therefore thiazides stimulate osteoblast differentiation and bone mineralization. Furthermore, they may induce the production of osteoblast markers, such as Runx2 and OP, the role of which was discussed above [112].
As far as the renin-angiotensin system is concerned, it seems that angiotensin II promotes bone loss by activating osteoclasts via RANKL induction, demonstrated by an increase in TRAP activity. Furthermore, the use of an angiotensin II type 1 receptor blockade (olmesartan) appeared to attenuate this process [113]. However, the evidence evaluating the role of angiotensin converting enzyme (ACE) inhibitors in preventing bone loss is controversial. Some studies associated their administration with an increase in hip and spine BMD [114] and others showed that ACE inhibition has no effect on the skeleton [115]. These different outcomes may be attributed to ACE polymorphisms. In particular, subjects presenting with the II + ID polymorphism exhibit a poor response to antihypertensive drug treatment with respect to bone mass, whereas those with the DD polymorphism seem to respond better [116]. Nonetheless, more confirmatory data are needed for these drugs to be indicated for both hypertension (a well-established risk factor for atherosclerosis) and osteoporosis.
Experimental therapies
New experimental therapeutic horizons have opened up, promoting the generation of dual-purpose treatment, which will retard the progression of atherosclerotic plaques and enhance bone density. Briefly, they involve: (i) inhibition of 12/15 lipoxygenase (ii) attenuation of osteocalcin expression (iii) modifying LRP5 (iv) deletion of Wnt antagonists (v) recombinant osteoprotegerin, and, (vi) teriparatide (the 1–34 N-terminal fragment of PTH, which is used as an anabolic therapy for severe osteoporosis and is shown to limit aortic valve calcification) [35]. However, these attractive and encouraging therapies are very distant from their final application in patients to achieve dual prevention from the adverse outcomes attributed to osteoporosis and atherosclerosis (Table 3).
Conclusions
Emerging data support the association between atherosclerosis and osteoporosis beyond the aging process. Except for the common risk factors that influence both cardiovascular risk and bone metabolism, their association could be attributed to several shared biochemical, molecular and cellular processes. However, further investigation and large randomized controlled trials are needed to confirm these relationships. A link between atherosclerosis and osteoporosis could potentially influence the pharmacological options for the prevention and treatment of these highly prevalent conditions.
References
Frye MA, Melton LJ 3rd, Bryant SC et al (1992) Osteoporosis and calcification of the aorta. Bone Miner 19:185–194
Vogt MT, San Valentin R, Forrest KY et al (1997) Bone mineral density and aortic calcification: the Study of Osteoporotic fractures. J Am Geriatr Soc 45:140–145
Sinnott B, Syed I, Sevrukov A et al (2006) Coronary calcification and osteoporosis in men and postmenopausal women are independent processes associated with aging. Calcif Tissue Int 78:195–202
Dent CE, Engelbrecht HE, Godfrey RC (1968) Osteoporosis of lumbar vertebrae and calcification of abdominal aorta in women living in Durban. Br Med J 4:76–79
Reid IR, Ames RW, Evans MC et al (1994) Determinants of the rate of bone loss in normal postmenopausal women. J Clin Endocrinol Metab 79:950–954
Tekin GO, Kekilli E, Yagmur J et al (2008) Evaluation of cardiovascular risk factors and bone mineral density in postmenopausal women undergoing coronary angiography. Int J Cardiol [Epub ahead of print]
Von der Recke P, Hansen MA, Hassager C (1999) The association between low bone mass at the menopause and cardiovascular mortality. Am J Med 106:273–278
Kado DM, Browner WS, Blackwell T et al (2000) Rate of bone loss is associated with mortality in older women: a prospective study. J Bone Miner Res 15:1974–1980
Browner WS, Seeley DG, Vogt TM et al (1991) Non-trauma mortality in elderly women with low bone mineral density. Study of Osteoporotic Fractures Research Group. Lancet 338:355–358
Marcovitz PA, Tran HH, Franklin BA et al (2005) Usefulness of bone mineral density to predict significant coronary artery disease. Am J Cardiol 96:1059–1063
Sennerby U, Farahmand B, Ahlbom A et al (2007) Cardiovascular diseases and future risk of hip fracture in women. Osteoporos Int 18:1355–1362
Kiel DP, Kauppila LI, Cupples LA et al (2001) Bone loss and the progression of abdominal aortic calcification over a 25 year period: the Framingham Heart Study. Calcif Tissue Int 68:271–276
Schulz E, Arfai K, Liu X et al (2004) Aortic calcification and the risk of osteoporosis and fractures. J Clin Endocrinol Metab 89:4246–4253
Hyder JA, Allison MA, Criqui MH et al (2007) Association between systemic calcified atherosclerosis and bone density. Calif Tissue Int 80:301–306
Mangiafico RA, Alagona C, Pennisi P et al (2008) Increased augmentation index and central aortic blood pressure in osteoporotic postmenopausal women. Osteoporos Int 19:49–56
Barengolts EI, Berman M, Kukreja SC et al (1998) Osteoporosis and coronary atherosclerosis in asymptomatic postmenopausal women. Calcif Tissue Int 62:209–213
Uyama O, Yoshimoto Y, Yamamoto Y et al (1997) Bone changes and carotid atherosclerosis in postmenopausal women. Stroke 28:1730–1732
Jorgensen L, Joakimsen O, Mathiesen EB et al (2006) Carotid plaque echogenicity and risk of nonvertebral fractures in women: a longitudinal population-based study. Calcif Tissue Int 79:207–213
Shaffer JR, Kammerer CM, Rainwater DL et al (2007) Decreased bone mineral density is correlated with increased subclinical atherosclerosis in older, but not younger, Mexican American women and men: the San Antonio Family Osteoporosis Study. Calcif Tissue Int 81:430–441
Van der Klift M, Pols HA, Hak AE et al (2002) Bone mineral density and the risk of peripheral arterial disease: the Rotterdam Study. Calcif Tissue Int 70:443–449
Jorgensen L, Engstad T, Jacobsen BK (2001) Bone mineral density in acute stroke patients: low bone mineral density may predict first stroke in women. Stroke 32:47–51
Browner WS, Pressman AR, Nevitt MC et al (1993) Association between low bone density and stroke in elderly women: the study of osteoporotic fractures. Stroke 24:940–946
NIH (2001) Consensus development panel on osteoporosis prevention, diagnosis, and therapy. JAMA 285:785–795
Jorgensen L, Joakimsen O, Rosvold Berntsen GK et al (2004) Low bone mineral density is related to echogenic carotid artery plaques: a population-based study. Am J Epidemiol 160:549–556
Farhat GN, Strotmeyer ES, Newman AB et al (2006) Volumetric and areal bone mineral density measures are associated with cardiovascular disease in older men and women: the health, aging and body composition study. Calcif Tissue Int 79:102–111
Johansson C, Black D, Johnell O et al (1998) Bone mineral density is a predictor of survival. Calcif Tissue Int 63:190–196
Cappuccio FP, Meilahn E, Zmuda JM et al (1999) High blood pressure and bone-mineral loss in elderly white women: a prospective study. Study of Osteoporotic Fractures Research Group. Lancet 354:971–975
Selby PL (1988) Osteopenia and diabetes. Diabetic Med 5:423–428
Stevenson JC, Lees B, Devenport M et al (1989) Determinants of bone density in normal women: risk factors for future osteoporosis? Br Med J 298:924–928
Kanders B, Dempster DW, Lindsay R (1988) Interaction of calcium nutrition and physical activity on bone mass in young women. J Bone Miner Res 3:145–149
Hofbauer LC, Brueck CC, Shanahan CM et al (2007) Vascular calcification and osteoporosis-from clinical observation towards molecular understanding. Osteoporos Int 18:251–259
Schmid K, McSharry WO, Pameijer CH et al (1980) Chemical and physiochemical studies on the mineral deposits of the human atherosclerotic aorta. Atherosclerosis 37:199–210
Shanahan CM, Cary NR, Metcalfe JC et al (1994) High expression of genes for calcification-regulating proteins in human atherosclerotic plaques. J Clin Invest 93:2393–2402
Luo G, Ducy P, McKee MD et al (1997) Spontaneous calcification of arteries and cartilage in mice lacking GLA protein. Nature 386:78–81
Hamerman D (2005) Osteoporosis and atherosclerosis: biological linkages and the emergence of dual-purpose therapies. QJM 98:467–484
Chen D, Zhao M, Mundy GR (2004) Bone morphogenetic proteins. Growth Factors 22:233–241
Galvin KM, Donovan MJ, Lynch CA et al (2000) A role for Smad6 in development and homeostasis of the cardiovascular system. Nature Genetics 24:171–174
Shao JS, Cheng SL, Pingsterhaus JM et al (2005) Msx2 promotes cardiovascular calcification by activating paracrine Wnt signals. J Clin Invest 115:1210–1220
Hsu H, Lacey DL, Dunstan CR et al (1999) Tumor necrosis factor receptor family member RANK mediates osteoclast differentiation and activation induced by osteoprotegerin ligand. Proc Natl Acad Sci USA 96:3540–3545
Collinp-Osdoby P, Rothe L, Anderson F et al (2001) Receptor activator of NF-kB and osteoprotegerin expression by human microvascular endothelial cells, regulation by inflammatory cytokines, and role in human osteoclastogenesis. J Biol Chem 276:20659–20672
Bucay N, Sarosi I, Dunstan CR et al (1998) Osteoprotegerin-deficient mice develop early onset osteoporosis and arterial calcification. Genes Dev 12:1260–1268
Browner WS, Lui LY, Cummings SR (2001) Association of serum osteoprotegerin levels with diabetes, stroke, bone density, fractures and mortality in elderly women. J Clin Endocrinol Metab 86:631–637
Schoppet M, Sattler AM, Schaefer JR et al (2003) Increased osteoprotegerin serum levels in men with coronary artery disease. J Clin Endocrinol Metab 88:1024–1028
Collins T, Cybulsky MI (2001) NF-kB: pivotal mediator or innocent bystander in atherogenesis? J Clin Invest 107:255–264
Tamai K, Semenov M, Kato Y et al (2000) LDL-receptor related proteins in Wnt signal transduction. Nature 407:530–535
Bienz M (1998) TCF: transcriptional activator or repressor? Curr Opin Cell Biol 10:366–372
Mikhaylova L, Malmquist J, Nurminskaya M (2007) Regulation of in vitro vascular calcification by BMP4, VEGF and Wnt3a. Calcif Tissue Int 81:372–381
Manolagas SC, Almeida M (2007) Gone with the Wnts: beta-catenin, T-cell factor, forkhead box O, and oxidative stress in age-dependent diseases of bone, lipid, and glucose metabolism. Mol Endocrinol 21:2605–2614
Mani A, Radhakrishnan J, Wang H (2007) LRP6 mutation in a family with early coronary disease and metabolic risk factors. Science 315:1278–1282
Banerjee C, McCabe LR, Choi JY et al (1997) Runt homology domain proteins in osteoblast differentiation: AML3/CBFA1 is a major component of a bone specific complex. J Cell Biochem 66:1–8
Byon CH, Javed A, Dai Q et al (2008) Oxidative stress induces vascular calcification through modulation of the osteogenic transcription factor Runx-2 by Akt signalling. J Biol Chem [Epub ahead of print]
Odelberg SJ, Kollhoff A, Keating MT (2000) Dedifferentiation of mammalian myotubes induced by msx1. Cell 103:1099–1109
Cheng SL, Shao JS, Charlton-Kachigian N et al (2003) Msx2 promotes osteogenesis and suppresses adipogenic differentiation of multipotent mesenchymal progenitors. J Biol Chem 278:45969–45977
Christiansen C, Lindsay R (1990) Estrogen, bone loss and preservation. Osteoporos Int 1:7–13
Mendelsohn ME, Karas RH (1999) The protective effects of estrogen on the cardiovascular system. N Engl J Med 340:1801–1811
Losordo DW, Kearney M, Kim EA et al (1994) Variable expression of the estrogen receptor in normal and atherosclerotic coronary arteries of premenopausal women. Circulation 89:1501–1510
Barrett-Connor E, Mueller JE, von Mühlen DG et al (2000) Low levels of esrtadiol are associated with vertebral fractures in older men, but not women: The Rancho Bernardo Study. J Clin Endocrinol Metab 85:219–223
Khosla S, Atkinson EJ, Melton LJ 3rd et al (1997) Effects of age and estrogen status on serum parathyroid hormone levels and biochemical markers of bone turnover in women: a population-based study. J Clin Endocrinol Metab 82:1522–1527
Stefenelli T, Mayr H, Bergler-Klein J et al (1993) Primary hyperparathyroidism: incidence of cardiac abnormalities and partial reversibility after successful parathyroidectomy. Am J Med 95:197–202
Hak AE, Polderman KH, Westendorp IC et al (2000) Increased plasma homocysteine after menopause. Atherosclerosis 149:163–168
Zhu X, Bonet B, Knopp RH (2000) Estradiol 17beta inhibition of LDL oxidation and endothelian cell cytotoxicity is opposed by progestins to different degrees. Atherosclerosis 148:31–41
Christen WG, Ajani UA, Glynn RJ et al (2000) Blood levels of homocysteine and increased risks of cardiovascular disease: causal or casual? Arch Intern Med 160:422–434
Miyao M, Morita H, Hosoi T et al (2000) Association of methylenetetrahydrofolate reductase (MTHFR) polymorphism with bone mineral density in postmenopausal Japanese women. Calcif Tissue Int 66:190–194
Van der Mooren MJ, Wouters MG, Blom HJ et al (1994) Hormone replacement therapy may reduce high serum homocysteine in postmenopausal women. Eur J Clin Invest 24:733–736
Parhami F, Jackson SM, Tintut Y et al (1999) Atherogenic diet and minimally oxidized low density lipoprotein inhibit osteogenic and promote adipogenic differentiation of marrow stromal cells. J Bone Miner Res 14:2067–2078
Demer LL (2002) Vascular calcification and osteoporosis: inflammatory responses to oxidized lipids. Int J Epidemiol 31:737–741
Kühn H, Belkner J, Suzuki H et al (1994) Oxidative modification of human lipoprotein by lipoxygenases of different positional specificities. J Lipid Res 35:1749–1759
Klein RF, Allard J, Avnur Z et al (2004) Regulation of bone mass in mice by the lipoxygenase gene Alox15. Science 303:229–232
Cornicelli JA, Trivedi BK (1999) 15-Lipoxygenase and its inhibitions: a novel therapeutic target for vascular disease. Curr Pharm Des 5:11–20
Lawson JA, Rokach J, FitzGerald GA (1999) Isoprostanes: formation, analysis and use as indices of lipid peroxidation in vivo. J Biol Chem 274:24441–24444
Tintut Y, Parhami F, Tsingotjidou A et al (2002) 8-Isoprostaglandin E2 enhances receptor-activated NFkappa B ligand (RANKL)-dependent osteoclastic potential of marrow hematopoietic precursors via the cAMP pathway. J Biol Chem 277:14221–14226
Mangiafico RA, Malaponte G, Pennisi P et al (2007) Increased formation of 8-iso-prostaglandin F(2alpha) is associated with altered bone metabolism and lower bone mass in hypercholesterolaemic subjects. J Intern Med 261:587–596
Pai JK, Pischon T, Ma J et al (2004) Inflammatory markers and the risk of coronary heart disease in men and women. N Eng J Med 351:2599–2610
Papanicolaou DA, Wilder RL, Manolagas SC et al (1998) The pathophysiologic roles of interleukin-6 in human disease. Ann Intern Med 128:127–137
Koh JM, Khang YH, Jung CH et al (2005) Higher circulating hsCRP levels are associated with lower bone mineral density in healthy pre- and postmenopausal women: evidence for a link between systemic inflammation and osteoporosis. Osteoporos Int 16:1263–1271
Kim BJ, Yu YM, Kim EN et al (2007) Relationship between serum hsCRP concentration and biochemical bone turnover markers in healthy pre- and postmenopausal women. Clin Endocrinol (Oxf) 67:152–158
Gallagher JC, Riggs BL, Eisman J et al (1979) Intestinal calcium absorption and serum vitamin D metabolites in normal subjects and osteoporotic patients. J Clin Invest 64:729–736
Merke J, Milde P, Lewicka S et al (1989) Identification and regulation of 1,25-dihydroxyvitamin D3 receptor activity and biosynthesis of 1,25-dihydroxyvitamin D3. Studies in cultured bovine aortic endothelial cells and human dermal capillaries. J Clin Invest 83:1903–1915
Merke J, Hofmann W, Goldschmidt D et al (1987) Demonstration of 1,25(OH)2 vitamin D3 receptors and actions in vascular smooth muscle cells in vitro. Calcif Tissue Int 41:112–114
Kammerer CM, Dualan AA, Samollow PB et al (2004) Bone mineral density, carotid artery intimal medial thickness, and the vitamin D receptor BsmI Polymorphism in Mexican American Women. Calcif Tissue Int 75:292–298
Gong G, Stern HS, Cheng SC et al (1999) The association of bone mineral density with vitamin D receptor gene polymorphisms. Osteoporos Int 9:55–64
Van Schooten FJ, Hirvonen A, Maas LM et al (1998) Putative susceptibility markers of coronary artery disease: association between VDR genotype, smoking, and aromatic DNA adduct levels in human right atrial tissue. FASEB J 12:1409–1417
Ortlepp JR, Lauscher J, Hoffmann R et al (2001) The vitamin D receptor gene variant is associated with the prevalence of type 2 diabetes mellitus and coronary artery disease. Diabet Med 18:842–845
Ortlepp JR, von Korff A, Hanrath P et al (2003) Vitamin D receptor gene polymorphism BsmI is not associated with the prevalence and severity of CAD in a large-scale angiographic cohort of 3441 patients. Eur J Clin Invest 33:106–109
Braam LA, Knapen MH, Geusens P et al (2003) Vitamin K1 supplementation retards bone loss in postmenopausal women between 50 and 60 years of age. Calcif Tissue Int 73:21–26
Price PA, Williamson MK, Lothringer JW (1981) Origin of the vitamin K-dependent bone protein found in plasma and its clearance by kidney and bone. J Biol Chem 256:12760–12766
Jie KG, Bots ML, Vermeer C et al (1996) Vitamin K status and bone mass in women with and without aortic atherosclerosis: a population-based study. Calcif Tissue Int 59:352–356
Baigent C, Keech A, Kearney PM et al (2005) Cholesterol Treatment Trialists’ (CTT) Collaborators. Efficacy and safety of cholesterol-lowering treatment: prospective meta-analysis of data from 90,056 participants in 14 randomised trials of statins. Lancet 366:1267–1278
Athyros VG, Kakafika AI, Papageorgiou AA et al (2007) GREACE Study Collaborative Group. Atorvastatin decreases triacylglycerol-associated risk of vascular events in coronary heart disease patients. Lipids 42:999–1009
Paraskevas KI, Athyros VG, Briana DD et al (2007) Statins exert multiple beneficial effects on patients undergoing percutaneous revascularization procedures. Curr Drug Targets 8:942–951
Meier CR, Schlienger RG, Kraenzlin ME et al (2000) HMG-CoA reductase inhibitors and the risk of fractures. JAMA 283:3205–3210
Paraskevas KI, Tzovaras AA, Briana DD et al (2007) Emerging indications for statins: a pluripotent family of agents with several potential applications. Curr Pharm Des 13:3622–3636
Liao JK (2002) Isoprenoids as mediators of the biological effects of statins. J Clin Invest 110:285–288
Alegret M, Silvestre JS (2006) Pleiotropic effects of statins and related pharmacological experimental approaches. Methods Find Exp Clin Pharmacol 28:627–656
Hughes A, Rogers MJ, Idris AI et al (2007) A comparison between the effects of hydrophobic and hydrophilic statins on osteoclast function in vitro and ovariectomy-induced bone loss in vivo. Calcif Tissue Int 81:403–413
Chan MH, Mak TW, Chiu RW et al (2001) Simvastatin increases serum osteocalcin concentration in patients treated for hypercholesterolaemia. J Clin Endocrinol Metab 86:4556–4559
Pérez-Castrillón JL, Vega G, Abad L et al (2007) Effects of Atorvastatin on vitamin D levels in patients with acute ischemic heart disease. Am J Cardiol 99:903–905
Mundy G, Garrett R, Harris S et al (1999) Stimulation of bone formation in vitro and in rodents by statins. Science 286:1946–1949
Bauer DC, Mundy GR, Jamal SA et al (2004) Use of statins and fracture: results of 4 prospective studies and cumulative meta-analysis of observational studies and controlled trials. Arch Intern Med 164:146–152
Reid IR, Hague W, Emberson J et al (2001) Effect of pravastatin on frequency of fracture in the LIPID study: secondary analysis of a randomised controlled trial. Long-term Intervention with Pravastatin in Ischaemic Disease. Lancet 357:509–512
Pedersen TR, Kjekshus J (2000) Statin drugs and the risk of fracture. 4S Study Group. JAMA 284:1921–1922
Uzzan B, Cohen R, Nicolas P et al (2007) Effects of statins on bone mineral density: a meta-analysis of clinical studies. Bone 40:1581–1587
Ylitalo R (2000) Bisphosphonates and atherosclerosis. Gen Pharmacol 35:287–296
Luckman SP, Hughes DE, Coxon FP et al (1998) Nitrogen-containing bisphosphonates inhibit the mevalonate pathway and prevent post-translational prenylation of GTP-binding proteins, including Ras. J Bone Miner Res 13:581–589
Koshiyama H, Nakamura Y, Tanaka S (2000) Decrease in carotid intima-media thickness after 1-year therapy with etidronate for osteopenia associated with type 2 diabetes. J Clin Endocrinol Metab 85:2793–2796
Bonnet N, Gadois C, McCloskey E et al (2007) Protective effect of beta blockers in postmenopausal women: influence on fractures, bone density, micro and macroarchitecture. Bone 40:1209–1216
Chenu C (2004) Role of innervation in the control of bone remodeling. J Musculoskelet Neuronal Interact 2:132–134
Togari A (2002) Adrenergic regulation of bone metabolism: possible involvement of sympathetic innervation of osteoblastic and osteoclastic cells. Microsc Res Tech 58:77–84
Meisinger C, Heier M, Lang O et al (2007) Beta-blocker use and risk of fractures in men and women from the general population: the MONICA/KORA Augsburg cohort study. Osteoporos Int 18:1189–1195
Reid IR, Ames RW, Orr-Walker BJ et al (2000) Hydrochlorothiazide reduces loss of cortical bone in normal postmenopausal women: a randomized controlled trial. Am J Med 109:362–370
Bolland MJ, Ames RW, Horne AM et al (2007) The effect of treatment with a thiazide diuretic for 4 years on bone density in normal postmenopausal women. Osteoporos Int 18:479–486
Dvorak MM, De Joussineau C, Carter DH et al (2007) Thiazide diuretics directly induce osteoblast differentiation and mineralized nodule formation by targeting a sodium chloride cotransporter in bone. J Am Soc Nephrol 18:2509–2516
Shimizu H, Nakagami H, Osako MK et al (2008) Angiotensin II accelerates osteoporosis by activating osteoclasts. FASEB J 6 [Epub ahead of print]
Lynn H, Kwok T, Wong SY et al (2006) Angiotensin converting enzyme inhibitor use is associated with higher bone mineral density in elderly Chinese. Bone 38:584–588
Stimpel M, Jee WS, Ma Y et al (1995) Impact of antihypertensive therapy on postmenopausal osteoporosis: effects of the angiotensin converting enzyme inhibitor moexipril, 17beta-estradiol and their combination on the ovariectomy-induced cancellous bone loss in young rats. J Hypertens 13:1852–1856
Pérez-Castrillón JL, Silva J, Justo I et al (2003) Effect of quinapril, quinapril-hydrochlorothiazide, and enalapril on bone mass of hypertensive subjects: relationship with angiotensin converting enzyme polymorphisms. Am J Hypertens 16:453–459
Conflicts of interest
This review was written independently; no company or institution supported it financially. Some of the authors have attended conferences, given lectures and participated in advisory boards or other trials sponsored by various pharmaceutical companies.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Anagnostis, P., Karagiannis, A., Kakafika, A.I. et al. Atherosclerosis and osteoporosis: age-dependent degenerative processes or related entities?. Osteoporos Int 20, 197–207 (2009). https://doi.org/10.1007/s00198-008-0648-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00198-008-0648-5