
Abstract
As a by-product of heme catabolism by the heme oxygenase system, carbon monoxide (CO) has been neglected for many years, and only recently has its role as an essential signaling molecule been appreciated. In the past decade, the use of CO gas in pre-clinical experimental models of disease has produced some remarkable data indicating that its therapeutic delivery to mammals could alleviate inflammatory processes and cardiovascular disorders. However, the inherent toxic nature of CO cannot be ignored, knowing that inhalation of uncontrolled amounts of this gas can ultimately lead to serious systemic complications and neuronal derangements. From a clinical perspective, a key question is whether a safe and therapeutically effective threshold of CO can be reached locally in organs and tissues without delivering potentially toxic amounts through the lung. The advent of CO-releasing molecules (CO-RMs), a group of compounds capable of carrying and liberating controlled quantities of CO in cellular systems, appears a plausible alternative in the attempt to overcome the limitations of CO gas. Although in its infancy and far from being used for clinical applications, the CO-RMs technology is supported by very encouraging biological results and reflected by the chemical versatility of these compounds and their endless potential to be transformed into CO-based pharmaceuticals.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Because of its bad reputation as an odorless, colorless and silent killer, carbon monoxide (CO) eluded for decades investigation by scientists, who thought that no biological relevance could emerge from exploring the physiological effects of such a toxic gas. This is because CO binds very strongly to the iron atoms in hemoglobin and can significantly reduce the oxygen-carrying capacity of this protein after inhalation at relevant doses and for prolonged periods of time, consequently leading to tissue hypoxia [1]. The affinity between CO and hemoglobin is, indeed, approximately 220 times stronger than the affinity of hemoglobin for oxygen, and the preferential binding of CO to heme iron with the subsequent formation of carbonmonoxy-hemoglobin (HbCO) may lead to CO poisoning in humans [2]. However, the perception that CO can exert only negative effects is challenged by studies corroborating cytoprotective and anti-oxidant activities of inducible heme oxygenase-1 (HO-1), the enzyme that produces CO in the body [3]. In this context, increased generation of endogenous CO in stressful conditions may reflect a dynamic and active involvement of this by-product in the protective response [4]. This notion dovetails with the fact that in the majority of experimental models where a beneficial participation of HO-1 has been demonstrated, CO “at appropriate doses” exerts a comparable protective effect on its own, even when heme oxygenase activity is totally abolished [4, 5]. The tissues of most mammals, including humans, have the ability to generate CO locally as cells express both inducible (HO-1) and constitutive (HO-2) heme oxygenase isoforms [3]. These enzymes, which utilize heme as substrate for the concomitant production of CO, iron and biliverdin, are present in all tissues examined so far, with high abundance in the brain (HO-2), liver (HO-2 and HO-1), spleen (HO-1), vascular endothelial cells and smooth muscle tissues (HO-1 and HO-2). Because a comprehensive report on the regulation and biological roles of heme oxygenase enzymes in response to stress can be found in this issue as part of a “Mini-series on Basic Research and Intensive Care Medicine” (see Bauer M. et al., “The heme oxygenase–carbon monoxide system: regulation and role in stress response and organ failure”; DOI 10.1007/s00134-008-1010-2), this article will focus on the bioactive and potential therapeutic properties of CO. Specifically, in the following sections we will: (1) review the pathophysiological effects and the signaling properties of CO in mammals; (2) assess the role of CO as a marker of disease in humans; (3) examine the therapeutic potential of CO gas inhalation; and (4) propose an alternative way of delivering CO to cells and tissues based on a new class of CO-releasing molecules (CO-RMs). The beneficial effects of CO will be analyzed with the perspective of using CO as a pharmaceutical for the treatment of diverse pathological conditions, with a particular emphasis on lung disease, systemic inflammation and cardiovascular disorders.
Carbon monoxide: toxic effects versus signaling actions
To reiterate the toxic effects of CO, an increase in the levels of HbCO in humans is generally perceived as an indication of impaired oxygen delivery to tissue. However, and contrary to what one would intuitively expect, we need to realize that CO poisoning is a poorly defined pathophysiological event, and the assessment of its injurious effects cannot rely solely on the levels of blood HbCO [2]. The data in the literature on this subject are difficult to interpret, as the quality of health outcomes in CO-poisoned patients usually does not correlate with HbCO levels, there is a lack of proper control groups in the studies, and the results are often confounded by different times of exposure to CO, the presence of anesthesia and the auto-regulatory mechanisms that may differ significantly among individuals [2]. For instance, persistent functional deficits or neuropsychiatric sequelae are experienced by 11–30% of patients who have had CO poisoning but this strictly depends on whether the event was acute (short exposure to high levels of CO) or chronic (long exposure to low levels of CO) [6]. The percentage of HbCO in blood still remains at present the best predictive marker for extrapolating the amount of CO present in the body. Based on this, the data reported so far in the literature indicate that a 15–20% proportion of HbCO is, in the majority of cases, not detrimental and can be considered the “biological threshold” for CO tolerance in humans beyond which severe CO-mediated injury is likely to occur.
Most studies conducted so far suggest that endogenously generated CO and exogenous CO gas, inhaled at doses whereby the oxygen-carrying capacity of hemoglobin is not severely compromised (HbCO < 20%), elicits protection and beneficial outcomes, covering a vast array of responses against multiple organ injury, inflammation, apoptosis, cell proliferation, vasoconstriction and both systemic and pulmonary hypertension [4, 7–9]. The initial evidence supporting a beneficial action of CO originated from studies on lung injury in animals [10] and was reproduced later in almost all tissues examined, including heart, liver, kidney, intestine and the reticulo-endothelial system [4, 7]. Although this scenario emphasizes the pleiotropic activities of CO and offers a wide range of practical applications to explore, it also indicates a complicated CO-dependent signaling network that varies in response to a specific stimulus and the type of tissue being considered. Research aimed at understanding the mechanisms and signaling pathways involved in CO-mediated activities is still in its infancy, and the mechanisms proposed explain only partially the physiological action of CO. It is possible that a concerted action of several crucial pathways mediate the cellular activities of CO in diverse conditions. For example, guanylate cyclase activation is a likely target of CO-mediated regulation of vessel tone and blood pressure [11, 12]. Similarly, mitogen-activated protein kinases (MAPKs), especially the P38 protein, phosphatidylinositol 3-kinase (PI3-K) and NFkB signaling pathways, have been implicated in the anti-apoptotic and anti-inflammatory function of CO [4, 7, 8], although the precise “molecular switch” responsible for this activation remains to be identified. Hence, the actions mediated by CO seem to invoke, directly or indirectly, different signaling mechanisms in different cell types and tissues.
Thus, the findings on cellular and animal models reflect favorably on the potential use of “small amounts” of CO gas as a therapeutic agent in stressful conditions, but at the same time the multiplicity of CO effects and its versatile action make its specific target(s) difficult to decipher. With the recent development of controlled methods for CO delivery (CO-releasing molecules or “CO-RMs”) to target different pathological conditions, [13–15], the prospect that CO could become a therapeutic option in the near future is feasible (see below).
Measurements of endogenous CO production in humans: role of CO as a marker of disease
With the recognized importance of the heme oxygenase system in protection against oxidative stress, scientists began to measure CO levels in different models of tissue injury in animal species. Generally, CO levels are found to correlate either with the degree of stress or with increased survival. Although still limited in number, human clinical trials report similar responses inasmuch as critically ill patients produce higher amounts of CO than healthy controls [16]. Scharte and colleagues examined in a recent study whether endogenous CO production correlated with the severity of disease in intensive care patients who were mechanically ventilated [17]. Interestingly, critically ill patients suffering cardiac disease and those subjected to dialysis exhibited higher amounts of CO than other critically ill patients. The levels of CO correlated also with serum bilirubin, the other catabolite of heme degradation by heme oxygenase, and with serum creatinine, in accordance with the severe renal failure developed by these patients [17]. The production of CO is also higher in septic patients than in control subjects and, importantly, the levels of CO measured at 1 day were more elevated in the survivors of sepsis than in non-survivors [18]. Another investigation found no correlation between exhaled CO and severity of illness or degree of inflammation, but observed a strong trend of higher levels of exhaled CO in the survivors group than in non-survivors [19]. A significant correlation between HbCO levels and increased leukocyte counts in intensive care patients was reported by Hunter and co-workers [20]. In particular, these authors noted that an increase in the white blood cell count is associated with an increase in the severity of illness and suggest that CO may play a direct role in the pathogenesis of the disease. Similar results have also been reported in the pediatric age group. Shi et al. [21] suggested that CO might be a mediator in the pathogenesis of neonatal sepsis after observing significantly higher levels of exhaled CO in full-term septic infants admitted to neonatal ICU than in healthy neonates. An interesting correlation between cGMP and HbCO levels in pre-term infants with respiratory distress syndrome has also been described, sustaining the possibility that CO-mediated increase in cGMP contributes to systemic vasodilatation and hypotension in pre-term infants [22].
Collectively, the studies highlighted above point to important issues and raise more questions on the significance of CO levels in a clinical setting. Can CO levels serve as a marker of disease progression or severity? Could CO measurements possess potentially useful prognostic value? Is increased CO production an indication of the involvement of the gas in the pathogenesis of disease, or is it a sign of augmented heme oxygenase activity in response to stress conditions? These points have special relevance to acute pathological conditions, where a fluctuation of CO production would be expected to occur during the course of the disease. Based on the evidence accumulated so far, we speculate that measurement of CO levels at the early stages of pathological states might be crucial in understanding and predicting outcomes such as survival in intensive care cases. There may also be specific diseases where measurements of CO levels provide important insights into the evolution of the disease state, in which case an effort is required to identify the clinical settings where detection of CO would be most useful. In this respect, the scientific literature indicates preferential pathologies to explore, such as hematological diseases characterized by hemolysis. A recent study showed that exhaled CO levels, measured with a commercially available end-tidal CO (ETCO) monitor, were higher in children with sickle cell disease than in controls and pointed to the use of the technique as a reliable method to monitor hemolysis in children [23]. Furthermore, the use of CO as a marker of inflammation may be appropriate in several inflammatory conditions of the respiratory tract and other systems, since exhaled CO levels reflect the severity of pulmonary inflammation in asthmatic patients (responding also to steroid therapy) [24], nasal CO levels are elevated in patients with seasonal allergic rhinitis [25], and CO levels measured in the lumen of the human colon increase in patients in the active stage of ulcerative colitis [26]. Exhaled CO is also higher than normal in prematurely born infants who develop bronchopulmonary dysplasia [27] and in patients with cystic fibrosis [28]. Most recently Tran and colleagues reported that HbCO levels is increased in cirrhotic patients, but no correlation was found with the severity of the disease [29]. In contrast, elevated arterial HbCO was found in critically ill patients, arguing that there might be an optimal therapeutic range for HO-1 induction [30].
Therapeutic potentials of CO gas inhalation
Effects of CO gas on lung disease
The fact that increased expression of stress-induced HO-1 was observed in broncho-alveolar epithelial and macrophage cells after acute lung injury such as hyperoxia [10], ischemia [31] and acute respiratory distress syndrome (ARDS) [32] suggests a contribution of the HO-1 pathway to pulmonary function during pathological states. Higher levels of exhaled CO were also reported in experimental ARDS [33] and after acute lung injury induced by extracorporeal circulation [34], indicating the actual availability of substrate for sustaining increased heme oxygenase activity in pulmonary tissue. Studies performed in lung epithelial cells [35] and fibroblasts [36] confirmed that HO-1 overexpression and/or CO gas treatment conferred resistance to oxidative damage, TNF-α-mediated apoptosis and ischemic lung injury [37]. Although not strictly the focus of this review, it should be mentioned that the other by-products of HO-1 activity (biliverdin and bilirubin) also exhibit significant cytoprotective and anti-oxidative effects both in the lung and cardiovascular system [38]; a key difference between biliverdin/bilirubin and CO might originate in the unique signaling (and ubiquitous) action of CO, compared to the direct oxidant-scavenging activity of the biliverdin/bilirubin couple.
CO gas administration was initially shown to provide protection of the lung against hyperoxic injury [10] in rats. The study is of particular importance because: (1) it was the first to consider inhalation of CO at low concentrations as a therapeutic strategy to combat acute diseases; and (2) it proposed anti-inflammatory and anti-apoptotic actions of CO as likely mechanisms involved in the observed protection. The beneficial properties of CO were then confirmed in models in vitro and in vivo, showing that CO reduces the inflammation associated with allergen-induced asthma in mice [39], protects against orthotopic lung transplantation [40] and lung injury caused by oxidants [41], and reverses established pulmonary hypertension [9]. Mitogen-activated protein kinases (p38, MKK3) [41], caspase-3 [37] and cGMP-dependent mechanisms [42] appear to mediate some of the cytoprotective effects elicited by CO gas in the lung. Furthermore, there is evidence that CO inhalation is effective against ischemic lung injury by attenuating deposition of fibrin in the microvasculature [31, 42], a crucial factor in the pathophysiology of this condition, and by reducing TNF-α production and neutrophil recruitment to alveoli in ventilator-induced injury [43]. Conversely, the positive view emerging from the use of CO inhalation as a potential therapeutic approach in lung disease has been challenged by Clayton and co-workers, who reported no significant benefit of CO treatment on hyperoxic acute lung injury and observed neurotoxicity at relatively low levels of CO exposure (200 and 500 ppm) [44].
CO gas and systemic inflammation
The anti-inflammatory action of CO is perhaps the most intriguing and potentially useful for the future therapeutic application of CO, inflammation being the underlying cause of a variety of chronic pathologies, including cardiovascular disease, diabetes, cancer and obesity, as well as representing an innate body response in acute conditions such as bacterial infection. In several in vitro cell culture models CO gas reduces the production of pro-inflammatory cytokines (TNF-α, IL-1β) induced by LPS and stimulates the release of IL-10, an anti-inflammatory molecule [8]. A similar effect, sometimes involving different cytokines (e.g. IL-2 or IL-6), is observed in animal experiments where inflammation is caused by LPS [8, 45], ischemia–reperfusion and organ transplantation [46] or by aeroallergens such as ovalbumin [39].
It is often stated that this beneficial outcome of CO mimics the anti-inflammatory activity of HO-1, which was originally identified as a key factor in the resolution of inflammation in carrageenan-mediated acute pleurisy [47] and was subsequently demonstrated to play a major role in counteracting inflammation in many animal models [7, 48] and in human HO-1 deficiency [49]. This view is also partially supported by experiments showing that in the absence of HO-1 (HO-1-deficient mice and cells, or inhibition of heme oxygenase activity), exogenously administered CO provides full protection by itself [48]. However, it is highly unlikely that the amount of CO synthesized following induction of HO-1 can reach the high levels used in studies employing CO gas, suggesting that the mimicking action between the HO-1/CO system and CO gas is questionable. This point is well exemplified by a study in which HbCO levels were measured following either HO-1 up-regulation or CO inhalation in a model of lung transplant in mice. Minamoto and colleagues [42] reported that treatment of mice with cobalt protoporphyrin (5 mg/kg), which significantly raised tissue HO-1 expression, resulted in HbCO levels of 0.4% (from a baseline of ∼0.2%), while exposure to 250 ppm CO caused an increase in HbCO to 17%. Both enhanced HO-1 protein and exogenous CO reduced to a similar extent the production of inflammatory cytokines and airway luminal occlusion after transplantation, but it could be argued that different cellular mechanisms were involved in the protection achieved by the two treatments, since the HbCO levels were so different. Looking at this issue from another angle, we predict that administration of a very small amount of CO gas that increases HbCO from 0.2% to 0.4% would probably not elicit any beneficial effect. Nevertheless, the ability of CO to exert therapeutic activities per se should continue to be explored regardless of whether it mimics HO-1 induction or not; the recent observations that CO inhalation (500 ppm for 1 h) at various time points after injection of a lethal dose of endotoxin rescues 20–90% of mice from fulminant hepatitis [50] emphasize the potential importance of CO therapy in an intensive care setting. However, it should be noted that inhalation of CO (500 ppm) in a pilot trial on human volunteers could not reduce the inflammatory response elicited by experimental endotoxemia [51], even though HbCO levels significantly increased to 7%. These results are in contrast to those of animal studies, in which even lower doses of CO (250 ppm) could decrease LPS-induced inflammation [52], underlining the need for a better characterization of CO action in humans.
CO gas and the cardiovascular system
A recent report demonstrated that chronic inhalation of CO gas in rats for 72 weeks at doses found in tobacco smoke (200 ppm), which results in a steady-state level of 14% HbCO, is associated with myocardial hypertrophy but is not responsible for the respiratory pathology, atherogenesis, weight loss and tumor development that often typify cigarette smoking [53]. It has to be noted that the majority of studies conducted in animal experimental models of disease used acute rather than chronic CO gas inhalation (250–500 ppm for 1–24 h) showing that this approach can reduce significantly ischemia–reperfusion injury in most of the vital organs, including the heart [46, 54, 55]. CO has been demonstrated to diminish ischemia–reperfusion injury associated with cardiac rejection after transplantation [5]. These protective effects of CO are related not only to its anti-apoptotic, anti-inflammatory and vasodilatory functions but also to its ability to suppress platelet aggregation and fibrinolysis. Furthermore, a newly identified role for CO in promoting cardiac mitochondrial biogenesis [56] indicates that the gas may regulate intracellular utilization of energy and participate in those pathologies characterized by mitochondrial dysfunction. This is consistent with the finding that CO inhalation improves cardiac energetics, thereby protecting the heart against ischemic injury in pigs [57]. Positive findings on the effects of CO inhalation on myocardial function in humans are scarce as the data once again derive primarily from subjects affected by CO poisoning, who inevitably show symptoms of cardiotoxicity such as arrhythmia, angina pectoris and tachycardia [6]. However, in this context two studies should be mentioned in which patients were transplanted using hearts from non-conventional donors such as those who died of CO poisoning [58]. The retrieval and use of non-conventional donors in the setting of transplantation is important because of the growing imbalance between the supply of and demand for donor organs. The studies showed that the transplanted patients had an overall satisfactory recovery, indicating that CO-poisoned hearts and possibly other CO-poisoned organs can be used for intrathoracic transplantation [58]. These data are also in line with the recent findings showing that ex-vivo treatment of different organs with cold preservation solutions containing CO-releasing agents (see section below) can improve their function at reperfusion [59].
The CO-mediated protection of the heart and other organs may be a reflection of the positive effects of this gas on vascular activities. A direct pharmacological action of CO in the cardiovascular system is, indeed, apparent when considering the modulation of vascular tone. Although NO plays a major role in controlling vessel contractility in normal conditions, a lesser role for endogenous CO is also present and may become more relevant when NO signaling pathways are disrupted [12]. The vasodilatory action of CO is associated with guanylate cyclase (sGC) activation and consequent production of cGMP as well as stimulation of various potassium channels [60], but other unidentified mechanisms might be involved. Interestingly, CO caused vasoconstriction rather than dilatation in gracilis muscle arterioles [61], suggesting that CO-mediated vascular responses are tissue-dependent. In general, however, CO elicits vasodilatation in all other vessels investigated, including the aorta and vessel of the cerebral circulation [60, 62]. By administering CO via intravenous injection of different doses of CO-saturated saline solutions, Hangai-Hoger and colleagues showed that CO produced vasodilatation and improved microvascular hemodynamics (vessel diameter, red blood cell velocity and functional capillary density) in the hamster window chamber model [63]. The possibility of developing CO as a true pharmaceutical is, however, exemplified by the development of CO-releasing molecules.
Carbon monoxide-releasing molecules: a crucial step towards the development of CO-based pharmaceuticals
At the time our group started to appreciate that transition metal carbonyl complexes commonly used in catalysis and synthetic chemistry can release CO under appropriate experimental conditions [64], the first indications of the cytoprotective effects of CO gas inhalation were published [10]. These notions, combined with the tangible prospect of chemically modifying transition metal carbonyls for pharmaceutical purposes, motivated us to investigate the biological action of this class of compounds. Three consequential steps led us to ascertain the feasibility of exploiting CO-releasing molecules (CO-RMs) for therapeutic purposes. First of all, two commercially available transition metal carbonyls, subsequently termed CORM-1 (Mn2CO10) and CORM-2 [Ru(CO)3Cl2-dimer], proved unequivocally to liberate CO when stimulated under appropriate physiological conditions and promoted vasodilatation and hypotension in vivo [65]. Notably, these two compounds are lipid-soluble and were tested using dimethylsulfoxide (DMSO) as both a solvent and a vehicle for our biological experiments [65]. Secondly, we succeeded in synthesizing the first prototypic example of a water-soluble transition metal carbonyl (CORM-3), which is stable in aqueous solutions but rapidly releases CO (t1/2 < 1 min) once in contact with biological fluids and cellular components [66]. At the same time, we discovered that boranocarbonate (CORM-A1), which does not contain a transition metal carbonyl but a carboxylic group that is converted to CO through hydrolysis, can slowly liberate CO (t1/2 = 21 min) under physiological conditions [67]. This compound causes gradual vasodilatory and hypotensive effects that are strictly controlled by pH and can be amplified with the concomitant use of guanylate cyclase sensitizers [67]. The third indication for a potential use of CO-RMs as pharmaceuticals originates from their multiple bioactivities, which are being progressively identified since more scientists now utilize CO-RMs as a mean to deliver CO in in vitro, ex vivo and in vivo experimental models of disease [13]. Table 1 summarizes the chemical properties and pharmacological activities of the most commonly studied CO-RMs. The protective and beneficial effects of CO-RMs reported to date include: cardioprotection against both ischemia and myocardial infarction [66, 68]; reduction of cardiac graft rejection and positive inotropic effects on the heart [66, 69]; attenuation of the acute inflammatory response both in vitro and in vivo and amelioration of neuro-inflammatory responses in microglia [70–74]; reduction of immunological histamine release from guinea pig mast cells and human neutrophils [75]; anti-hypertensive effects and inhibition of platelet aggregation [65, 67, 76]; vasodilatation and anti-apoptotic effects in the cerebral circulation [77–79]; alleviation of hepatic leukocyte sequestration and systemic inflammatory response during severe burn injury [80]; mitigation of photocarcinogenesis in the skin [81]; improved kidney function following cold ischemia occurring during organ preservation and protection against cisplatin-induced nephrotoxicity [59, 82] The advent of CO-RMs has also provided a tool to explore the interaction of CO with various cellular targets and investigate its mechanism(s) of action [14, 83, 84]. In fact, the liberation of CO from CO-RMs affects the activity and function of several heme- and metal-dependent proteins that are crucially involved in processes controlling cellular homeostasis [14]. Among these are the production of reactive oxygen species, cell proliferation, angiogenesis and mitochondrial respiration [56, 83, 85–87]. Notably, in human airway smooth muscle cells and neutrophils CO liberated from CO-RMs inhibits the activity of NADPH-oxidase, a heme-dependent enzyme responsible for the production of superoxide anion and a major player in triggering and propagating oxidative stress [75, 83]. Other metal-containing proteins present in cells may have a preferential affinity for CO, and this is an area for future studies needed to identify the most likely targets that transduce the CO signals into beneficial effects.
CO as therapeutic agent: the challenges after the promises
A key question that arises primarily among clinicians is whether CO gas inhalation or CO-RMs will ever be utilized as therapeutic strategic approaches. CO gas inhalation as therapy in diseases has been discussed in a comprehensive article published elsewhere [4, 7, 88]. Although the use of small amounts of CO gas in medicine is feasible, one could intuitively argue that gaseous compounds in general are difficult to manipulate and to deliver directly to living cells or organism in an accurate, safe and measurable fashion. Thus, can CO-RMs overcome these obstacles? A conclusive answer to this question cannot be formulated at present since the assessment of a specific therapeutic role for a given CO-RM is still under scrutiny; however, the premise is that CO-RMs represent a good alternative to CO gas both from a pharmaceutical perspective and in terms of specificity of action. First of all, CO liberated from CO-RMs can be precisely controlled and delivered at given concentrations through all possible routes of administration, unlike CO gas, which can be delivered effectively only by inhalation. Secondly, not only should administration of CO-RMs mitigate the overall adverse systemic effects of CO inhalation but their use as specific CO carriers is likely to bypass more effectively the biological trap represented by deoxyhemoglobin, which is inevitably and rapidly converted to HbCO in the lung following CO gas inhalation (see Fig. 1). Published data indicate that HbCO does not increase to “dangerous” levels (less than 10%) when CORM-3 and CORM-A1 are used at doses that are pharmacologically effective in reducing myocardial infarction and improving renal hemodynamics after acute renal failure, respectively [68, 89, 90]. This optimistic view on the use of CO carriers is also based on the chemical versatility represented by transition metal carbonyls and boranocarbonate-containing CO-RMs [15]. The results reported in the literature also confirm how the kinetics of CO release can be tuned by modifying the ligand coordinated to the metal center or by changing the pH [67, 91]. Moreover, from a medicinal chemistry perspective, working with metallo-based compounds in drug development offers a unique advantage due to their great adaptability and because the method of their synthesis is usually very reliable and the ligand substitution to the metal center as well as their redox potentials can be finely adjusted to a specific need. Major progress in the development of pharmaceuticals based on CO-RMs technology will rely primarily on formulating compounds that are rather stable in vivo and can be triggered to release CO locally when needed or used to precisely channel CO to the site of injury. In addition, the product after CO release should be non-toxic or of low and known toxicity and, ideally, biologically relevant. Pharmacokinetics, efficacy profiles and other essential aspects of CO-RMs will have to be addressed, but the possibilities are endless for CO-RMs to become a new class of pharmaceuticals.

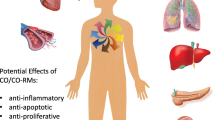
Carbon monoxide-releasing molecules (CO-RMs), which carry one or more CO groups, travel in the bloodstream, where they transport and release CO to tissue. CO (or CO-RMs) can then enter cells and trigger mechanisms that prevent or attenuate hypertension, inflammation and ischemic events. The advantage of this approach is twofold, as: (1) CO-RMs directly bypass the pulmonary system, where CO would mainly bind to haemoglobin, thereby reducing the oxygen supply to tissue, and (2) CO-RMs can be chemically engineered to trigger the specific delivery of small amounts of CO into tissues, providing pharmacological actions during a disease state
Conclusions
Despite its well-known toxic effects when inhaled at high concentrations, unexpected but consistent beneficial effects of CO gas are being reported, with the proposition of using CO therapy in the clinical setting. However, the administration route for gaseous compounds is restricted to inhalation through the lung, with difficulties in controlling the absorption, distribution and specificity of CO. The characterization and implementation of CO carriers (CO-RMs) that deliver CO in a more controlled and effective fashion has opened a concrete opportunity for the design of CO-based pharmaceuticals for future therapeutic applications. The success of the CO-RMs technology will depend crucially on the synergism between chemists, biologists and pharmacologists in their combined efforts to generate novel molecules that can mimic more closely the biological action of the heme oxygenase/CO system.
References
Piantadosi CA (2002) Biological chemistry of carbon monoxide. Antioxid Redox Signal 4:259–270
Gorman D, Drewry A, Huang YL, Sames C (2003) The clinical toxicology of carbon monoxide. Toxicology 187:25–38
Maines MD (1997) The heme oxygenase system: a regulator of second messenger gases. Annu Rev Pharmacol Toxicol 37:517–554
Kim HP, Ryter SW, Choi AM (2006) CO as a Cellular Signaling Molecule. Annu Rev Pharmacol Toxicol 46:411–449
Sato K, Balla J, Otterbein L, Smith RN, Brouard S, Lin Y, Csizmadia E, Sevigny J, Robson SC, Vercellotti G, Choi AM, Bach FH, Soares MP (2001) Carbon monoxide generated by heme oxygenase-1 suppresses the rejection of mouse-to-rat cardiac transplants. J Immunol 166:4185–4194
Rosenthal LD (2006) Carbon monoxide poisoning. Immediate diagnosis and treatment are crucial to avoid complications. Am J Nurs 106:40–46
Ryter SW, Alam J, Choi AM (2006) Heme oxygenase-1/carbon monoxide: from basic science to therapeutic applications. Physiol Rev 86:583–650
Otterbein LE, Bach FH, Alam J, Soares M, Tao Lu H, Wysk M, Davis RJ, Flavell RA, Choi AM (2000) Carbon monoxide has anti-inflammatory effects involving the mitogen-activated protein kinase pathway. Nature Med 6:422–428
Zuckerbraun BS, Chin BY, Wegiel B, Billiar TR, Czsimadia E, Rao J, Shimoda L, Ifedigbo E, Kanno S, Otterbein LE (2006) Carbon monoxide reverses established pulmonary hypertension. J Exp Med 203:2109–2119
Otterbein LE, Mantell LL, Choi AMK (1999) Carbon monoxide provides protection against hyperoxic lung injury. Am J Physiol 276:L688–L694
Ndisang JF, Tabien HE, Wang R (2004) Carbon monoxide and hypertension. J Hypertens 22:1057–1074
Motterlini R, Gonzales A, Foresti R, Clark JE, Green CJ, Winslow RM (1998) Heme oxygenase-1-derived carbon monoxide contributes to the suppression of acute hypertensive responses in vivo. Circ Res 83:568–577
Motterlini R, Mann BE, Foresti R (2005) Therapeutic applications of carbon monoxide-releasing molecules (CO-RMs). Expert Opin Investig Drugs 14:1305–1318
Boczkowski J, Poderoso JJ, Motterlini R (2006) CO-metal interaction: vital signaling from a lethal gas. Trends Biochem Sci 31:614–621
Alberto R, Motterlini R (2007) Chemistry and biological activities of CO-releasing molecules (CORMs) and transition metal complexes. Dalton Trans 17:1651–1660
Meyer J, Prien T, Van Aken H, Bone HG, Waurick R, Theilmeier G, Booke M (1998) Arterio-venous carboxyhemoglobin difference suggests carbon monoxide production by human lungs. Biochem Biophys Res Commun 244:230–232
Scharte M, von Ostrowski TA, Daudel F, Freise H, Van Aken H, Bone HG (2006) Endogenous carbon monoxide production correlates weakly with severity of acute illness. Eur J Anaesthesiol 23:117–122
Zegdi R, Perrin D, Burdin M, Boiteau R, Tenaillon A (2002) Increased endogenous carbon monoxide production in severe sepsis. Intensive Care Med 28:793–796
Morimatsu H, Takahashi T, Maeshima K, Inoue K, Kawakami T, Shimizu H, Takeuchi M, Yokoyama M, Katayama H, Morita K (2006) Increased heme catabolism in critically ill patients: correlation among exhaled carbon monoxide, arterial carboxyhemoglobin and serum bilirubin IX{alpha} concentrations. Am J Physiol Lung Cell Mol Physiol 290:L114–L119
Hunter K, Mascia M, Eudaric P, Simpkins C (1994) Evidence that carbon monoxide is a mediator of critical illness. Cell Mol Biol (Noisy-le-grand) 40:507–510
Shi Y, Pan F, Li H, Pan J, Qin S, Jiang D, Shen C (2000) Plasma carbon monoxide levels in term newborn infants with sepsis. Biol Neonate 78:230–232
van Bel F, Latour V, Vreman HJ, Wong RJ, Stevenson DK, Steendijk P, Egberts J, Krediet TG (2005) Is carbon monoxide-mediated cyclic guanosine monophosphate production responsible for low blood pressure in neonatal respiratory distress syndrome? J Appl Physiol 98:1044–1049
Sylvester KP, Patey RA, Rafferty GF, Rees D, Thein SL, Greenough A (2005) Exhaled carbon monoxide levels in children with sickle cell disease. Eur J Pediatr 164:162–165
Zayasu K, Sekizawa K, Okinaga S, Yamaya M, Ohrui T, Sasaki H (1997) Increased carbon monoxide in exhaled air of asthmatic patients. Am J Respir Crit Care Med 156:1140–1143
Andersson JA, Uddman R, Cardell LO (2002) Increased carbon monoxide levels in the nasal airways of subjects with a history of seasonal allergic rhinitis and in patients with upper respiratory tract infection. Clin Exp Allergy 32:224–227
Takagi T, Naito Y, Tsuboi H, Isozaki Y, Katada K, Suzuki T, Terao K, Handa O, Kokura S, Ichikawa H, Yoshida N, Okuyama Y, Yagi N, Ueda H, Yoshikawa T (2006) Increased intestinal luminal carbon monoxide gas in patients with ulcerative colitis. Aliment Pharmacol Ther 24 Suppl 4:233–238
May C, Patel S, Peacock J, Milner A, Rafferty GF, Greenough A (2007) End-tidal carbon monoxide levels in prematurely born infants developing bronchopulmonary dysplasia. Pediatr Res 61:474–478
Terheggen-Lagro SW, Bink MW, Vreman HJ, van der Ent CK (2003) End-tidal carbon monoxide corrected for lung volume is elevated in patients with cystic fibrosis. Am J Respir Crit Care Med 168:1227–1231
Tran TT, Martin P, Ly H, Balfe D, Mosenifar Z (2007) Carboxyhemoglobin and its correlation to disease severity in cirrhotics. J Clin Gastroenterol 41:211–215
Melley DD, Finney SJ, Elia A, Lagan AL, Quinlan GJ, Evans TW (2007) Arterial carboxyhemoglobin level and outcome in critically ill patients. Crit Care Med 35:1882–1887
Fujita T, Toda K, Karimova A, Yan SF, Naka Y, Yet SF, Pinsky DJ (2001) Paradoxical rescue from ischemic lung injury by inhaled carbon monoxide driven by derepression of fibrinolysis. Nat Med 7:598–604
Mumby S, Upton RL, Chen Y, Stanford SJ, Quinlan GJ, Nicholson AG, Gutteridge JM, Lamb NJ, Evans TW (2004) Lung heme oxygenase-1 is elevated in acute respiratory distress syndrome. Crit Care Med 32:1130–1135
Liu HL, Zhao JY, Chen L (2004) Changes of carbon monoxide, nitric oxide levels and heme oxygenase system in acute respiratory distress syndrome induced by oleic acid. Zhonghua Yu Fang Yi Xue Za Zhi 38:240–243
Zegdi R, Fabre O, Lila N, Fornes P, Cambillau M, Shen M, Herve P, Carpentier A, Fabiani JN (2003) Exhaled carbon monoxide and inducible heme oxygenase expression in a rat model of postperfusion acute lung injury. J Thorac Cardiovasc Surg 126:1867–1874
Lee PJ, Alam J, Wiegand GW, Choi AMK (1996) Overexpression of heme oxygenase-1 in human pulmonary epithelial cells results in cell growth arrest and increased resistance to hyperoxia. Proc Natl Acad Sci USA 93:10393–10398
Tulis DA, Durante W, Peyton KJ, Evans AJ, Schafer AI (2001) Heme oxygenase-1 attenuates vascular remodeling following balloon injury in rat carotid arteries. Atherosclerosis 155:113–122
Zhang X, Shan P, Otterbein LE, Alam J, Flavell RA, Davis RJ, Choi AM, Lee PJ (2003) Carbon monoxide inhibition of apoptosis during ischemia-reperfusion lung injury is dependent on the p38 mitogen-activated protein kinase pathway and involves caspase 3. J Biol Chem 278:1248–1258
Foresti R, Green CJ, Motterlini R (2004) Generation of bile pigments by heme oxygenase: a refined cellular stratagem in response to stressful insults. Biochem Soc Symp 71:177–192
Chapman JT, Otterbein LE, Elias JA, Choi AM (2001) Carbon monoxide attenuates aeroallergen-induced inflammation in mice. Am J Physiol Lung Cell Mol Physiol 281:L209–L216
Song R, Kubo M, Morse D, Zhou Z, Zhang X, Dauber JH, Fabisiak J, Alber SM, Watkins SC, Zuckerbraun BS, Otterbein LE, Ning W, Oury TD, Lee PJ, McCurry KR, Choi AM (2003) Carbon monoxide induces cytoprotection in rat orthotopic lung transplantation via anti-inflammatory and anti-apoptotic effects. Am J Pathol 163:231–242
Otterbein LE, Otterbein SL, Ifedigbo E, Liu F, Morse DE, Fearns C, Ulevitch RJ, Knickelbein R, Flavell RA, Choi AM (2003) MKK3 Mitogen-activated protein kinase pathway mediates carbon monoxide-induced protection against oxidant-induced lung injury. Am J Pathol 163:2555–2563
Mishra S, Fujita T, Lama VN, Nam D, Liao H, Okada M, Minamoto K, Yoshikawa Y, Harada H, Pinsky DJ (2006) Carbon monoxide rescues ischemic lungs by interrupting MAPK-driven expression of early growth response 1 gene and its downstream target genes. Proc Natl Acad Sci USA 103:5191–5196
Dolinay T, Szilasi M, Liu M, Choi AM (2004) Inhaled carbon monoxide confers antiinflammatory effects against ventilator-induced lung injury. Am J Respir Crit Care Med 170:613–620
Clayton CE, Carraway MS, Suliman HB, Thalmann ED, Thalmann KN, Schmechel DE, Piantadosi CA (2001) Inhaled carbon monoxide and hyperoxic lung injury in rats. Am J Physiol Lung Cell Mol Physiol 281:L949–L957
Mazzola S, Forni M, Albertini M, Bacci ML, Zannoni A, Gentilini F, Lavitrano M, Bach FH, Otterbein LE, Clement MG (2005) Carbon monoxide pretreatment prevents respiratory derangement and ameliorates hyperacute endotoxic shock in pigs. FASEB J 19:2045–2047
Kohmoto J, Nakao A, Kaizu T, Tsung A, Ikeda A, Tomiyama K, Billiar TR, Choi AM, Murase N, McCurry KR (2006) Low-dose carbon monoxide inhalation prevents ischemia/reperfusion injury of transplanted rat lung grafts. Surgery 140:179–185
Willis D, Moore AR, Frederick R, Willoughby DA (1996) Heme oxygenase: a novel target for the modulation of inflammatory response. Nature Med 2:87–90
Otterbein LE (2002) Carbon monoxide: innovative anti-inflammatory properties of an age-old gas molecule. Antioxid Redox Signal 4:309–319
Yachie A, Niida Y, Wada T, Igarashi N, Kaneda H, Toma T, Ohta K, Kasahara Y, Koizumi S (1999) Oxidative stress causes enhanced endothelial cell injury in human heme oxygenase-1 deficiency. J Clin Invest 103:129–135
Tsui TY, Obed A, Siu YT, Yet SF, Prantl L, Schlitt HJ, Fan ST (2007) Carbon monoxide inhalation rescues mice from fulminant hepatitis through improving hepatic energy metabolism. Shock 27:165–171
Mayr FB, Spiel A, Leitner J, Marsik C, Germann P, Ullrich R, Wagner O, Jilma B (2005) Effects of carbon monoxide inhalation during experimental endotoxemia in humans. Am J Respir Crit Care Med 171:354–360
Sarady JK, Zuckerbraun BS, Bilban M, Wagner O, Usheva A, Liu F, Ifedigbo E, Zamora R, Choi AM, Otterbein LE (2004) Carbon monoxide protection against endotoxic shock involves reciprocal effects on iNOS in the lung and liver. FASEB J 18:854–856
Sorhaug S, Steinshamn S, Nilsen OG, Waldum HL (2006) Chronic inhalation of carbon monoxide: effects on the respiratory and cardiovascular system at doses corresponding to tobacco smoking. Toxicology 228:280–290
Neto JS, Nakao A, Kimizuka K, Romanosky AJ, Stolz DB, Uchiyama T, Nalesnik MA, Otterbein LE, Murase N (2004) Protection of transplant-induced renal ischemia/reperfusion injury with carbon monoxide. Am J Physiol Renal Physiol 287:F979–F989
Nakao A, Choi AM, Murase N (2006) Protective effect of carbon monoxide in transplantation. J Cell Mol Med 10:650–671
Suliman HB, Carraway MS, Tatro LG, Piantadosi CA (2006) A new activating role for CO in cardiac mitochondrial biogenesis. J Cell Sci 120:299–308
Lavitrano M, Smolenski RT, Musumeci A, Maccherini M, Slominska E, Di Florio E, Bracco A, Mancini A, Stassi G, Patti M, Giovannoni R, Froio A, Simeone F, Forni M, Bacci ML, D'Alise G, Cozzi E, Otterbein LE, Yacoub MH, Bach FH, Calise F (2004) Carbon monoxide improves cardiac energetics and safeguards the heart during reperfusion after cardiopulmonary bypass in pigs. FASEB J 18:1093–1095
Luckraz H, Tsui SS, Parameshwar J, Wallwork J, Large SR (2001) Improved outcome with organs from carbon monoxide poisoned donors for intrathoracic transplantation. Ann Thorac Surg 72:709–713
Sandouka A, Fuller BJ, Mann BE, Green CJ, Foresti R, Motterlini R (2006) Treatment with carbon monoxide-releasing molecules (CO-RMs) during cold storage improves renal function at reperfusion. Kidney Int 69:239–247
Wang R, Wang ZZ, Wu LY (1997) Carbon monoxide-induced vasorelaxation and the underlying mechanisms. Br J Pharmacol 121:927–934
Johnson FK, Johnson RA (2003) Carbon monoxide promotes endothelium-dependent constriction of isolated gracilis muscle arterioles. Am J Physiol Regul Integr Comp Physiol 285:R536–R541
Foresti R, Hammad J, Clark JE, Johnson RA, Mann BE, Friebe A, Green CJ, Motterlini R (2004) Vasoactive properties of CORM-3, a novel water-soluble carbon monoxide-releasing molecule. Br J Pharmacol 142:453–460
Hangai-Hoger N, Tsai AG, Cabrales P, Suematsu M, Intaglietta M (2007) Microvascular and systemic effects following top load administration of saturated carbon monoxide-saline solution. Crit Care Med 35:1123–1132
Herrmann WA (1990) 100 Years of metal carbonyls. A serendipitous chemical discovery of major scientific and industrial impact. J Organomet Chem 383:21–44
Motterlini R, Clark JE, Foresti R, Sarathchandra P, Mann BE, Green CJ (2002) Carbon monoxide-releasing molecules: characterization of biochemical and vascular activities. Circ Res 90:E17–E24
Clark JE, Naughton P, Shurey S, Green CJ, Johnson TR, Mann BE, Foresti R, Motterlini R (2003) Cardioprotective actions by a water-soluble carbon monoxide-releasing molecule. Circ Res 93:e2–e8
Motterlini R, Sawle P, Bains S, Hammad J, Alberto R, Foresti R, Green CJ (2005) CORM-A1: a new pharmacologically active carbon monoxide-releasing molecule. FASEB J 19:284–286
Guo Y, Stein AB, Wu WJ, Tan W, Zhu X, Li QH, Dawn B, Motterlini R, Bolli R (2004) Administration of a CO-releasing molecule at the time of reperfusion reduces infarct size in vivo. Am J Physiol Heart Circ Physiol 286:H1649–H1653
Musameh MD, Fuller BJ, Mann BE, Green CJ, Motterlini R (2006) Positive inotropic effects of carbon monoxide-releasing molecules (CO-RMs) in the isolated perfused rat heart. Br J Pharmacol 149:1104–1112
Sawle P, Foresti R, Mann BE, Johnson TR, Green CJ, Motterlini R (2005) Carbon monoxide-releasing molecules (CO-RMs) attenuate the inflammatory response elicited by lipopolysaccharide in RAW264.7 murine macrophages. Br J Pharmacol 145:800–810
Bani-Hani MG, Greenstein D, Mann BE, Green CJ, Motterlini R (2006) Modulation of thrombin-induced neuroinflammation in BV-2 microglia by a carbon monoxide-releasing molecule (CORM-3). J Pharmacol Exp Ther 318:1315–1322
Urquhart P, Rosignoli G, Cooper D, Motterlini R, Perretti M (2007) Carbon monoxide-releasing molecules modulate leukocyte-endothelial interactions under flow. J Pharmacol Exp Ther 321:656–662
Freitas A, Alves-Filho JC, Secco DD, Neto AF, Ferreira SH, Barja-Fidalgo C, Cunha FQ (2006) Heme oxygenase/carbon monoxide-biliverdin pathway down regulates neutrophil rolling, adhesion and migration in acute inflammation. Br J Pharmacol 149:345–354
Megias J, Busserolles J, Alcaraz MJ (2007) The carbon monoxide-releasing molecule CORM-2 inhibits the inflammatory response induced by cytokines in Caco-2 cells. Br J Pharmacol 150:977–986
Vannacci A, Giannini L, Fabrizi F, Uliva C, Mastroianni R, Masini E, Motterlini R, Mannaioni PF (2007) Effects of the carbon monoxide releasing molecule CORM-3 in a coincubation model of rat mast cells with human neutrophils. Inflamm Res 56:S13–S14
Chlopicki S, Olszanecki R, Marcinkiewicz E, Lomnicka M, Motterlini R (2006) Carbon monoxide released by CORM-3 inhibits human platelets by a mechanism independent of soluble guanylate cyclase. Cardiovasc Res 71:393–401
Fiumana E, Parfenova H, Jagger JH, Leffler CW (2003) Carbon monoxide mediates vasodilator effects of glutamate in isolated pressurized cerebral arterioles of newborn pigs. Am J Physiol Heart Circ Physiol 284:H1073–H1079
Parfenova H, Basuroy S, Bhattacharya S, Tcheranova D, Qu Y, Regan RF, Leffler CW (2005) Glutamate induces oxidative stress and apoptosis in cerebral vascular endothelial cells: contributions of HO-1 and HO-2 to cytoprotection. Am J Physiol Cell Physiol 290:C1399–C1410
Li MH, Cha YN, Surh YJ (2006) Carbon monoxide protects PC12 cells from peroxynitrite-induced apoptotic death by preventing the depolarization of mitochondrial transmembrane potential. Biochem Biophys Res Commun 342:990
Sun BW, Chen ZY, Chen X, Liu C (2007) Attenuation of leukocytes sequestration by carbon monoxide-releasing molecules: liberated carbon monoxide in the liver of thermally injured mice. J Burn Care Res 28:173–181
Allanson M, Reeve VE (2007) Carbon monoxide signalling reduces photocarcinogenesis in the hairless mouse. Cancer Immunol Immunother 56:1807–1815
Tayem Y, Johnson TR, Mann BE, Green CJ, Motterlini R (2006) Protection against cisplatin-induced nephrotoxicity by a carbon monoxide-releasing molecule. Am J Physiol Renal Physiol 290:F789–F794
Taille C, El-Benna J, Lanone S, Boczkowski J, Motterlini R (2005) Mitochondrial respiratory chain and NAD(P)H oxidase are targets for the antiproliferative effect of carbon monoxide in human airway smooth muscle. J Biol Chem 280:25350–25360
Bolognesi M, Sacerdoti D, Piva A, Di Pascoli M, Zampieri F, Quarta S, Motterlini R, Angeli P, Merkel C, Gatta A (2007) Carbon monoxide-mediated activation of large-conductance calcium-activated potassium channels contributes to mesenteric vasodilatation in cirrhotic rats. J Pharmacol Exp Ther 321:187–194
Srisook K, Han SS, Choi HS, Li MH, Ueda H, Kim C, Cha YN (2006) CO from enhanced HO activity or from CORM-2 inhibits both O2- and NO production and downregulates HO-1 expression in LPS-stimulated macrophages. Biochem Pharmacol 71:307–318
Sandouka A, Balogun E, Foresti R, Mann BE, Johnson TR, Tayem Y, Green CJ, Fuller B, Motterlini R (2005) Carbon monoxide-releasing molecules (CO-RMs) modulate respiration in isolated mitochondria. Cell Mol Biol 51:425–432
Jozkowicz A, Huk I, Nigisch A, Weigel G, Dietrich W, Motterlini R, Dulak J (2003) Heme oxygenase-1 and angiogenic activity of endothelial cells: stimulation by carbon monoxide, inhibition by tin protoporphyrin IX. Antioxid Redox Signal 5:155–162
Ryter SW, Otterbein LE (2004) Carbon monoxide in biology and medicine. BioEssays 26:270–280
Vera T, Henegar JR, Drummond HA, Rimoldi JM, Stec DE (2005) Protective effect of carbon monoxide-releasing compounds in ischemia-induced acute renal failure. J Am Soc Nephrol 16:950–958
Ryan MJ, Jernigan NL, Drummond HA, McLemore GR Jr, Rimoldi JM, Poreddy SR, Gadepalli RS, Stec DE (2006) Renal vascular responses to CORM-A1 in the mouse. Pharmacol Res 54:24–29
Sawle P, Hammad J, Fairlamb IJ, Moulton B, O'Brien CT, Lynam JM, Duhme-Klair AK, Foresti R, Motterlini R (2006) Bioactive properties of iron-containing carbon monoxide-releasing molecules (CO-RMs). J Pharmacol Exp Ther 318:403–410
Arregui B, Lopez B, Salom MG, Valero F, Navarro C, Fenoy FJ (2004) Acute renal hemodynamic effects of dimanganese decacarbonyl and cobalt protoporphyrin. Kidney Int 65:564–574
Botros FT, Navar LG (2006) Interaction between endogenously-produced carbon monoxide and nitric oxide in regulation of renal afferent arterioles. Am J Physiol Heart Circ Physiol 291:H2772–H2778
Deshane J, Chen S, Caballero S, Grochot-Przeczek A, Was H, Li CS, Lach R, Hock TD, Chen B, Hill-Kapturczak N, Siegal GP, Dulak J, Jozkowicz A, Grant MB, Agarwal A (2007) Stromal cell-derived factor 1 promotes angiogenesis via a heme oxygenase 1-dependent mechanism. J Exp Med 204:605–618
Stein AB, Guo Y, Tan W, Wu WJ, Zhu X, Li Q, Luo C, Dawn B, Johnson TR, Motterlini R, Bolli R (2005) Administration of a CO-releasing molecule induces late preconditioning against myocardial infarction. J Mol Cell Cardiol 38:127–134
Acknowledgements
Dr. Roberto Motterlini is funded by the Henry Smith Charity and the Dunhill Medical Trust.
Author information
Authors and Affiliations
Corresponding author
Additional information
This article is discussed in the editorial available at: http://dx.doi.org/10.1007/s00134-008-1013-z.
Rights and permissions
About this article
Cite this article
Foresti, R., Bani-Hani, M.G. & Motterlini, R. Use of carbon monoxide as a therapeutic agent: promises and challenges. Intensive Care Med 34, 649–658 (2008). https://doi.org/10.1007/s00134-008-1011-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00134-008-1011-1