
Abstract
Vascular diseases account for a significant proportion of preventable deaths, particularly in developed countries. Our understanding of diseases that alter the structure and function of blood vessels such as vascular calcification and vascular stiffness has grown enormously such that we now appreciate them to be active processes that can be modified. Interest has also grown in examining the links between other diseases of ageing such as the loss of bone (osteoporosis) and muscle (sarcopenia) with the development and progression of vascular disease as these three disease states commonly co-occur in older age. Cardiovascular disease (including calcification and arterial stiffness) is highly prevalent in older populations and it appears that its progression is accelerated in patients with osteoporosis, fracture, sarcopenia and in those who are functionally impaired. Biological and clinical evidence supports a view that vascular disease (calcification/stiffness) may be both a cause and consequence of diseases of ageing including musculoskeletal decline. This review provides an overview of the development of vascular calcification and stiffness and explores the molecular and physiological mechanisms linking osteoporosis and sarcopenia to vascular disease development. This review also examines clinical evidence supporting the association of muscle and bone loss with vascular disease and concludes by reviewing the interventional and therapeutic potential of bone-active minerals and hormones (calcium and vitamin D) on cardiovascular disease biology, given that these represent potential interventions to target multiple body systems. Overall, this review will aim to highlight the underappreciated burden of cardiovascular disease in individuals in the context of musculoskeletal diseases.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Part 1 Introduction: Osteoporosis, Sarcopenia and Vascular Disease: Shared Risk Factors and Clinical Associations
The loss of bone and muscle mass and the development of cardiovascular disease appear to be the inevitable consequences of ageing. Each of these body systems declines through defined mechanisms; yet, we are increasingly understanding the shared nature of diseases that affect the skeleton, muscles and the vascular system. It is uncertain if bone and muscle diseases (which usually occur concomitantly) arise as a consequence of prevalent but subclinical cardiovascular disease or that cardiovascular disease contributes to, and potentially accelerates, the development of musculoskeletal conditions. In short, are musculoskeletal disorders and cardiovascular diseases biologically linked or are their manifestations in older age just the inevitable confluence of age-related diseases? The following review is an exploration of (1) bone disease and its links to cardiovascular disease, (2) muscle disease and its links to cardiovascular disease and (3) potential treatment strategies that target all systems.
Osteoporosis
Definition
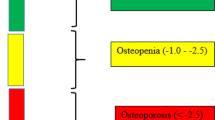
Osteoporosis is a systemic skeletal disorder characterised by the loss of bone mass and deterioration in bone microarchitecture. These structural and molecular changes in bone biology combine to compromise bone strength leading to skeletal fragility and increased susceptibility of fracture [1]. Recovery from a fracture is costly, takes many months and indeed some never truly recover as they may lose independence and the risk of mortality greatly increases following a fracture [2]. Bone is a dynamic organ that is constantly remodelling in response to the stresses of living. During ageing, gradual bone loss occurs from the relative increase of bone resorbing osteoclast activity to new bone-forming osteoblast activity. Due to the increased osteoclast activity relative to osteoblast activity, there is a deficit in new bone formation and this deficit compounds over time leading to skeletal fragility [3]. Clinically, osteoporosis is diagnosed either from sustaining a minimal trauma fracture (that is a fracture occurring following a fall from a standing height or from a minimal force applied such as bumping into a desk) or if the T score of bone mineral density (BMD) test from scans of the hip and lumbar spine by dual-energy x-ray absorptiometry (DXA) fall below − 2.5 (Fig. 1). In the context of BMD testing, a T score is a statistical measure of the relative distance of measured BMD from the BMD of a young, healthy person. T scores between − 2.5 and − 1.0 are considered to be osteopenic, that is, evidence of bone loss but not severe enough to greatly increase fracture risk but closer monitoring is warranted. Scores above − 1.0 are considered “normal” or “ideal”.

Classification of bone mineral density based on T score
In Australia, recent data suggest that approximately 2% of men and approximately 10% of women have osteoporosis based on hip BMD [4]. Considering the lumbar spine as well (hip or spine DXA-derived BMD), these numbers increased to 6% in women and 22% in men. Historical data has shown that in Australia, the (age-adjusted) rate of hip fractures in women is 25 per 10,000 person-years and 9 in men; similarly, the rate of vertebral fractures is 19 per 10,000 person-years and 7 in men [5]. Overall, this represents a substantial disease burden, and indeed in Australia, it is estimated that hip fractures cost an average of $23,000, and potentially over $33,000 in health care expenditure [6]. These data are replicated globally, where large-scale analyses have shown that there are approximately 3.5 million new fragility fractures every year in the European Union costing €37 billion annually and there are similar trends in the USA [7, 8].
Management
Management of osteoporosis usually begins with preventative strategies such as addressing modifiable lifestyle elements through promoting more physical activity and adequate intakes of calcium and vitamin D. Calcium and vitamin D have been shown to have only modest benefits in terms of increasing bone mass and reducing fracture rates [9]. They are nonetheless the standard, first-line recommendation for individuals with concerns over their bone health largely due to their low cost and proven safety, though calcium alone (either through supplements or through dietary means) has raised concerns about the potential for increased cardiovascular events [10, 11]. Pharmacological interventions to treat bone loss broadly fall into two categories: anti-resorptives and anabolics. Anti-resorptive medications seek to shift the bone resorption-formation balance toward more formation by inhibiting elements of bone resorption. There are two main types of anti-resorptives: bisphosphonates and denosumab. Bisphosphonates are a class of compounds so-named as the basic structure of the chemical has two phosphate groups and two substituents and combinations of which distinguishes the bisphosphonate type [12]. Bisphosphonates produce their anti-resorptive effects by binding, through the two phosphate groups, with high affinity to bone matrix which is rich in calcium. Bisphosphonates are structurally similar to pyrophosphate, the mineralised constituent of bone matrix. Bisphosphonates are taken up by osteoclasts as part of the resorption process and disrupt the enzyme farnesyl diphosphate synthase in the HMG-CoA reductase pathway (also known as the mevalonate pathway) in the osteoclast causing cell apoptosis from an inability to transcribe proteins necessary for cell membrane formation [13]. Denosumab is a human antibody to the receptor activator of nuclear factor kappa-β ligand (or RANKL). RANKL binds to receptor activator of nuclear factor kappa-β (RANK) on the surface progenitor cells of osteoclasts (pre-osteoclasts) promoting their differentiation into mature osteoclasts. Inhibition of the process restricts osteoclast maturation and ultimately reduces bone resorption [14]. Thus, denosumab mimics the action of osteoprotegerin (OPG), a natural inhibitor of RANK-RANKL interactions. The other category of osteoporosis medications is anabolic therapies which seek to promote bone formation. Teriparatide is a recombinant protein of human parathyroid hormone (PTH) consisting of the first 34 amino acids of the N-terminus (the active part of the compound). Teriparatide works by transiently stimulating osteoblast activity by increasing serum calcium concentration [15, 16]. Chronically elevated calcium levels (typically seen in cases of hyperparathyroidism) result in reduced BMD; thus, teriparatide is administered intermittently to stimulate the inhibition of osteoblast apoptosis. Strontium ranelate is another anabolic agent and is unique in that it has the dual-action of also inhibiting bone resorption [17]. Strontium ranelate’s mechanism of action is to stimulate calcium sensing receptors leading to the maturation of pre-osteoblasts to osteoblasts capable of bone formation. It also stimulates osteoblasts to secrete OPG which inhibits osteoclast maturation as outlined above.
Risk Factors for Osteoporosis and Relationship with Cardiovascular Disease and Mortality
The risk factors for osteoporosis include a family history of fracture, advanced age, history of falls, smoking, inadequate calcium and vitamin D intake, low amounts of physical activity and alcohol consumption [18]. These risk factors and their contribution to fracture risk have been incorporated into risk assessment tools such as FRAX® and the Garvan Fracture Risk Calculator [19, 20]. Interestingly, obesity (commonly co-exiting with these risk factors) is not a classic risk factor for osteoporosis and indeed seems “protective” against low bone density. The traditional explanation for this is that as bone is a dynamic organ, its cells are responsive to loading and other stresses from the environment [21]. In the context of obesity, the increased weight of the individual will create increased loading on the bones which over time translates into a higher bone density. Bone density is not the only feature of bone strength. Indeed, the bone microarchitecture of obese individuals is compromised and this has been demonstrated clinically and pre-clinically [22, 23]. Importantly, the majority of fractures in the population actually occur in overweight or obese individuals [24]. Obesity increases the risk of falls, mobility limitation and functional impairment, which may contribute to the risk for loss of bone density and increased fractures. Often, this is attributable to the phenomenon whereby as we age, muscle tissue is steadily lost and fat tends to accumulate (within and between the muscle fibres known as inter-intramuscular adipose tissue) such that relatively, the proportion of fat to muscle in the limbs increases with age [25]. Therefore, the remaining muscle mass may be insufficient to move the obese individual’s frame. Previous literature has demonstrated that in community-dwelling older adults, increasing visceral fat percentage and total body fat percentage was negatively correlated with muscle density (a proxy measure of inter-intramuscular adipose tissue) [26]. That is to say, the more adipose tissue in the body, the more appears in the muscle (lower muscle density is indicative of higher amounts of fat accumulation). Additionally, in another sample of community-dwelling older adults, calf-muscle density positively correlated with cortical volumetric BMD in the proximal tibia (an area rich in cortical bone) in men and was positively associated with cortical volumetric BMD and cortical area in the proximal tibia in obese women after multivariable adjustment [27]. That is to say, with increasing muscle density (hence less fat infiltration), there is greater amounts of cortical bone mass. It is therefore important to also consider obesity as a risk factor for poor bone health. If obesity is incorporated into the milieu of factors predisposing to osteoporosis and fracture, this consequently means that the majority of risk factors predisposing to cardiovascular, and in particular vascular diseases, are also risk factors for osteoporosis (Fig. 2). It is unsurprising then that much epidemiological evidence supports a shared bone-vascular disease axis. Numerous observational studies have shown that low bone density can predict cardiovascular disease and events [28, 29]. Equally, the risk of falls and fractures is increased in individuals with high blood pressure and elevated central stiffness [30]. Given this seemingly bi-directional relationship, it is difficult to determine what is the cause and what is the effect. Taking a life-course approach to understanding the development of osteoporosis and vascular disease would point to auxiliary factors that influence both bone heath and vascular health potentially earlier in life that may help explain their co-existence in older age. During ageing, and particularly in the growth phase of life, muscle mass and function are a crucial determinant of bone mass and strength and are also associated with a more favourable cardiovascular health profile [31]. Thus, understanding the relationship between the musculature and the vasculature may help unlock critical new pathways to further our knowledge of the bone-vascular axis (Table 1).

Shared and unique risk factors of osteoporosis and vascular disease
Sarcopenia
Definition
Sarcopenia describes the progressive loss of muscle mass and function leading to eventual frailty and increased risk of falls and fracture [40]. Previously, it was thought that the onset of physical frailty was an inevitable consequence of ageing. As life expectancy increases, particularly in developed countries, increasing attention has been paid to increasing the quality of life in older age as we are now living a substantial portion of our lives in this age group. One of the areas to address this need is in maintaining good physical function in older age. Foundational to this strategy is to preserve muscle mass and quality during ageing, where like bone, the intention is to capitalise on the growth phase of life to achieve the highest peak muscle mass achievable. As such, our understanding of the impact of age-related decline in muscle mass and quality has greatly increased. There are currently a number of “consensus” definitions for sarcopenia, but despite this heterogeneity, sarcopenia is recognised as a robust predictor of falls and mobility limitation [41]. The European Working Group on Sarcopenia in Old People (EWGSOP) has gained preference in the field and using this definition, prevalence of sarcopenia in older adults has ranged from 5 to 22%, where age and ethnicity significantly impact on disease prevalence [42, 43]. Recently, sarcopenia was given an ICD-10 code (M62.84) which many believe will facilitate recognition by caregivers and the uptake of intervention strategies. The development of sarcopenia is complex, but it is understood to involve a range of factors including sedentary lifestyle and micronutrient deficiencies such as vitamin D, amino acids and also macronutrients such as proteins which are required to maintain muscle tone and growth [44]. At the molecular level, the gradual loss in the regenerative capacity of muscle tissue is one of the better described pathways of muscle loss. Increased oxidative stress and chronic inflammation contribute to the differentiation of regenerative satellite cells into non-contractile, non-functional adipose tissue [45]. Recent data has suggested that this intra-/intermuscular adipose tissue has utility in predicting non-skeletal outcomes such as cardiovascular disease and is independently associated with inflammation [46, 47].
Management
Muscle, like bone, is a dynamic organ and responds to use and disuse. As such, the most effective strategy to limit the loss of muscle mass and strength is through exercise training. Exercise has proven effective across all ages throughout the life-course including in the very old and frail. Whilst bone responds most favourably to loading or impact type activities, most forms of physical activity (aerobic, resistance or weight training, incidental activities) appear to have positive effects on the musculature [48]. In older adults, walking is predominant as the most common source of physical activity. It has been observed that individuals reporting the highest amounts of physical activity also have greater amounts of lean mass and muscular strength [49]. Consequently, interventional studies involving aerobic, resistance and multimodal (combination of different forms exercise into the one regime) have proven benefits in improving muscle mass and function, even in the very old at high fracture risk [50, 51].
Nutrient consumption is also an important contributor to muscle mass and strength. Whilst total caloric consumption does not necessarily decline with advanced age, it is the composition of that consumption that contributes to muscle loss [52]. Importantly, protein represents an increasingly smaller proportion of daily dietary intakes as one ages. Protein provides the necessary amino acids required for building new muscle and it has been shown cross-sectionally that low protein consumption is associated with lower muscle mass in older age [53, 54]. Additionally, calcium and vitamin D are critical for muscle function. Calcium is required for neuromuscular transmission and vitamin D has a multitude of functions including, primarily, supporting calcium uptake and also has roles in muscle tone [55, 56]. Calcium and vitamin D intake also appear to decline with advancing age further contributing to musculoskeletal decline and indeed sarcopenia is highly prevalent in individuals with low intakes of these micronutrients [57]. Interventional studies have demonstrated that protein supplementation may be able to support muscle mass increases in the elderly and that calcium and/or vitamin D supplementation may also have a positive effect on sarcopenia [58, 59]. Indeed, combining these nutrients, for example, protein supplementation combined with vitamin D supplementation, has resulted in significant gains in skeletal muscle mass, muscle strength and some markers of inflammation in sarcopenic older adults [60, 61]. There is also some evidence to suggest that that combining nutrient supplementation with an exercise component can result in beneficial effects on sarcopenia [62, 63].
Relationship Between Sarcopenia and Cardiovascular Disease and Mortality
As muscle and bone share an intimate relationship, many of the risk factors linking bone and vascular diseases are similarly shared between sarcopenia and vascular disease [64]. Furthermore, similar to the relationship between osteoporosis and cardiovascular disease, much epidemiological evidence has demonstrated that sarcopenia and the components of sarcopenia individually are associated with cardiovascular disease and mortality. For example, in a cohort of 4252 community-dwelling older men, sarcopenia was associated with increased risk of mortality [hazard ratio (HR): 1.41; 95% confidence interval (CI) = 1.22–1.63] and those who were obese also had increased risk of mortality [1.21; 1.03–1.42]. Interestingly, there appeared to be a synergistic effect of the coincidence of sarcopenia and obesity on mortality with an approximately 72% increased risk of all-cause mortality [1.35–2.18] [65]. Other longitudinal studies have demonstrated that in older adults [n = 1512], the risk of cardiovascular mortality was greatest in those with low skeletal muscle mass [2.16: 1.51–3.08] [66]. These data indicate that muscle (as well as bone explored above) has independent effects on the development of cardiovascular diseases which would point to shared mechanisms (mechanisms common to both muscle and bone health and disease may be important for cardiovascular disease). Furthermore, nominal strategies that address osteoporosis and sarcopenia such as improving bone and muscle mass through exercise and micronutrient supplementation also have well-established profound effects on the cardiovascular system. This then leads one to speculate that we may be observing related processes: that there exists pathobiological links between the development of cardiovascular diseases, osteoporosis and sarcopenia. By understanding the commonalities between the skeletal, muscular and vascular systems, we may be able to identify common pathways which lead to diseases in all three systems and thus be able to develop a strategy to target these pathways and thus all three systems. In designing these strategies, it is important also to look at the nature of cardiovascular disease in older age as, generally, a large proportion of these cardiovascular disease effects the function and structure of blood vessels, are usually the result of chronic lifetime exposure to risk factors that compound to eventually manifest in older age.
Part 2: Diseases of Blood Vessels
What Is Vessel Disease?
Definition
The vasculature is a complex network of conduit vessels. The structure (including size, shape and cellular architecture) of each vessel ultimately governs its purpose and function. Thus, diseases that affect the structure and function of blood vessels are inherently detrimental to human health. Indeed, vessel disease is the third largest cause of non-coronary cardiovascular deaths in Australia totalling 4085 of the 17,426 (~ 23%) of non-coronary deaths in Australia in 2018 [67, 68]. Vascular disease can be broadly defined as diseases that alter the micro- and macro-anatomical composition of the vessel affecting its usual function and performance. Principally, the ageing aorta suffers two main fates: the vessel can become calcified and stiffen over time or the vessel walls can become weakened, expand (aneurysm) and eventually rupture. Indeed, the aorta is one of the first sites in which these structural and functional changes occur. Understanding of the fundamental biology and anatomy of the aorta is important if one is to understand the potential links of vascular disease to musculoskeletal diseases in ageing.
Anatomy and Biology of the Aorta
The aorta serves two important cardio-physiological functions: one as a major conduit for blood flow and another as a buffer against large transient increases in pressure created during systole, also known as the Windkessel effect. The Windkessel function of the aorta ensures smoothing of arterial pressure throughout the cardiac cycle. The aorta itself is muscular, has a normal diameter of around 30 mm and has its own blood supply. The structure of the aortic wall comprises three distinct layers (Fig. 3) [69]. The innermost layer (tunica intima) is a single-cell thick lining of endothelial cells that are highly responsive to stimuli including circulating factors and also the sheer stresses from blood flow. The middle layer (tunica media) is the thickest layer of the aortic wall. The predominant cell type in this layer is the vascular smooth muscle cells (VSMC). These are a highly specialised cell type that are capable of contraction as necessary during systole and have the ability to trans-differentiate into a proliferative and osteochondrogenic phenotype [70,71,72]. Interlaced between the VSMC are fibres of elastin and collagen. Elastin lamellae are concentrically arranged and are visible under light microscope, offering the aorta elastic abilities, stretching and relaxing according to tension and compression stresses throughout the cardiac cycle. Collagen fibres are arranged transversely along the length of the aorta providing structural integrity to the vessel and experience high tensional stress during diastole to promote blood flow. The outermost layer (tunica adventitia or externa) is mainly composed of collagen supported by some elastin fibres. Its main function is to anchor the vessel to surrounding structures. Importantly, the elastin-collagen ratio changes along the length of the aorta [73]. There is a higher elastin content in more proximal regions of the aorta and the content of collagen progressively increases in the more distal and descending regions of the aorta. This is significant because given the differences in elastic potential across the aorta, some regions are more susceptible to vascular damage than others [74].

Layered structure of an artery
Fate of the Ageing Aorta: Vascular Calcification and Stiffness
Of the fates of the ageing aorta, calcification is more a common disease than aneurysm; it is prevalent in up to 60% of healthy older individuals whereas the prevalence of aortic aneurysm is approximately 5% of older adults [75, 76]. Vascular calcification is a macro-anatomical disease of blood vessels that has a long history in humans. In the Tyrolean Ice Mummy discovered in northern Italy, an autopsy of the remains detected substantial calcium deposits in the abdominal aorta [77]. The remains are thought to be close to 5300 years old. Vascular calcification is considered to be a multifaceted disease, but in general is defined by the layer in which the calcification appears. Intimal calcification, occurring in the intimal layer of the aortic wall, is associated with atherosclerosis which is characterised by lipid-laden plaques that have become deposited in the aortic wall and surrounded by a highly inflammatory environment including leukocytes and other immune cells; predominantly macrophages [78]. This type of calcification represents the prototypical lesion to which the majority of historical investigation of calcified arteries has been dedicated. The calcification is phenotypically eccentric and occludes the lumen. Medial calcification, sometimes also referred to as Mönckeberg’s syndrome or medial calcific sclerosis, occurs in the medial layer of the aorta and this type of calcification is driven largely by VSMC deposition of mineral in the wall [79]. The calcification present is usually concentric and associated with inflammation in the adventitia. Site is significant when it comes to vascular calcification. Calcification of large, central arteries and smaller, peripheral arteries is associated with different disease states, but generally, calcification in larger vessels is indicative of calcification in smaller vessels [80]. Calcification in the abdominal aorta is one of the first sites in which calcification develops and thus may act as a sentinel site for more generalised vascular disease [81]. As such, a majority of the discussion in this review will be dedicated to the understanding of abdominal aortic calcification (AAC).
The abdominal aorta is unique in its biology. As alluded above, the higher collagen to elastin content in this region of the vessel renders it susceptible to vascular injury. Localised disruption in the aortic wall from collagen and/or elastin degradation which is mediated by resident matrix metalloproteinases 2 and 9 can weaken the vessel wall and promote dilatation and aneurysm development [82]. Degradation of aortic wall products promotes macrophage (and other leukocytes) homing at this site to clear potentially immune-reactive particles [83]. Ineffective clearance of these products (apoptotic bodies) appears to be a preferential site for nucleation of calcium-phosphate crystals by pro-inflammatory macrophages. More generalised damage such as the cross-linking of collagen fibres which seems to occur with chronic hyperglycemia or as part of a chronic immune process can stiffen the aorta (arteriosclerosis) compromising the aorta’s Windkessel function. Bioengineering studies have demonstrated that increased collagen content and the presence and composition of fibrotic/atherosclerotic lesions alter (increases) the strain modulus of blood vessels [84]. It naturally follows that calcification in and on the aortic wall would result in a loss of compliance (stiffening), resulting in increased pressure retention throughout the vasculature manifesting as elevated systemic blood pressure (hypertension). Ultimately, this may raise blood pressure and contribute to ventricular hypertrophy which has profound implications for cardiac risk [85, 86]. Therefore, calcification in the aorta may be seen as a cause of adverse haemodynamics resulting in elevated cardiovascular risk.
Stiffening may also precede calcification. Stiffness may directly impact on blood flow characteristics and affect target organs and tissues which, over time, result in the deposition and accumulation of ectopic minerals to fortify damaged vessels leading to calcification [87]. This theory would suggest that aortic calcification may in fact be a consequence of adverse haemodynamics. Increases in systemic blood pressure may expose endothelial cells, which are highly mechanosensitive, to excess or abnormal wall sheer stresses leading to their dysfunction (characterised by increased cell surface expression of integrins and other chemotactic markers) [88, 89]. Sustained excess sheer stresses on the endothelium may result in vascular injury which is the key trigger for promoting an immune-inflammatory response. Part of this response is to repair and remodel the aortic wall which may involve degradation of the elastin and collagen fibres exposing previously intracellular/intramural substances to the circulating immune system. The result of this is further inflammation and cellular infiltration and ultimately plaque formation. Macrophages are a key cellular infiltrate and as aforementioned, ineffective clearance of apoptotic bodies can trigger calcium-phosphate crystal formation. Generally, calcification of plaques is viewed as an attempt by the body to protect or stabilise the plaque and ‘hide’ potentially immunoreactive substances under a robust layer of fibrotic/calcific cap [90]. Given this pathobiological background, there appears a number of commonalities as well as differences between these two related vascular features, calcification and stiffness, and these are summarised below (Table 2).
Imaging and Quantifying Calcification and Arterial Stiffness
Investigation of aortic disease is part of routine clinical practice for those at high vascular risk. Imaging of the aorta can be achieved through a number of modalities including magnetic resonance imaging (MRI), quantitative computed tomography (qCT), lateral spine radiography and lateral spine densitometry. Functional assessment of the aorta can be achieved by functional MRI (fMRI), fludeoxyglucose F18 (18F-FDG) positron emission tomography (PET) or more commonly, Doppler ultrasound. Doppler ultrasound imaging is inexpensive and can be utilised in a variety of vascular regions including the abdominal aorta, it has the advantage of being able to view vessel with and without evidence of plaque. Recently however, densitometry is gaining popularity as the imaging modality of choice due to the low radiation exposure and rapid image acquisition and the opportunity to screening for cardiovascular disease at the time of bone density and vertebral fracture assessment [91]. Next-generation densitometric devices have a lateral enable function which allows image acquisition with a patient lying in a supine position facilitating image acquisition in older individuals and individuals who may have difficulty in laying in a foetal position. Once detected, a semi-quantitative scoring system is used to quantify the extent of calcification. The most commonly used scoring system is the AAC24 where scores can range from 0 to 24 [92]. This system involves examining the extent of calcific deposits on the anterior and posterior aspects of the aortic wall in the L1–L4 vertebrae (Fig. 4). In this technique, for each vertebral segment, calcified deposits are scored 0 for no evidence of calcification; 1—if one-third or less of the wall is calcified, 2—between one-third and two-thirds of the walls are calcified and 3—more than two-thirds of the walls are calcified. The scores for the anterior and posterior walls are summed meaning each vertebral segment contributes a maximum of score of six to the final score [92]. The final aortic calcification score (ACS) is a composite of all four vertebral segments ranging from a minimum “0” to a maximum of “24”. Scores are non-normally distributed, and thus, ACS is considered a non-continuous outcome. It is important to consider the severity of calcification. Based on the ACS, severity of calcification is defined according to three groupings: “no calcification” (ACS = 0), “moderate calcification” (ACS between 1 and 5) and “severe calcification” (ACS ≥ 6).

AAC24 scoring system
Vascular calcification adversely affects the elastic properties of blood vessels and though the stiffening of vessels cannot be seen, it can be quantified. There are a number of parameters which serve as useful estimates of vessel stiffness. Pulse pressure (PP) is the difference between the systolic (pressure at ventricular contraction) and the diastolic (pressure between contractions) blood pressure. A narrow PP usually reflects central vascular stiffness most likely related to calcification of the aorta. This can be measured from a standard office blood pressure monitor (sphygmomanometer). Pulse wave velocity (PWV) is the composite of the forward pressure wave created by ventricular contraction and a reflected wave from a distal site (Fig. 5) [93]. The gold standard measurement of arterial stiffness is the carotid–femoral PWV determined by tonometry or by intra-aorta catheter. This is estimated using the foot-to-foot velocity method whereby transcutaneously, the right common carotid artery and the right femoral artery and the time delay (or transit time, Dt) are measured (in seconds, s) between the feet of the two waveforms. A variety of different waveforms can be used including Doppler, pressure and distension. The distance (L) covered by the waves between these two sites is measured (in metres, m), and PWV is then calculated as 1/4 L/Dt with the unit m/s. The less compliant the arteries, the faster the reflected wave returns augmenting systolic pressure interpreted as an increased PWV. The extent of this augmentation in systolic pressure is called the augmentation index (AI). These measures of arterial stiffness can also be quantified using oscillometric devices which have shown strong agreement with classic techniques [94].

Arterial stiffness: example wave forms explaining derivation of PWV (left) and AI (right)—adapted from Laurent et al. (2006) [95]
Cardiovascular Risk Related to Vascular Calcification and Stiffness
Vascular calcification and stiffness are well-established markers of cardiovascular disease and can both independently predict future cardiovascular events and mortality [93, 96, 97]. Whilst this finding has been demonstrated in multiple populations, our understanding of how these vascular features impose an increased risk is not precisely understood. It is generally accepted that calcification and stiffness place an extra burden on the heart as it must work harder to overcome peripheral resistance. Overtime, remodelling of the heart to cope with chronic over-burden increases the risk of adverse cardiac events. Several studies exist which support this understanding (Table 3). A study in older men with high cardiac risk but preserved ejection fraction, calcification in both the thoracic and abdominal aorta was associated with a number of echocardiographic parameters and the total aortic calcification was positively associated higher left ventricular mass [85]. This finding supports the view that calcification in the large vessels, particularly more central vessels, create extra peripheral resistance for which the heart has to work against in order to maintain adequate supply and tissue perfusion. Though, other studies investigating calcification in coronary arteries have not shown this suggesting that calcification may have a site-specific effect on left ventricular mass [98]. This may be explained by the aorta receiving a large volume of blood and pressure direct from the heart during each cardiac cycle and as such stiffness in the aorta would have a more pronounced effect on overall cardiac work. It would be informative to understand if other conditions of ageing associated with increased cardiovascular risk and mortality (particularly those that appear to co-exist with vascular calcification and stiffness) if they share similar associations with cardiac remodelling and altered function or performance.
Part 3: Associations of Osteoporosis with Vascular Calcification and Stiffness
Mechanisms
Bone loss is a feature of ageing and commonly co-exists with vascular calcification in elderly populations [99]. Epidemiological evidence has pointed towards a potential shared development [100]. Recently, much evidence has come to light that the factors known to influence bone mass have direct roles in the development of calcification.
Wingless-related integration site (Wnt)/beta (β)-catenin signalling is a central pathway of bone formation. Wnt and β-catenin are functionally connected elements of a signalling cascade that is necessary for the commencement of osteoblast differentiation and proper bone formation through regulating gene expression, most notably the RunX2 gene [101, 102]. Conditional knockdown of these elements or upregulation in factors known to suppress these signalling elements can result in ectopic bone formation or mineralisation of tissue and this has been demonstrated in vitro and in vivo [103,104,105,106]. As such, there is biological and clinical interest into how Wnt/β-catenin signalling elements including its regulators and suppressors may contribute to the development of vascular calcification given the bone-like appearance and features of vascular calcification.
The differentiation of vascular smooth muscle cells (VSMCs) into bone-like cells capable of laying down bone matrix is a key mechanism of vascular calcification. VSMCs are the cellular components of the normal blood vessel wall. These cells provide structural integrity and can also regulate the diameter of the vessels by contracting and relaxing dynamically in response to vasoactive stimuli. VSMC are unique in that given the appropriate signals they can undergo trans-differentiation into a cell that displays surface markers such as RANK and release factors normally attributable to bone cells such as RANKL, osteocalcin and alkaline phosphatase [71, 107, 108]. Wnt/β-catenin signalling is important in the process of VSMC trans-differentiation. Wnt/β-catenin signalling can promote osteogenesis by directly stimulating Runx2 gene expression and this process is seen in VSMCs of the arterial wall [109]. It has been demonstrated in a rodent model of vascular calcification that Runx2 expression is induced and β-catenin is activated by a high-phosphate environment [110]. Furthermore, a specific Wnt signalling molecule, Wnt3a, upregulated osteocalcin expression in VSMCs and promoted calcium deposition in the VSMCs. In cultured cells of the aortic tunica media of these rats, β-catenin was activated and Runx2 mRNA levels correlated with the abundance of β-catenin in the aortic walls. Whilst activation of β-catenin by Wnt induced Runx2 expression, inhibition of Wnt/β-catenin by natural inhibitors of Wnt signalling such as Dickkopf-1 (DKK-1) and sclerostin can attenuate Runx2 induction and thus limit the progression to calcification. Taken together, these observations demonstrate that Runx2 (from VSMC) may mediate the action of Wnt/β-catenin signalling in promoting vascular calcification (Fig. 6). Given the importance of these factors to bone development and skeletal maintenance over the life course, the relevance of bone loss to the development of vascular calcification is of clinical interest. Currently however, there are no clinical data evaluating directly the effect of modulating the Wnt signalling pathway in the context of vascular calcification. Trials examining the anti-sclerostin antibody, romosozumab, have demonstrated efficacy in terms of fracture risk reduction and BMD improvements [111, 112]. The trial in men revealed an increase in adjudicated cardiovascular events [n = 8/163 (4.9%) in romosozumab treated versus n = 2/81 (2.5%) in placebo-treated men]. This difference appeared to be driven by a significant difference in the number of cardiac ischaemic events in the romosozumab-treated men [n = 3/163 (1.8%) versus n = 0/81 (0%)] and given the nature of ischaemic events, likely atherosclerotic in origin, we can draw inferences about potentially pro-atherosclerotic effects on sclerostin inhibition. Despite lumbar radiography being conducted on trial participants, no data are yet available on the effect of romosozumab on aortic calcification [111].

Role of vitamin D, DKK-1 and VSMC in arterial calcification and remodelling. Low vitamin D is common in the elderly and more pronounced with increasing adiposity. Wnt signalling inhibitors are vitamin D-regulated elements, and thus without natural inhibitors, Wnt is allowed to signal. Intracellularly, VSMC increased the expression of the gene RunX2 which elicits a multitude of effects including increasing calcium efflux from the cell, increasing expression of RANK and increasing the secretion of osteocalcin. The sum total of this process is that the VSMC undergoes trans-differentiation into a bone-like cell capable of laying down bone matrix promoting arterial remodelling and eventual calcification of the vessel
Epidemiological Insights into the Bone-Vascular Axis
These observations have been supported in human studies. In a cross-sectional analysis of elderly women who took part in a randomised controlled trial of calcium supplementation for fracture reduction, circulating levels of the Wnt antagonist DKK-1 were evaluated [113]. The study cohort was stratified into quartiles of circulating DKK-1 concentration and the proportion of women with severe AAC on lateral spine radiographs was lowest in the highest quartile of DKK-1, indicating an inverse relationship. Additionally, compared to the highest quartile of DKK-1, the lowest and the second lowest quartiles had approximately two-fold increased likelihood of having any AAC and near two-fold increased likelihood of severe AAC. These associations were independent of important cardiovascular risk factors including previous hospitalisation for vascular disease, renal function and anti-hypertension medication use. In another study of post-menopausal women, DKK-1 was inversely associated with AAC on a 24-point scoring scale, and women with a score of one or less (indicating low or no AAC) had significantly less circulating DKK-1. Additionally, women with high pulse wave velocity (a measure of arterial stiffness) had higher levels of sclerostin [114]. Sclerostin is encoded by the SOST gene and is produced by osteocytes. Sclerostin has profound anti-anabolic effects on bone formation through inhibition of elements of the Wnt signalling pathway including low-density lipoprotein receptor-related proteins-4, -5 and -6. In a murine model of aortic aneurysm and atherosclerosis, transgenic mice for the SOST gene were protected from plaque development and the aortae showed differential expression of proteins known to be involved in the degradation of blood vessel matrix [115]. These observations demonstrate that established modulators of Wnt signalling may protect against the progression of calcification possibly by inhibiting the upregulation of β-catenin activity. This clinical and pre-clinical evidence supports a role for regulators of bone mineral metabolism in the development and progression of vascular calcification and stiffness.
DKK-1 and sclerostin are vitamin D-regulated genes, and supplementation appears to modify their serum levels. Therefore, vitamin D may be a clinical link between bone metabolism and vascular calcification and stiffness (Fig. 6). A small sample of young individuals (average age 32 years) with vitamin D deficiency (25-hydroxyvitamin D concentration < 20 ng/mL) received three once-monthly intramuscular injections of 300,000 IU of vitamin D. It was observed that there was a statistically significant difference between pre-treatment and post-treatment values in 25(OH)D levels, parathyroid hormone (PTH) and sclerostin levels demonstrating their responsiveness to vitamin D [116]. In a similar study of older individuals (mean age 61 years), a single intramuscular dose of 300,000 IU of vitamin D increased serum levels of DKK-1 and sclerostin and the changes in sclerostin were maintained after 3 months. Further in regression analysis, only the change in serum vitamin D was found to be a significant predictor of serum DDK-1 levels at 3 months [117].This would suggest that that vitamin D deficiency may be implicated in adversely affecting the Wnt/β-catenin signalling pathways which consequently provides the conditions for VSMC differentiation and calcification. However, it has yet to be conclusively demonstrated that vitamin D supplementation can modify the potential vascular effects of Wnt/β-catenin signalling pathways including, ultimately, vascular calcification. Given the chronic nature of vascular calcification, it may be unfeasible to test the effect of vitamin D supplementation in a randomised setting. Thus, much literature has been devoted to understanding the effects of vitamin D supplementation on surrogate markers of calcification including endothelial function and arterial stiffness. Several randomised trials have examined the effects of vitamin D supplementation on markers of arterial stiffness across multiple populations including type 2 diabetes [118], hypertension [119] and chronic kidney disease [120] coincidently all these co-morbidities are populations in which aortic calcification is highly prevalent (Table 2). These clinical data have provided inconsistent evidence, and thus, the utility, from a clinical and public health perspective, of vitamin D supplementation in improving vascular disease is unknown. Synthesis of this evidence would be of enormous benefit in terms of determining what factor contributed to null/negative findings such as dosing strength and length of supplementation as well as identifying the particular patient groups which may likely benefit most from a specific supplementation regimen. Given the majority of these studies did not enrol individuals with known vitamin D deficiency/insufficiency, future trials specifically recruiting these individuals are needed. Vitamin D supplementation in non-vitamin D-deficient populations has proved futile whereas supplementation in deficient populations has led to improved outcomes [121].
The relationships between DDK-1, sclerostin and vitamin D with calcification provide mechanistic insight supporting observations linking low bone mineral density (BMD) and fractures with adverse cardiovascular disease outcomes. In a study of 3676 healthy older women, it was demonstrated that yearly BMD loss at the femoral neck was greatest in those in the highest quartile of systolic blood pressure (SBP) [36] and in another study of 3151 healthy older men and women, hypertension was more prevalent in those who had a history of hip fracture (31.3%) compared to those who had not previously fractured (22.5%). In multivariable regression, previous wrist fractures were associated with increased likelihood of hypertension (odds ratio (OR) = 1.48; 95% confidence interval [CI] = 1.10, 1.99) though hip fractures were marginally non-significant (1.19; 0.98, 1.45) [29]. These data support our understanding of a shared relationship between bone and vascular disease; however, the direction of the relationship (if bone loss precedes vascular disease or vascular disease promotes bone loss) is not understood. Indeed, there appears to be bi-directional relationships between osteoporosis, fracture and cardiovascular disease (Table 1). In a large study of all 31,936 twins born in Sweden between 1914 and 1994, it was shown that cardiovascular disease increases the risk of fractures. In multivariable models, the hazard ratio (HR) for hip fracture after a diagnosis of heart failure was 4.40 (3.43,5.63); after a stroke, 5.09 (4.18,6.20); after a diagnosis of peripheral atherosclerosis, 3.20 (2.28,4.50); and after an ischemic heart disease event, 2.32 (1.91,2.84) [30]. This finding was replicated in men, where in a large cohort study of 113,600 individuals including 60,637 men, atrial fibrillation was associated with a near doubled risk of incident fractures [35]. Given the orthodox understanding that loss of bone mass occurs concomitantly with the development and progression of aortic calcification and considering the potential impact of aortic calcification on cardiac function leading to cardiovascular events, it then follows that low bone mass would be associated with poor cardiac function. This has yet to be shown clinically and would enhance our understanding of why cardiovascular diseases are highly prevalent in individuals with low bone mass and in those who have fractured.
If We Stop Bone Loss, Can We Reduce Vascular Disease?
Although negative media attention about the risks of osteoporosis treatment, prescriptions for bisphosphonates in Australia remain high [122]. A meta-analysis of 61 trials examining the effects of bisphosphonates on any cardiovascular outcome identified two studies (totalling n = 152 patients) that included a measure of vascular calcification. Bisphosphonates were found to produce a significant reduction in vascular calcification (mean difference between treated and control = − 11.52 units [95% CI: − 16.51, − 6.52; p < 0.01; heterogeneity between studies: I2 = 13%]) [123]. Despite low statistical heterogeneity, the studies themselves were quite distinct; the larger of the two trials was conducted in 108 Japanese patients with hypercholesterolemia randomised into three treatment groups: atorvastatin (a cholesterol-lowering agent) combined with etidronate (a type of bisphosphonate) or either alone [124]. It was found that atorvastatin plus etidronate combination therapy for 12 months significantly reduced both thoracic and abdominal aortic plaques, whereas atorvastatin monotherapy reduced only thoracic aortic plaques and etidronate monotherapy reduced only abdominal aortic plaques. The preferential regression of plaques by etidronate in the abdominal aorta may be explained by the fact that thoracic plaque is more commonly associated with fatty streaks, whereas abdominal plaques are commonly more calcified. Indeed, in this study, calcified abdominal plaques were more prevalent than calcified thoracic plaques [28.7% vs. 13.9%, respectively]. The other trial involving bisphosphonates was conducted in a cohort of 50 individuals with chronic kidney disease stages 3–4 [125]. It was shown that after 18 months of treatment with alendronate (a type of bisphosphonate), there was no difference in the frequency of progression of vascular calcification between treated and placebo groups (change in Hounsfield units: − 24.2 [95% CI: − 77.0, 28.6; p = 0.4]). Given that these studies are relatively small and restricted to patient populations, further research is required to determine effects of bisphosphonates on cardiovascular health.
There is one study that has examined the effect of denosumab on aortic calcification in a randomised controlled trial design. This trial was a secondary evaluation of the larger “Fracture Reduction Evaluation of Denosumab in Osteoporosis Every 6 Months (FREEDOM)” study [126]. Over 7000 women aged 60–90 received denosumab or placebo every 6 months for 36 months. A subset of women with a mean age of 74 years at baseline [placebo: n = 501 and denosumab: n = 544] had lateral spine imaging evaluated for AAC. It was demonstrated that after 3 years of follow-up, the frequency of progression of AAC did not differ between denosumab [22% with AAC progression] or placebo [22%] groups [p = 0.98] [127]. This finding was consistent even when the analysis was restricted to those who had severe AAC at baseline (i.e. those most likely to provide evidence for AAC progression). In this restricted analysis, 15% showed progression in denosumab groups versus 19% in placebo-treated group. Outside of randomised studies, there also exists a case report of a 57-year-old Japanese women with multiple myeloma and chronic kidney disease [stage not reported] who was treated for 10 months with denosumab [128]. She developed rapidly progressive vascular and soft tissue calcification to which her physicians attributed to the large doses of vitamin D and calcium she was treated with to correct severe hypocalcaemia secondary to denosumab treatment. These reports, taken together, would thus cast doubt on the increasingly accepted bone-vascular axis. It appears that strategies with known effects on bone do not appear to influence vascular calcification biology. Thus, other strategies which have known effects on bone and vascular health need to be identified.
Part 4: Associations of Sarcopenia with Vascular Calcification and Stiffness
Mechanisms Potentially Linking Sarcopenia to Vascular Calcification and Stiffness
Many factors are thought to contribute to age-related declines in muscle mass and quality. Cellular and molecular triggers of muscle loss include myocyte apoptosis, alterations in muscle protein turnover, suboptimal utilisation of dietary amino acids for protein synthesis and impaired satellite cell function and regeneration [40]. These triggers are responsible for oxidative stress, mitochondrial dysfunction, inflammation, hormonal changes, fibre disorganisation and neuromuscular imbalances which can lead to the preferential loss of fast motor units. Disuse and generally low levels of physical activity (which are common in the elderly) are key contributors to these adverse changes in muscle biology [129]. Significantly, inflammation and increased oxidative stress can promote the differentiation of regenerative satellite cells into non-contractile non-functional adipose tissue [so called intra-/inter-muscular adipose tissue (IMAT)] [25]. A higher amount of IMAT therefore increases the load burden of remaining muscle and is independently associated with poor physical performance in older adults [45]. Also, obesity has been associated with inflammation, muscle triglyceride content and serum IL-17 levels as well as the development of early atherosclerosis which provides evidence for shared mechanisms between vascular calcification and muscle loss [130]. Interestingly, fat infiltration into skeletal muscle (one of the features of IMAT) also increases the risk of cardiovascular mortality, suggestive of an interplay between the musculature and vasculature [46] (Table 4). However, this study did not provide data on the type of cardiovascular mortality, and therefore, no comment can be made as to the potential association and involvement of IMAT (which may be indicative of a more generalised disease in skeletal muscle) and atherosclerotic vascular disease.
Epidemiological Insights into the Muscle-Vascular Axis
Epidemiological evidence suggests that low muscle mass and low muscle quality (poor strength and/or function) increase the risk of cardiovascular mortality [65, 66, 138, 139]. Despite this, there exists few clinical studies on a potential muscle-vascular disease relationship (Table 4). The first account of an association between muscle (lean) mass and vascular calcification was reported in a modest cohort [n = 168] of middle-aged to elderly men [131]. Body composition was determined by dual-energy x-ray absorptiometry [DXA] and aortic calcification was detected on lateral spine radiographs. It was shown that aortic calcification was negatively correlated with total body lean mass and there was an even more robust correlation with peripheral lean mass considering the peripheral limbs (otherwise described as appendicular lean mass, ALM). In multivariable regression analysis accounting for age, prevalent cardiovascular disease and serum total cholesterol, there was an inverse association between peripheral lean mass and aortic calcification severity (β = − 0.153, p = 0.049). This observation was subsequently supported by a large cross-sectional study of healthy adult men and women (mean age approximately 40 years) [136]. In this cohort, individuals in the lowest quintile of skeletal muscle mass had the greatest prevalence of coronary artery calcification (CAC) and greatest CAC score. On multivariable regression analysis, every standard deviation decrease in skeletal muscle mass was associated with an approximately 46% increased likelihood of having greater coronary calcium. This association was consistent in individuals with either high (score > 100) or low (score ≤ 100) coronary calcium score suggestive that muscle loss may be associated with both the early development and ongoing progression of disease. The amount of muscle mass is strongly determined by myostatin, a protein of the transforming growth factor-β superfamily [140]. Myostatin has also been implicated, pre-clinically, in glucose homeostasis and atherosclerotic/calcific plaque development by promoting mesenchymal stem cells to undergo osteogenic differentiation [141, 142]. Complementing the observation that low muscle mass is associated with more vascular calcification; in a large sample of middle-aged men, higher circulating levels of myostatin were associated with lower odds of having AAC, detected via lateral spine assessments on DXA, after adjustment for multiple risk factors [132]. This finding was consistent in a sub-group analysis of men aged over 60 and indeed AAC prevalence was lowest in the highest quartile of serum myostatin relative to the lower three tertiles combined. Interestingly, AAC prevalence was lowest in individuals in the lowest quartile of C-reactive protein (CRP) which is a biomarker of systemic inflammation. This possibly suggests that both increases in inflammation and a lack of muscle synthesis promoters play a role in aortic calcification. Overall, this study provided a potential biological explanation for a muscle-vascular disease association, though it is uncertain whether these factors causatively effect vascular calcification or that these complimentary observations are age-driven associations. Other epidemiological studies in cohorts of men and women have demonstrated null associations between abdominal muscle mass and calcification in various vascular beds including the ascending and descending aorta and coronary arteries [133].Therefore, there could be some age-specific effects not noted in earlier studies or, given the younger age of the cohort in question (approximately 64 years), any association between muscle mass and vascular calcification may be less pronounced. This hypothesis is supported by two studies conducted in institutionalised nonagenarians [134, 137]. Low muscle mass and fatty infiltration into muscle were characteristic features of the frail individuals in the cohort. Compared to robust (high functioning) individuals, frail individuals (low functioning) had greater CAC scores but this association did not reach statistical significance [134]. However, extra-coronary calcification was significantly higher in frail individuals suggesting that the association between muscle and vascular disease is more pronounced in older age [137]. Other evidence also points to factors relating to muscle function and quality (supporting the frail/robust findings) having an association with vessel disease (Table 5). No study has directly examined associations of measures of physical function with aortic calcification. There appears to be more consistent associations with vascular measurements in women than in men. In a multiethnic study of post-menopausal women, abdominal muscle at baseline was associated with longitudinal changes in CAC and this association differed by ethnicity [135]. Despite these observations, no study exists which directly measures muscle mass or muscle quality (strength or function) at a clinically relevant site such as peripheral limbs (important to physical function and independence) in both men and women and determines the association with AAC.
Role of Exercise in Modulating Muscle-Vascular Axis
Compared to the relative lack of efficacy of osteoporosis medications on vascular disease measures, strategies that address poor muscle mass and function have appeared to exert more favourable effects on vascular calcification and stiffness. There is extensive literature examining the role of exercise on sarcopenia and its components [150]. The consistent message is that power-training (or resistance or loading-type exercise) is the best strategy to combat sarcopenia as this type of intervention is sufficient to induce myogenesis in the elderly [151]. Equally, there is extensive literature examining the benefits of exercises on vascular function as measured through estimating arterial stiffness [152, 153]. Exercise is thought to deliver these vascular adaptions by augmenting NO-dependent vasodilation, which affects arterial function [154]. To date, no study has directly examined the effect of an exercise intervention, in older people on the progression or severity of AAC. This is despite studies determining direct links between muscle mass and aortic calcification [131].
Observational studies have supported interventional trials in that vascular health is better in individuals (including older adults) who engage in higher amounts of physical exertion (structured activities or incidental activities of daily living) [155]. Furthermore, observational studies have illustrated that important determinants of poor muscle mass and physical function also are independent risk factors for increased arterial stiffness (Table 5). For example, obesity has independently been associated with increased arterial stiffness and with sarcopenia in older adults [156]. Accordingly, a meta-analysis of weight loss trials has demonstrated that weight reduction can result in significant improvement in a number of arterial parameters [157]. The difficulty with weight loss is that although it may have profound benefits on the vascular system, it may lead to unfavourable musculoskeletal outcomes as is seen in patients who have had weight loss surgery [158]. Therefore, a more generalised strategy that targets all systems concurrently would be the ideal strategy to ensure vascular risk reduction whilst maintaining high musculoskeletal functioning. This strategy would thus target the shared risk factors of these conditions (Fig. 2). Currently, no trials exist examining the impact of lifestyle interventions which have benefits on multiple risk factors, including exercise programmes on advanced atherosclerotic vascular disease. Studies have, however, explored micronutrients such as calcium and vitamin D (critical in both the musculoskeletal and vascular systems) in their relation to vascular risk.
Part 5: Calcium-Vitamin D Links to Vascular Calcification and Stiffness
Vitamin D and calcium appear to have numerous roles in musculoskeletal ageing and vascular disease. As outlined above, vitamin D responsive genes are critical to osteogenesis and have a role in vascular calcification. Calcium salts (calcium phosphate, calcium carbonate) are the main mineral constituent of bone and appear in ectopic calcification as well. Mineral perturbations, such as those seen in chronic kidney disease and modelled in vitro in the setting of high circulating phosphate, are associated with increased mineralisation of vascular tissue. This has resulted in two major themes of clinical interest that have predominated the literature without resolution:
-
(i)
If vitamin D responsive elements are responsible for calcification in non-skeletal sites and low vitamin D (either deficiency or insufficiency) is highly prevalent in individuals with calcification, does supplementation of vitamin D result in a decrease in markers of calcification and vascular disease more generally?
-
(ii)
If calcium is the predominant component of calcification, do excessively high intakes of calcium (through dietary or supplemental sources) result in excess calcification and vascular disease more generally?
Vitamin D
Vitamin D is a pleiotropic steroid hormone whose primary action is to facilitate calcium uptake in the small intestine, fortifying the collagenous fibres. Vitamin D also has a number of non-skeletal effects that may favourably influence the cardiovascular system such as down-regulation of the renin-angiotensin system, enhancing insulin sensitivity and modulating inflammation [56]. The receptor for vitamin D exists on skeletal muscle myocytes and promotes protein synthesis and trophism by negatively regulating myostatin [159]. The vascular endothelium is also responsive to vitamin D. Endothelial cells express the vitamin D receptor and disruption in vitamin D signalling in the presence of calcium increases thrombogenesis, enhances VSMC proliferation and inflammation; and in cardiac myocytes, deletion of vitamin D may promote calcium absorption suggestive of a role in ectopic calcification [160, 161]. Importantly, vitamin D has a role in regulating the Wnt/β-catenin signalling pathway. Whilst these pre-clinical data support a role for vitamin D in the pathogenesis of vascular disease, the evidence for a clinical role for vitamin D supplementation in the treatment of vascular diseases is less clear. Hypovitaminosis D (defined as serum calcifediol/25-hydroxyvitamin D (25OHD) concentration below 50 nmol/L [= 20 ng/mL] as according to The Endocrine Society guidelines) has been the subject of intense investigation. Most prospective studies have reported moderate to strong inverse associations between vitamin D concentrations and cardiovascular diseases, serum lipid concentrations, inflammation, glucose metabolism disorders, weight gain, infectious diseases, multiple sclerosis, mood disorders, declining cognitive function, impaired physical functioning and all-cause mortality [121].
A large body of observational evidence supports a view that there is an inverse relationship between circulating vitamin D and cardiovascular disease and this has been reviewed elsewhere [162]. What has been less established is the therapeutic potential of vitamin D supplementation directly on vascular markers (e.g. calcification), but also indirectly on the underlying mechanisms (e.g. inflammation). This is despite the general inverse relationship between vitamin D and cardiovascular disease is consistently evident in specific disease states such as heart failure [163] and specific disease manifestations such as increased arterial stiffness [164]. Interventional studies have provided mixed results regarding supplemental effects on disease markers and underlying mechanisms. For example, in a randomised study of over 300 older adults, 12 months of oral 4000 international units (IU)/day of vitamin D3 did not produce any beneficial effects on blood pressure, heart rate or arterial stiffness (estimated by the augmentation index, stiffness index and reflection index—all markers of the arterial pressure waveform) [165]. Though, through a secondary analysis of another randomised controlled trial of vitamin D (monthly 100,000 IU) on cardiovascular outcomes including the arterial stiffness measures augmentation index (between group difference in change from baseline: − 5.7% [95%CI: − 10.8, − 0.6] and pulse wave velocity [− 0.3 m/s; − 0–0.6, − 0.1], it appears that the beneficial effects of vitamin D are limited to those who have underlying vitamin D deficiency [166]. This is consistent with pre-clinical evidence for the role of vitamin D in regulating genes involved in pathogenic pathways of vascular disease. Furthermore, vitamin D3 supplementation of daily 4000 IU for 12 months in older patients with clinical heart failure (an ejection fraction of less than 45%) improved cardiac function (overall approximate 6% improvement in ejection fraction [95%CI = 3.20–8.95]) and reversed left ventricular remodelling [167]. It was hypothesised that reductions in inflammation, which is a leading driver of cardiac remodelling and reduced cardiac function, was responsible for these improvements. Vitamin D has immunomodulatory effects, but it has yet to be conclusively demonstrated that vitamin D can improve the inflammatory status in individuals with known cardiovascular disease. Furthermore, given the potential beneficial effect of vitamin D on heart function, no trial to date has examined if vitamin D supplementation can affect the development and/or progression of vascular calcification, something which may precede clinical heart disease such as heart failure or elevated blood pressure.
Calcium
There is great biological and clinical debate surrounding the potential adverse cardiovascular effects of calcium including the role of circulating calcium and calcium intake from dietary and supplemental sources. Several systematic reviews have previously been dedicated to the topic [168, 169]. At the molecular level, calcium is required for the production of action potentials at the neuromuscular junction for muscle contraction and is an important intracellular signalling molecule and intercellular second messenger for downstream gene transcription. This is particularly relevant regarding the trans-differentiation of VSMC. Given the standard public health message for healthy ageing (which includes musculoskeletal and cardiovascular health) encourages adequate calcium intake from dietary sources, the following will be dedicated to understanding the effect of dietary calcium in vascular disease [170].
A recent meta-analysis proposed that increasing calcium intake through dietary sources is safe and tolerable [168]. Causing much controversary was the publication of a report from a large population study conducted in Sweden in postmenopausal women which concluded that those who consumed more than 1400 mg/day of calcium from dietary sources were at an increased risk of all-cause, cardiovascular and myocardial infarction mortality, but not stroke, sparking much commentary [171]. This finding went against conventional wisdom that higher calcium intake from dietary sources is reflective of a health-conscious lifestyle and favourable health outcomes [170]. More recent studies from other countries including Australia have reported contrasting findings to that of the highly publicised Swedish report indicating that there might be ethnic/regional specific effects of dietary calcium on cardiovascular and other mortality outcomes [172]. Furthermore, given postmenopausal accelerated bone loss, the main focus of bone preservation has focused on women and thus most studies (including the Swedish report) have enrolled only women. Therefore, more studies directly examining potential sex-specific effects of dietary calcium intake are needed.
It is assumed that underlying vessel diseases account for a large proportion of the cardiovascular deaths in these epidemiological studies. There is scant literature regarding the association of dietary calcium on markers of vascular disease. In the Multiethnic Study of Atherosclerosis, higher estimated dietary calcium intake was associated with a decreased risk for developing CAC [relative risk (RR) = 0.73(0.57–0.93)] though other studies have suggested there is no association [173, 174]. However, total calcium intake including from supplemental sources appeared to increase the risk for incident CAC [RR = 1.22(1.07–1.39)] [174]. This would suggest that there may be some adverse effect of calcium at the extremes of plausible intake levels, but this has yet to be shown regarding cardiovascular mortality or vascular disease markers including aortic calcification.
Part 6: Conclusion and Proposed Disease Model
This review explored three fundamental features of ageing: muscle loss, bone loss and vascular disease. There are several biologically and clinically plausible connections linking musculoskeletal decline and vascular disease and numerous key research questions are offered throughout this review highlighting areas for further development in our understanding of how these ageing aspects are potentially connected. These research questions can be answered in both interventional and observational settings. Summarising the available evidence, a conceptual disease model is proposed (Fig. 7).

Conceptual disease model. At the microsystemic level, inflammation and the action of small minerals and other micronutrients promote cellular stresses and damage which, if sustained, manifest at the macrosystemic level as tissue loss (bone and muscle) and evidence vascular damage (calcification). The development, progression and acceleration of these disease states may eventually manifest clinically (and indeed become obvious to the individual) evidenced by fractures, poor physical function/functional decline and an inability to perform activities of daily living as well as decreased cardiovascular health
In this model, central to the manifestations of ageing [poor physical function, adverse skeletal outcomes including fractures and overt cardiovascular disease including heart failure and cardiac events] is the bi-directional nature of the relationship between the musculoskeletal system and the vasculature. Factors that influence the fate of muscle mass and bone mass also appear to influence vessel disease—suggestive of shared biology and fate. The underlying disease state in the microenvironment has direct effects on the musculoskeletal system and the vasculature (the macrosystemic level) and high-quality randomised trials targeting these elements as primary end-points and linking them to hard outcomes are needed to firmly understand disease progression by confirming observational and pre-clinical evidence. Concerning the bone-vascular axis, given the relative lack of efficacy of bone-active medications on calcification there an opportunity for other interventions which target shared risk factors more generally. Calcification is a modifiable process, and this improvement through strategies with known effects on bone and muscle (and the risk factors for low bone and muscle) such as exercise (which is well established to promote favourable musculoskeletal) may also promote improvements in calcification. These interventions can be fortified by addressing underlying micronutrient deficiencies which together may be at the heart of unhealthy ageing.
References
Kanis JA. Diagnosis of osteoporosis and assessment of fracture risk. Lancet. 2002;359:1929–36.
Center JR, Nguyen TV, Schneider D, Sambrook PN, Eisman JA. Mortality after all major types of osteoporotic fracture in men and women: an observational study. Lancet. 1999;353:878–82.
Seeman E. Invited review: pathogenesis of osteoporosis. J Appl Physiol. 2003;95(5):2142–51.
Wade SW, Strader C, Fitzpatrick LA, Anthony MS, O’Malley CD. Estimating prevalence of osteoporosis: examples from industrialized countries. Arch Osteoporos. 2014;9(1):182.
Sanders KM, Seeman E, Ugoni AM, et al. Age-and Gender-Specific Rate of Fractures in Australia: A Population-Based Study https://search-proquest-com.ezproxy.lib.monash.edu.au/docview/848553449?accountid=12528. Accessed 20 Aug 2018.
Mithal A, Bansal B, Kyer C, Ebeling P. The Asia-Pacific Regional Audit-Epidemiology, Costs, and Burden of Osteoporosis in India 2013: A Report of International Osteoporosis Foundation. Indian J Endocrinol Metab. 2014;18:449.
Hernlund E, Svedbom A, Ivergård M, et al. Osteoporosis in the European Union: medical management, epidemiology and economic burden. A report prepared in collaboration with the International Osteoporosis Foundation (IOF) and the European Federation of Pharmaceutical Industry Associations (EFPIA). Arch Osteoporos. 2013;8:136.
Burge R, Dawson-Hughes B, Solomon DH, Wong JB, King A, Tosteson A. Incidence and economic burden of osteoporosis-related fractures in the United States, 2005-2025. J Bone Miner Res. 2007;22:465–75.
Dawson-Hughes B, Harris SS, Krall EA, Dallal GE. Effect of calcium and vitamin D supplementation on bone density in men and women 65 years of age or older. N Engl J Med. 1997;337:670–6.
Bolland MJ, Barber PA, Doughty RN, et al. Vascular events in healthy older women receiving calcium supplementation: randomised controlled trial. BMJ. 2008;336:262–6.
Bolland MJ, Avenell A, Baron JA, et al. Effect of calcium supplements on risk of myocardial infarction and cardiovascular events: meta-analysis. BMJ. 2010;341:c3691.
Licata AA. Discovery, clinical development, and therapeutic uses of bisphosphonates. Ann Pharmacother. 2005;39(4):668–77.
Rodan GA, Fleisch HA. Bisphosphonates: mechanisms of action. J Clin Invest. 1996;97:2692–6.
Lacey DL, Boyle WJ, Simonet WS, Kostenuik PJ, Dougall WC, Sullivan JK, et al. Bench to bedside: elucidation of the OPG–RANK–RANKL pathway and the development of denosumab. Nat Rev Drug Discov. 2012;11(5):401–19.
Satterwhite J, Heathman M, Miller PD, Marín F, Glass EV, Dobnig H. Pharmacokinetics of teriparatide (rhPTH[1-34]) and calcium pharmacodynamics in postmenopausal women with osteoporosis. Calcif Tissue Int. 2010;87:485–92.
Brixen KT, Christensen B, Ejersted C, Langdahl BL. Teriparatide (biosynthetic human parathyroid hormone 1-34): a new paradigm in the treatment of osteoporosis. Pharmacol Toxicol. 2004;94(6):260–70.
Brennan T, Rybchyn M, Green W, Atwa S, Conigrave A, Mason R. Osteoblasts play key roles in the mechanisms of action of strontium ranelate. Br J Pharmacol. 2009;157:1291–300.
Kanis JA, McCloskey EV. Risk factors in osteoporosis. Maturitas. 1998;30(3):229–33.
Kanis JA, Harvey NC, Johansson H, Odén A, McCloskey EV, Leslie WD. Overview of fracture prediction tools. J Clin Densitom. 2017;20:444–50.
Nguyen TV, Eisman JA. Fracture risk assessment: from population to individual. J Clin Densitom. 2017;20:368–78.
Iwaniec UT, Turner RT. Influence of body weight on bone mass, architecture and turnover. J Endocrinol. 2016;230:R115–30.
Kerckhofs G, Durand M, Vangoitsenhoven R, Marin C, van der Schueren B, Carmeliet G, et al. Changes in bone macro- and microstructure in diabetic obese mice revealed by high resolution microfocus X-ray computed tomography. Sci Rep. 2016;6(1):35517.
Liu C-T, Broe KE, Zhou Y, Boyd SK, Cupples LA, Hannan MT, et al. Visceral adipose tissue is associated with bone microarchitecture in the Framingham osteoporosis study. J Bone Miner Res. 2017;32(1):143–50.
Nielson CM, Srikanth P, Orwoll ES. Obesity and fracture in men and women: an epidemiologic perspective. J Bone Miner Res. 2012;27:1–10.
Marcus RL, Addison O, Kidde JP, Dibble LE, Lastayo PC. Skeletal muscle fat infiltration: impact of age, inactivity, and exercise. J Nutr Health Aging. 2010;14:362–6.
Scott D, Shore-Lorenti C, McMillan LB, et al. Calf muscle density is independently associated with physical function in overweight and obese older adults. J Musculoskelet Neuronal Interact. 2018;18:9–17.
Scott D, Shore-Lorenti C, McMillan L, et al. Associations of components of sarcopenic obesity with bone health and balance in older adults. Arch Gerontol Geriatr. 2018;75:125–31.
Szulc P, Samelson EJ, Kiel DP, Delmas PD. Increased bone resorption is associated with increased risk of cardiovascular events in men: the MINOS study. J Bone Miner Res. 2009;24:2023–31.
Yang S, Chen A, Wu T. Association of history of fracture with prehypertension and hypertension: a retrospective case-control study. BMC Musculoskelet Disord. 2015;16:86.
Sennerby U, Melhus H, Gedeborg R, et al. Cardiovascular diseases and risk of hip fracture. JAMA. 2009;302:1666.
Fricke O, Beccard R, Semler O, Schoenau E. Analyses of muscular mass and function: the impact on bone mineral density and peak muscle mass. Pediatr Nephrol. 2010;25:2393–400.
Tsuda K, Nishio I, Masuyama Y. Bone mineral density in women with essential hypertension. Am J Hypertens. 2001;14:704–7.
Jorgensen L, Joakimsen O, Rosvold Berntsen GK, Heuch I, Jacobsen BK. Low bone mineral density is related to echogenic carotid artery plaques: a population-based study. Am J Epidemiol. 2004;160:549–56.
Tanko LB, Christiansen C, Cox DA, Geiger MJ, McNabb MA, Cummings SR. Relationship between osteoporosis and cardiovascular disease in postmenopausal women. J Bone Miner Res. 2005;20(11):1912–20.
Wong CX, Gan SW, Lee SW, et al. Atrial fibrillation and risk of hip fracture: a population-based analysis of 113,600 individuals. Int J Cardiol. 2017;243:229–32.
Cappuccio FP, Meilahn E, Zmuda JM, Cauley JA. High blood pressure and bone-mineral loss in elderly white women: a prospective study. Study of Osteoporotic Fractures Research Group. Lancet. 1999;354:971–5.
Sennerby U, Farahmand B, Ahlbom A, Ljunghall S, Michaelsson K. Cardiovascular diseases and future risk of hip fracture in women. Osteoporos Int. 2007;18:1355–62.
Bagger YZ, Tanko LB, Alexandersen P, Qin G, Christiansen C. Radiographic measure of aorta calcification is a site-specific predictor of bone loss and fracture risk at the hip. J Intern Med. 2006;259:598–605.
Jorgensen L, Joakimsen O, Mathiesen EB, et al. Carotid plaque echogenicity and risk of nonvertebral fractures in women: a longitudinal population-based study. Calcif Tissue Int. 2006;79:207–13.
Scott D, Blizzard L, Fell J, Jones G. The epidemiology of sarcopenia in community living older adults: what role does lifestyle play? J Cachexia Sarcopenia Muscle. 2011;2(3):125–34.
Bischoff-Ferrari HA, Orav JE, Kanis JA, et al. Comparative performance of current definitions of sarcopenia against the prospective incidence of falls among community-dwelling seniors age 65 and older. Osteoporos Int. 2015;26:2793–802.
Patel HP, Syddall HE, Jameson K, Robinson S, Denison H, Roberts HC, et al. Prevalence of sarcopenia in community-dwelling older people in the UK using the European working group on sarcopenia in older people (EWGSOP) definition: findings from the Hertfordshire cohort study (HCS). Age Ageing. 2013;42(3):378–84.
Yamada M, Nishiguchi S, Fukutani N, et al. Prevalence of sarcopenia in community-dwelling Japanese older adults. J Am Med Dir Assoc. 2013;14:911–5.
Waters DL, Baumgartner RN, Garry PJ. Sarcopenia: current perspectives. J Nutr Health Aging. 2000;4:133–9.
Delmonico MJ, Harris TB, Visser M, Park SW, Conroy MB, Velasquez-Mieyer P, et al. Longitudinal study of muscle strength, quality, and adipose tissue infiltration. Am J Clin Nutr. 2009;90(6):1579–85.
Miljkovic I, Kuipers AL, Cauley JA, et al. Greater skeletal muscle fat infiltration is associated with higher all-cause and cardiovascular mortality in older men. J Gerontol A Biol Sci Med Sci. 2015;70:1133–40.
Scott D, Trbojevic T, Skinner E, Clark RA, Levinger P, Haines TP, et al. Associations of calf inter- and intra-muscular adipose tissue with cardiometabolic health and physical function in community-dwelling older adults. J Musculoskelet Neuronal Interact. 2015;15(4):350–7.
Rosique-Esteban N, Babio N, Díaz-López A, et al. Leisure-time physical activity at moderate and high intensity is associated with parameters of body composition, muscle strength and sarcopenia in aged adults with obesity and metabolic syndrome from the PREDIMED-Plus study. Clin Nutr. 2018; published online June 6. https://doi.org/10.1016/j.clnu.2018.05.023.
Kim J, Lee Y, Kye S, Chung Y-S, Kim K-M. Association between healthy diet and exercise and greater muscle mass in older adults. J Am Geriatr Soc. 2015;63:886–92.
Reid KF, Martin KI, Doros G, et al. Comparative effects of light or heavy resistance power training for improving lower extremity power and physical performance in mobility-limited older adults. J Gerontol A Biol Sci Med Sci. 2015;70:374–80.
PETERSON MD, SEN A, GORDON PM. Influence of resistance exercise on lean body mass in aging adults. Med Sci Sport Exerc. 2011;43:249–58.
Campbell WW, Trappe TA, Wolfe RR, Evans WJ. The recommended dietary allowance for protein may not be adequate for older people to maintain skeletal muscle. J Gerontol A Biol Sci Med Sci. 2001;56:M373–80.
Morris MS, Jacques PF. Total protein, animal protein and physical activity in relation to muscle mass in middle-aged and older Americans. Br J Nutr. 2013;109:1294–303.
Granic A, Mendonça N, Sayer AA, et al. Low protein intake, muscle strength and physical performance in the very old: The Newcastle 85+ Study. Clin Nutr. 2017; published online Nov 16. https://doi.org/10.1016/j.clnu.2017.11.005.
Gonzalez-Freire M, de Cabo R, Studenski SA, Ferrucci L. The neuromuscular junction: aging at the crossroad between nerves and muscle. Front Aging Neurosci. 2014;6:208.
Rosen CJ, Adams JS, Bikle DD, Black DM, Demay MB, Manson JAE, et al. The nonskeletal effects of vitamin D: an Endocrine Society scientific statement. Endocr Rev. 2012;33(3):456–92.
Scott D, Blizzard L, Fell J, Giles G, Jones G. Associations between dietary nutrient intake and muscle mass and strength in community-dwelling older adults: the Tasmanian older adult cohort study. J Am Geriatr Soc. 2010;58:2129–34.
Rousseau A-F, Foidart-Desalle M, Ledoux D, Remy C, Croisier JL, Damas P, et al. Effects of cholecalciferol supplementation and optimized calcium intakes on vitamin D status, muscle strength and bone health: a one-year pilot randomized controlled trial in adults with severe burns. Burns. 2015;41(2):317–25.
van Dronkelaar C, van Velzen A, Abdelrazek M, van der Steen A, Weijs PJM, Tieland M. Minerals and Sarcopenia; The Role of Calcium, Iron, Magnesium, Phosphorus, Potassium, Selenium, Sodium, and Zinc on Muscle Mass, Muscle Strength, and Physical Performance in Older Adults: A Systematic Review. J Am Med Dir Assoc. 2018;19:6–11.e3.
Bo Y, Liu C, Ji Z, et al. A high whey protein, vitamin D and E supplement preserves muscle mass, strength, and quality of life in sarcopenic older adults: A double-blind randomized controlled trial. Clin Nutr. 2018; published online Jan 9. https://doi.org/10.1016/j.clnu.2017.12.020.
Bauer JM, Verlaan S, Bautmans I, et al. Effects of a vitamin D and leucine-enriched whey protein nutritional supplement on measures of sarcopenia in older adults, the PROVIDE study: a randomized, double-blind, placebo-controlled trial. J Am Med Dir Assoc. 2015;16:740–7.
Beaudart C, Dawson A, Shaw SC, et al. Nutrition and physical activity in the prevention and treatment of sarcopenia: systematic review. Osteoporos Int. 2017;28:1817–33.
Antoniak AE, Greig CA. The effect of combined resistance exercise training and vitamin D 3 supplementation on musculoskeletal health and function in older adults: a systematic review and meta-analysis. BMJ Open. 2017;7:e014619.
Trajanoska K, Schoufour JD, Darweesh SK, et al. Sarcopenia and its clinical correlates in the general population: the Rotterdam study. J Bone Miner Res. 2018;33:1209–18.
Atkins JL, Whincup PH, Morris RW, Lennon LT, Papacosta O, Wannamethee SG. Sarcopenic obesity and risk of cardiovascular disease and mortality: a population-based cohort study of older men. J Am Geriatr Soc. 2014;62:253–60.
Chuang S-Y, Chang H-Y, Lee M-S, Chia-Yu Chen R, Pan W-H. Skeletal muscle mass and risk of death in an elderly population. Nutr Metab Cardiovasc Dis. 2014;24:784–91.
Cardiovascular disease: Australian facts 2011. Canberra, 2011. https://www.aihw.gov.au/getmedia/9621f6a8-f076-4e3e-a9c7-dece59ff0d74/12116.pdf.aspx?inline=true. Accessed 9 Jan 2018.
Australian Institute of Health and Welfare. Cardiovascular Disease in Australia 2018. 2018. https://www.aihw.gov.au/reports/heart-stroke-vascular-disease/cardiovascular-health-compendium/data. Accessed 7 Jul 2018.
Tsamis A, Krawiec JT, Vorp DA. Elastin and collagen fibre microstructure of the human aorta in ageing and disease: a review. J R Soc Interface. 2013;10:20121004.
Speer MY, Yang H-Y, Brabb T, et al. Smooth muscle cells give rise to osteochondrogenic precursors and chondrocytes in calcifying arteries. Circ Res. 2009;104:733–41.
Zhu D, Mackenzie NCW, Millán JL, Farquharson C, MacRae VE. The appearance and modulation of osteocyte marker expression during calcification of vascular smooth muscle cells. PLoS One. 2011;6:e19595.
Pugliese G, Iacobini C, Fantauzzi CB, Menini S. The dark and bright side of atherosclerotic calcification. Atherosclerosis. 2015;238. https://doi.org/10.1016/j.atherosclerosis.2014.12.011.
Wolinsky H, Glagov S. A lamellar unit of aortic medial structure and function in mammals. Am Hear Assoc http://circres.ahajournals.org/content/circresaha/20/1/99.full.pdf. Accessed 26 Mar 2018.
Ooshima A, Fuller GC, Cardinale GJ, Spector S, Udenfriend S. Increased collagen synthesis in blood vessels of hypertensive rats and its reversal by antihypertensive agents. Proc Natl Acad Sci U S A. 1974;71:3019–23.
Shang X, Scott D, Hodge A, et al. Adiposity assessed by anthropometric measures has a similar or greater predictive ability than dual-energy X-ray absorptiometry measures for abdominal aortic calcification in community-dwelling older adults. Int J Card Imaging. 2016;32:1451–60.
Norman PE, Jamrozik K, Lawrence-Brown MM, et al. Population based randomised controlled trial on impact of screening on mortality from abdominal aortic aneurysm. BMJ. 2004;329:1259.
Murphy WA, Zur ND, Gostner P, Knapp R, Recheis W, Seidler H. The Iceman: Discovery and Imaging. Radiology. 2003;226:614–29.
Virchow R. Cellular pathology. As based upon physiological and pathological histology. Lecture XVI--atheromatous affection of arteries. 1858. Nutr Rev. 1989;47:23–5.
Lehto S, Niskanen L, Suhonen M, Rönnemaa T, Laakso M. Medial artery calcification. A neglected harbinger of cardiovascular complications in non-insulin-dependent diabetes mellitus. Arterioscler Thromb Vasc Biol. 1996;16:978–83.
Lewis JR, Schousboe JT, Lim WH, Wong G, Zhu K, Lim EM, et al. Abdominal aortic calcification identified on lateral spine images from bone densitometers are a marker of generalized atherosclerosis in elderly women. Arterioscler Thromb Vasc Biol. 2016;36(1):166–73.
Lewis J, Schousboe J, Lim W, et al. Abdominal aortic calcification identified on images from bone densitometers and long-term cardiovascular outcomes in elderly women. J Bone Miner Res Conf. 2016;31. https://doi.org/10.1002/jbmr.3107.
Ishii T, Asuwa N. Collagen and elastin degradation by matrix metalloproteinases and tissue inhibitors of matrix metalloproteinase in aortic dissection. Hum Pathol. 2000;31:640–6.
Rattazzi M, Rosenfeld ME. The multifaceted role of macrophages in cardiovascular calcification. Atherosclerosis. 2018; published online Feb 1. https://doi.org/10.1016/J.ATHEROSCLEROSIS.2018.01.046.
Loree HM, Tobias BJ, Gibson LJ, Kamm RD, Small DM, Lee RT. Mechanical properties of model atherosclerotic lesion lipid pools. Arterioscler Thromb. 1994;14:230–4.
Cho I-J, Chang H-J, Park H-B, et al. Aortic calcification is associated with arterial stiffening, left ventricular hypertrophy, and diastolic dysfunction in elderly male patients with hypertension. J Hypertens. 2015;33:1633–41.
Wagenseil JE, Mecham RP. Elastin in large artery stiffness and hypertension. J Cardiovasc Transl Res. 2012;5:264–73.
Guo J, Fujiyoshi A, Willcox B, et al. Increased Aortic Calcification Is Associated With Arterial Stiffness Progression in Multiethnic Middle-Aged Men. Hypertension. 2017;69:102–8.
Chien S. Mechanotransduction and endothelial cell homeostasis: the wisdom of the cell. Am J Physiol Heart Circ Physiol. 2007;292:H1209–24.
Yang JW, Cho KI, Kim JH, et al. Wall shear stress in hypertensive patients is associated with carotid vascular deformation assessed by speckle tracking strain imaging. Clin Hypertens. 2014;20:10.
Nicoll R, Henein M. Arterial calcification: a new perspective? Int J Cardiol. 2017;228:11–22.
Schousboe JT, Taylor BC, Kiel DP, Ensrud KE, Wilson KE, McCloskey EV. Abdominal aortic calcification detected on lateral spine images from a bone densitometer predicts incident myocardial infarction or stroke in older women. J Bone Miner Res. 2008;23(3):409–16.
Kauppila LI, Polak JF, Cupples LA, Hannan MT, Kiel DP, Wilson PW. New indices to classify location, severity and progression of calcific lesions in the abdominal aorta: a 25-year follow-up study. Atherosclerosis. 1997;132:245–50.
Laurent S, Boutouyrie P, Asmar R, et al. Aortic stiffness is an independent predictor of all-cause and cardiovascular mortality in hypertensive patients. Hypertension. 2001;37:1236–41.
Hametner B, Wassertheurer S, Kropf J, Mayer C, Eber B, Weber T. Oscillometric estimation of aortic pulse wave velocity. Blood Press Monit. 2013;18:173–6.
Laurent S, Cockcroft J, Van Bortel L, et al. Expert consensus document on arterial stiffness: methodological issues and clinical applications. Eur Heart J. 2006;27:2588–605.
Lewis JR, Schousboe JT, Lim WH, et al. J Bone Miner Res. 2018; published online Feb 14. https://doi.org/10.1002/jbmr.3405.
Witteman JM, Kok F, Van Saase JCM, Valkenburg H. Aortic calcification as a predictor of cardiovascular mortality. Lancet. 1986;328:1120–2.
Nielsen ML, Pareek M, Gerke O, et al. Uncontrolled hypertension is associated with coronary artery calcification and electrocardiographic left ventricular hypertrophy: a case-control study. J Hum Hypertens. 2015;29:303–8.
Issever AS, Kentenich M, Kohlitz T, Diederichs G, Zimmermann E. Osteoporosis and atherosclerosis: a post-mortem MDCT study of an elderly cohort. Eur Radiol. 2013;23(10):2823–9.
Kiel DP, Kauppila LI, Cupples LA, Hannan MT, O’Donnell CJ, Wilson PW. Bone loss and the progression of abdominal aortic calcification over a 25 year period: the Framingham heart study. [erratum appears in Calcif tissue Int. 2004 Feb;74(2):208]. Calcif Tissue Int. 2001;68:271–6.
Cai T, Sun D, Duan Y, Wen P, Dai C, Yang J, et al. WNT/β-catenin signaling promotes VSMCs to osteogenic transdifferentiation and calcification through directly modulating Runx2 gene expression. Exp Cell Res. 2016;345(2):206–17.
Vega OA, Lucero CMJ, Araya HF, et al. Wnt/β-catenin signaling activates expression of the bone-related transcription factor RUNX2 in select human osteosarcoma cell types. J Cell Biochem. 2017;118:3662–74.
Shi L, Cai G, Shi J, et al. Ossification of the posterior ligament is mediated by osterix via inhibition of the β-catenin signaling pathway. Exp Cell Res. 2016;349:53–9.
Moon YJ, Yun C-Y, Lee J-C, Kim JR, Park B-H, Cho E-S. Maturation of cortical bone suppresses periosteal osteoprogenitor proliferation in a paracrine manner. J Mol Histol. 2016;47:445–53.
Kook S-H, Heo JS, Lee J-C. Crucial roles of canonical Runx2-dependent pathway on Wnt1-induced osteoblastic differentiation of human periodontal ligament fibroblasts. Mol Cell Biochem. 2015;402:213–23.
Yu S, Yerges-Armstrong LM, Chu Y, Zmuda JM, Zhang Y. AP2 suppresses osteoblast differentiation and mineralization through down-regulation of Frizzled-1. Biochem J. 2015;465:395–404.
Yuan L-Q, Zhu J-H, Wang H-W, et al. RANKL is a downstream mediator for insulin-induced osteoblastic differentiation of vascular smooth muscle cells. PLoS One. 2011;6:e29037.
Malyankar UM, Scatena M, Suchland KL, Yun TJ, Clark EA, Giachelli CM. Osteoprotegerin is an α v β 3 -induced, NF-κB-dependent survival factor for endothelial cells. J Biol Chem. 2000;275:20959–62.
Sun Y, Byon CH, Yuan K, et al. Smooth muscle cell-specific Runx2 deficiency inhibits vascular calcification. Circ Res. 2012;111:543–52.
Zhang K, Zhang Y, Feng W, et al. Interleukin-18 Enhances Vascular Calcification and Osteogenic Differentiation of Vascular Smooth Muscle Cells Through TRPM7 Activation. Arterioscler Thromb Vasc Biol. 2017;37(10):1933–43.
Cosman F, Crittenden DB, Adachi JD, et al. Romosozumab treatment in postmenopausal women with osteoporosis. N Engl J Med. 2016;375:1532–43.
Lewiecki EM, Blicharski T, Goemaere S, Lippuner K, Meisner PD, Miller PD, et al. A phase III randomized placebo-controlled trial to evaluate efficacy and safety of Romosozumab in men with osteoporosis. J Clin Endocrinol Metab. 2018;103(9):3183–93.
Touw WA, Ueland T, Bollerslev J, Schousboe JT, Lim WH, Wong G, et al. Association of Circulating Wnt Antagonists with Severe Abdominal Aortic Calcification in elderly women. J Endocr Soc. 2017;1(1):26–38.
Hampson G, Edwards S, Conroy S, Blake GM, Fogelman I, Frost ML. The relationship between inhibitors of the Wnt signalling pathway (Dickkopf-1(DKK1) and sclerostin), bone mineral density, vascular calcification and arterial stiffness in post-menopausal women. Bone. 2013;56:42–7.
Krishna SM, Seto S-W, Jose RJ, et al. Wnt signaling pathway inhibitor Sclerostin inhibits angiotensin II–induced aortic aneurysm and AtherosclerosisHighlights. Arterioscler Thromb Vasc Biol. 2017;37:553–66.
Acıbucu F, Dokmetas H, Acıbucu D, Kılıclı F, Aydemir M, Cakmak E. Effect of vitamin D treatment on serum Sclerostin level. Exp Clin Endocrinol Diabetes. 2017;125:634–7.
Sankaralingam A, Roplekar R, Turner C, Dalton RN, Hampson G. Changes in Dickkopf-1 (DKK1) and Sclerostin following a loading dose of vitamin D 2 (300,000 IU). J Osteoporos. 2014;2014:682763.
Breslavsky A, Frand J, Matas Z, Boaz M, Barnea Z, Shargorodsky M. Effect of high doses of vitamin D on arterial properties, adiponectin, leptin and glucose homeostasis in type 2 diabetic patients. Clin Nutr. 2013;32:970–5.
Larsen T, Mose FH, Bech JN, Hansen AB, Pedersen EB. Effect of cholecalciferol supplementation during winter months in patients with hypertension: a randomized, placebo-controlled trial. Am J Hypertens. 2012;25:1215–22.
Dreyer G, Tucker AT, Harwood SM, Pearse RM, Raftery MJ, Yaqoob MM. Ergocalciferol and microcirculatory function in chronic kidney disease and concomitant vitamin D deficiency: an exploratory, double blind, Randomised Controlled Trial. PLoS One. 2014;9:e99461.
Rejnmark L, Bislev LS, Cashman KD, et al. Non-skeletal health effects of vitamin D supplementation: a systematic review on findings from meta-analyses summarizing trial data. PLoS One. 2017;12:e0180512.
Sambrook PN, Chen JS, Simpson JM, March LM. Impact of adverse news media on prescriptions for osteoporosis: effect on fractures and mortality. Med J Aust. 2010;193(3):154–6.
Kranenburg G, Bartstra JW, Weijmans M, et al. Bisphosphonates for cardiovascular risk reduction: a systematic review and meta-analysis. Atherosclerosis. 2016;252:106–15.
Kawahara T, Nishikawa M, Kawahara C, Inazu T, Sakai K, Suzuki G. Atorvastatin, etidronate, or both in patients at high risk for atherosclerotic aortic plaques: a randomized, controlled trial. Circulation. 2013;127:2327–35.
Toussaint ND, Lau KK, Strauss BJ, Polkinghorne KR, Kerr PG. Effect of alendronate on vascular calcification in CKD stages 3 and 4: a pilot randomized controlled trial. Am J Kidney Dis. 2010;56:57–68.
Cummings SR, Martin JS, McClung MR, et al. Denosumab for prevention of fractures in postmenopausal women with osteoporosis. N Engl J Med. 2009;361:756–65.
Samelson EJ, Miller PD, Christiansen C, et al. RANKL inhibition with denosumab does not influence 3-year progression of aortic calcification or incidence of adverse cardiovascular events in postmenopausal women with osteoporosis and high cardiovascular risk. J Bone Miner Res. 2014;29:450–7.
Ueki K, Yamada S, Tsuchimoto A, et al. Rapid progression of vascular and soft tissue calcification while being managed for severe and persistent hypocalcemia induced by Denosumab treatment in a patient with multiple myeloma and chronic kidney disease. Intern Med. 2015;54:2637–42.
Scott D, Blizzard L, Fell J, Jones G. Prospective associations between ambulatory activity, body composition and muscle function in older adults. Scand J Med Sci Sports. 2011;21:e168–75.
Tarantino G, Costantini S, Finelli C, et al. Is serum Interleukin-17 associated with early atherosclerosis in obese patients? J Transl Med. 2014;12:214.
Alexandersen P, Tankó LB, Bagger YZ, Jespersen J, Skouby SO, Christiansen C. Associations between aortic calcification and components of body composition in elderly men*. Obesity. 2006;14:1571–8.
Szulc P, Hofbauer LC, Rauner M, Goettsch C, Chapurlat R, Schoppet M. Serum myostatin levels are negatively associated with abdominal aortic calcification in older men: the STRAMBO study. Eur J Endocrinol. 2012;167:873–80.
Jensky NE, Allison MA, Loomba R, et al. Null association between abdominal muscle and calcified atherosclerosis in community-living persons without clinical cardiovascular disease: the multi-ethnic study of atherosclerosis. Metabolism. 2013;62:1562–9.
Idoate F, Cadore EL, Casas-Herrero A, et al. Adipose tissue compartments, muscle mass, muscle fat infiltration, and coronary calcium in institutionalized frail nonagenarians. Eur Radiol. 2015;25:2163–75.
Wassel CL, Laughlin GA, Saad SD, Araneta MRG, Wooten W, Barrett-Connor E, et al. Associations of abdominal muscle area with 4-year change in coronary artery calcium differ by ethnicity among post-menopausal women. Ethn Dis. 2015;25(4):435–42.
Ko B-J, Chang Y, Jung H-S, et al. Relationship between low relative muscle mass and coronary artery calcification in healthy AdultsSignificance. Arterioscler Thromb Vasc Biol. 2016;36:1016–21.
Idoate F, Cadore EL, Casas-Herrero A, et al. Noncoronary vascular calcification, bone mineral density, and muscle mass in institutionalized frail nonagenarians. Rejuvenation Res. 2017;20:298–308.
Spahillari A, Mukamal KJ, DeFilippi C, et al. The association of lean and fat mass with all-cause mortality in older adults: the cardiovascular health study. Nutr Metab Cardiovasc Dis. 2016;26:1039–47.
Chuang S-Y, Hsu Y-Y, Chen RC-Y, Liu W-L, Pan W-H. Abdominal obesity and low skeletal muscle mass jointly predict Total mortality and cardiovascular mortality in an elderly Asian population. J Gerontol A Biol Sci Med Sci. 2016;71(8):1049–55.
Elliott B, Renshaw D, Getting S, Mackenzie R. The central role of myostatin in skeletal muscle and whole body homeostasis. Acta Physiol. 2012;205:324–40.
Hamrick MW, Shi X, Zhang W, Pennington C, Thakore H, Haque M, et al. Loss of myostatin (GDF8) function increases osteogenic differentiation of bone marrow-derived mesenchymal stem cells but the osteogenic effect is ablated with unloading. Bone. 2007;40(6):1544–53.
Artaza JN, Singh R, Ferrini MG, Braga M, Tsao J, Gonzalez-Cadavid NF. Myostatin promotes a fibrotic phenotypic switch in multipotent C3H 10T1/2 cells without affecting their differentiation into myofibroblasts. J Endocrinol. 2008;196:235–49.
Saely CH, Dyballa T, Vonbank A, et al. Type 2 diabetes but not coronary atherosclerosis is an independent determinant of impaired mobility in angiographied coronary patients. Diabetes Res Clin Pract. 2008;82:185–9.
Abizanda Soler P, Paterna Mellinas G, Martín Sebastiá E, Casado Moragón L, López Jiménez E, Martínez SE. Aterosclerosis subclínica, un predictor de limitación funcional al año en ancianos con alto nivel funcional: estudio Albacete. Rev Esp Geriatr Gerontol. 2010;45:125–30.
Den Ouden MEM, Schuurmans MJ, Arts EMA, et al. Atherosclerosis and physical functioning in older men, a longitudinal study. J Nutr Health Aging. 2013;17:97–104.
Park J, Park H. Muscle strength and carotid artery flow velocity is associated with increased risk of atherosclerosis in adults. Cardiol J. 2017;24:385–92.
Shimizu Y, Sato S, Koyamatsu J, et al. Handgrip strength and subclinical carotid atherosclerosis in relation to platelet levels among hypertensive elderly Japanese. Oncotarget. 2017;8:69362–9.
Suwa M, Imoto T, Kida A, Yokochi T, Iwase M, Kozawa K. Association of body flexibility and carotid atherosclerosis in Japanese middle-aged men: a cross-sectional study. BMJ Open. 2018;8:e019370.
Everson-Rose SA, Mendes de Leon CF, Roetker NS, Lutsey PL, Alonso A. Subclinical cardiovascular disease and changes in self-reported mobility: multi-ethnic study of atherosclerosis. Journals Gerontol A Biol Sci Med Sci. 2018;73:218–24.
Jadczak AD, Makwana N, Luscombe-Marsh N, Visvanathan R, Schultz TJ. Effectiveness of exercise interventions on physical function in community-dwelling frail older people. JBI Database System Rev Implement Rep. 2018;16:752–75.
Lai C-C, Tu Y-K, Wang T-G, Huang Y-T, Chien K-L. Effects of resistance training, endurance training and whole-body vibration on lean body mass, muscle strength and physical performance in older people: a systematic review and network meta-analysis. Age Ageing. 2018;47(3):367–73.
Rossow LM, Fahs CA, Thiebaud RS, et al. Arterial stiffness and blood flow adaptations following eight weeks of resistance exercise training in young and older women. Exp Gerontol. 2014;53:48–56.
Shiotsu Y, Watanabe Y, Tujii S, Yanagita M. Effect of exercise order of combined aerobic and resistance training on arterial stiffness in older men. Exp Gerontol. 2018;111:27–34.
Green D, Cheetham C, Mavaddat L, Watts K, Best M, Taylor R, et al. Effect of lower limb exercise on forearm vascular function: contribution of nitric oxide. Am J Physiol Heart Circ Physiol. 2002;283(3):H899–907.
Endes S, Schaffner E, Caviezel S, et al. Long-term physical activity is associated with reduced arterial stiffness in older adults: longitudinal results of the SAPALDIA cohort study. Age Ageing. 2016;45:110–5.
Wildman RP, Mackey RH, Bostom A, Thompson T, Sutton-Tyrrell K. Measures of obesity are associated with vascular stiffness in young and older adults. Hypertension. 2003;42:468–73.
Jarrell JC, Morin J, Vasquez K, Cuneo GJ, Kossodo S, Peterson JD. Imaging of Cathepsin K activity in rodent models of bone turnover and soft tissue calcification. Mol Imaging Biol. 2012;1:S1741.
Shanbhogue VV, Støving RK, Frederiksen KH, Hanson S, Brixen K, Gram J, et al. Bone structural changes after gastric bypass surgery evaluated by HR-pQCT: a two-year longitudinal study. Eur J Endocrinol. 2017;176(6):685–93.
Szulc P, Schoppet M, Goettsch C, et al. Endocrine and clinical correlates of Myostatin serum concentration in men—the STRAMBO study. J Clin Endocrinol Metab. 2012;97:3700–8.
Aihara K, Azuma H, Akaike M, et al. Disruption of nuclear vitamin D receptor gene causes enhanced Thrombogenicity in mice. J Biol Chem. 2004;279:35798–802.
Chen S, Law CS, Grigsby CL, et al. Cardiomyocyte-specific deletion of the vitamin D receptor gene results in cardiac HypertrophyClinical perspective. Circulation. 2011;124:1838–47.
Norman PE, Powell JT. Vitamin D and cardiovascular disease. Circ Res. 2014;114:379–93.
Zittermann A, Schleithoff SS, Tenderich G, Berthold HK, Korfer R, Stehle P. Low vitamin D status: a contributing factor in the pathogenesis of congestive heart failure? J Am Coll Cardiol. 2003;41:105–12.
Al Mheid I, Patel R, Murrow J, et al. Vitamin D status is associated with arterial stiffness and vascular dysfunction in healthy humans. J Am Coll Cardiol. 2011;58:186–92.
Tomson J, Hin H, Emberson J, et al. Effects of vitamin D on blood pressure, arterial stiffness, and cardiac function in older people after 1 year: BEST-D (biochemical efficacy and safety trial of vitamin D). J Am Heart Assoc. 2017;6:e005707.
Sluyter JD, Camargo CA, Stewart AW, et al. Effect of monthly, high-dose, Long-Term Vitamin D Supplementation on Central Blood Pressure Parameters: A Randomized Controlled Trial Substudy. J Am Heart Assoc. 2017;6:e006802.
Witte KK, Byrom R, Gierula J, et al. Effects of vitamin D on cardiac function in patients with chronic HF: the VINDICATE study. J Am Coll Cardiol. 2016;67:2593–603.
Chung M, Tang AM, Fu Z, Wang DD, Newberry SJ. Calcium intake and cardiovascular disease risk. Ann Intern Med. 2016;165:856.
Reid IR, Gamble GD, Bolland MJ. Circulating calcium concentrations, vascular disease and mortality: a systematic review. J Intern Med. 2016;279(6):524–40.
Harvey NC, Biver E, Kaufman J-M, Bauer J, Branco J, Brandi ML, et al. The role of calcium supplementation in healthy musculoskeletal ageing. Osteoporos Int. 2017;28(2):447–62.
Michaelsson K, Melhus H, Warensjo Lemming E, Wolk A, Byberg L. Long term calcium intake and rates of all cause and cardiovascular mortality: community based prospective longitudinal cohort study. BMJ. 2013;346:f228.
Khan B, Nowson CA, Daly RM, et al. Higher dietary calcium intakes are associated with reduced risks of fractures, cardiovascular events, and mortality: a prospective cohort study of older men and women. J Bone Miner Res. 2015;30:1758–66.
Kim JH, Yoon JW, Kim KW, et al. Increased dietary calcium intake is not associated with coronary artery calcification. Int J Cardiol. 2012;157:429–31.
Anderson JJB, Kruszka B, Delaney JAC, et al. Calcium intake from diet and supplements and the risk of coronary artery calcification and its progression among older adults: 10-year follow-up of the multi-ethnic study of atherosclerosis (MESA). J Am Heart Assoc. 2016;5:e003815.
Funding
No funding was required in undertaking this work.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
The authors declare that they have no conflict of interest.
Ethics Approval
No ethics committee approval was required in undertaking this work.
Informed Consent
No informed consent was required in undertaking this work as work did not involve any human participants or animals.
Rights and permissions
About this article

Cite this article
Rodriguez, A.J., Scott, D. & Ebeling, P.R. Exploring the Links Between Common Diseases of Ageing—Osteoporosis, Sarcopenia and Vascular Calcification. Clinic Rev Bone Miner Metab 17, 1–23 (2019). https://doi.org/10.1007/s12018-018-9251-2
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12018-018-9251-2