
Abstract
Aims/hypothesis
O-GlcNAcylation is characterised by the addition of N-acetylglucosamine to various proteins by O-GlcNAc transferase (OGT) and serves in sensing intracellular nutrients by modulating various cellular processes. Although it has been speculated that O-GlcNAcylation is associated with glucose metabolism, its exact role in whole body glucose metabolism has not been fully elucidated. Here, we investigated whether loss of O-GlcNAcylation globally and in specific organs affected glucose metabolism in mammals under physiological conditions.
Methods
Tamoxifen-inducible global Ogt-knockout (Ogt-KO) mice were generated by crossbreeding Ogt-flox mice with R26-Cre-ERT2 mice. Liver, skeletal muscle, adipose tissue and pancreatic beta cell-specific Ogt-KO mice were generated by crossbreeding Ogt-flox mice with Alb-Cre, Mlc1f-Cre, Adipoq-Cre and Pdx1 PB-CreER™ mice, respectively. Glucose metabolism was evaluated by i.p. glucose and insulin tolerance tests.
Results
Tamoxifen-inducible global Ogt-KO mice exhibited a lethal phenotype from 4 weeks post injection, suggesting that O-GlcNAcylation is essential for survival in adult mice. Tissue-specific Ogt deletion from insulin-sensitive organs, including liver, skeletal muscle and adipose tissue, had little impact on glucose metabolism under physiological conditions. However, pancreatic beta cell-specific Ogt-KO mice displayed transient hypoglycaemia (Ogt-flox 5.46 ± 0.41 vs Ogt-βKO 3.88 ± 0.26 mmol/l) associated with about twofold higher insulin secretion and accelerated adiposity, followed by subsequent hyperglycaemia (Ogt-flox 6.34 ± 0.32 vs Ogt-βKO 26.4 ± 2.37 mmol/l) with insulin depletion accompanied by beta cell apoptosis.
Conclusions/interpretation
These findings suggest that O-GlcNAcylation has little effect on glucose metabolism in insulin-sensitive tissues but plays a crucial role in pancreatic beta cell function and survival under physiological conditions. Our results provide novel insight into O-GlcNAc biology and physiology in glucose metabolism.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Post-translational modifications to intracellular proteins regulate a variety of physiological cellular functions. Growing evidence indicates that dysfunction of post-translational modifications, including phosphorylation, ubiquitination and acetylation, disrupts cellular homeostasis and is strongly associated with the pathogenesis of numerous diseases, including metabolic disorders [1,2,3]. Thus, elucidation of the relationship between post-translational modifications and cellular physiology may be a first step to better understand disease pathogenesis and provide insight into the development of novel therapeutics.
O-GlcNAcylation has been analysed as an important regulator of glucose metabolism [4,5,6]. In a branch of the glycolytic pathway, fructose-6-phosphate is converted into uridine diphosphate N-acetylglucosamine (UDP-GlcNAc) through multiple enzymes in the hexosamine biosynthetic pathway. In this process, not only glucose but also glutamine and acetyl-CoA are integrated to form UDP-GlcNAc. O-GlcNAc transferase (OGT) is essential for O-GlcNAcylation and attaches O-GlcNAc to proteins at specific serine or threonine residues [7]. Thus, O-GlcNAcylation has recently received remarkable attention as ‘an intracellular nutrient sensor’ that can regulate diverse cellular processes in response to nutritional status [8].
O-GlcNAcylation is conserved throughout species. O-GlcNAc modification in Caenorhabditis elegans plays an important role in longevity, stress and immunity [9]. In drosophila, knockdown of Ogt reduced IGF homologue expression, thereby affecting organism growth and glucose metabolism [10]. These observations indicate a potential role for O-GlcNAcylation in glucose metabolism in mammals. Indeed, previous studies in cultured cells have demonstrated that O-GlcNAcylation is involved in glucose metabolism in the liver [11], skeletal muscle [12] and adipose tissues [13], and accelerates insulin secretion in pancreatic beta cells [14]. However, little is known about the physiological roles of this post-translational modification in glucose metabolism in vivo because congenital Ogt-knockout (KO) mice are embryonic lethal [15].
In this study, to reveal the physiological role of O-GlcNAcylation in glucose metabolism in living mice, tamoxifen (TM)-inducible global Ogt-KO mice were analysed. In addition, mice lacking Ogt in glucose metabolism-related tissues, including the liver, skeletal muscle, adipose tissues and pancreatic beta cells, were analysed.
Methods
Animal experiments
All animal handling and experimentation were conducted according to the guidelines of the Research Center for Animal Life Science at Shiga University of Medical Science. All experimental protocols were approved by the Gene Recombination Experiment Safety Committee and the Research Center for Animal Life Science at Shiga University of Medical Science. Global and tissue-specific Ogt-KO mice were generated using the Cre-LoxP system. Ogt-flox (Ogt f/f) female mice [15] (Jackson Laboratory, Bar Harbor, ME, USA) were crossbred with R26-Cre-ERT2 [16] (Jackson Laboratory) to generate global Ogt-KO mice, and with Alb-Cre [17] (Jackson Laboratory), Mlc1f-Cre [18], Adipoq-Cre [19] and Pdx1 PB-CreER™ mice [20] to generate liver (Ogt-LKO), skeletal muscle (Ogt-MKO), adipose tissue (Ogt-FKO) and beta cell (Ogt-βKO) specific Ogt-KO mice, respectively. In Ogt-KO and Ogt-βKO mice, TM (Sigma Aldrich, St Louis, MO, USA) was injected i.p. for five consecutive days (0.15 g kg−1 day−1) at approximately 10–12 weeks of age. Only male mice were used for all experiments. Except for this, all samples were included for further analysis.
Islet isolation study
Primary mouse islets from Ogt-flox and Ogt-βKO mice were isolated by collagenase digestion as described previously with slight modifications [21].
Blood analysis
Blood glucose concentrations were measured with GDH-PQQ glucose test strips (Glutest Sensor, Sanwa Kagaku, Nagoya, Japan). Plasma insulin and glucagon levels were measured by ELISA (Morinaga, Tokyo, Japan, and Mercodia, Uppsala, Sweden).
Insulin and glucose tolerance tests
Mice were i.p. injected with 0.5 U/kg insulin or 1 g/kg glucose after fasting for 6–8 h. Blood glucose levels were determined 15, 30, 60, 90 and 120 min after injection.
Histological analyses
Fixed specimens embedded in paraffin were sectioned (3 μm thickness). Antibodies to O-linked N-acetylglucosamine (1:200, ab2739, Abcam, Cambridge, UK), insulin (1:500, ab7842, Abcam) and Ki67 (1:100, ab16667, Abcam) were used. Transmission electron microscopic analysis was performed with the Hitachi H-7500 (Hitachi, Tokyo, Japan). Cell apoptosis was analysed using the TACS2 TdT In Situ Apoptosis Detection Kit (Trevigen, Gaithersburg, MD, USA).
Total RNA preparation and qRT-PCR analysis
Total RNA was extracted from tissues using the RNeasy Kit (Qiagen, Valencia, CA, USA). cDNA was synthesised using reverse transcription reagents (Takara Bio, Otsu, Japan). Transcript abundance was assessed by real-time PCR on an Applied Biosystems 7500 Real-Time PCR System (PerkinElmer Applied Biosystems, Foster City, CA, USA) with SYBR Green (Bio-Rad Laboratories, Hercules, CA, USA). Analytical data were adjusted with 36B4 mRNA expression as an internal control. Primer sequences are provided in the electronic supplementary material (ESM) Table 1.
Western blot analysis
For western blot analysis, proteins were resolved by SDS-PAGE and transferred to nitrocellulose membranes. The membranes were then incubated with the following antibodies: OGT (1:1000, O6264, Sigma Aldrich); MGEA5 (3:10,000, ab124807, Abcam); RL2 (1:1000, MA1-072, Thermo Scientific, MA, USA); Actin (1:10,000, sc-1616, Santa Cruz Biotechnology, CA, USA) and β Actin (1:10,000, A5316, Sigma Aldrich). After additional washing, the membranes were incubated with horseradish peroxidase-linked secondary antibodies before chemiluminescence detection.
Statistical analysis
Data are expressed as means ± SEM. MANOVA followed by the Student’s t test was used for time course analysis. Student’s t tests were used to assess differences between two groups. The logrank test was performed for survival curves. A p value <0.05 was considered to be statistically significant.
Results
Ogt deletion in adult mice resulted in a lethal phenotype accompanied by body weight loss and hypoglycaemia
We first clarified the physiological role of O-GlcNAcylation in glucose metabolism in adult mice. Congenital global Ogt deletion is embryonic lethal, indicating that Ogt is critical for development in mice [15]. TM-inducible global Ogt-KO mice were generated by crossbreeding Ogt-flox female mice [15] with R26-Cre-ERT2 male mice [16] (Fig. 1a). At 10 weeks of age, Ogt deletion was induced by i.p. TM injection (Fig. 1a). Immunohistochemical analysis demonstrated that O-GlcNAcylation was present in both the nucleus and cytosol of liver, skeletal muscle, adipose tissue cells and particularly pancreatic islets, and O-GlcNAcylation was absent from these tissues in Ogt-KO mice (Fig. 1b). In addition, western blot analysis revealed decreases in Ogt and O-GlcNAcylation (RL2) levels in other tissues with varying magnitudes (ESM Fig. 1a). Following TM injection, Ogt-KO mice gradually lost weight accompanied by loss of body fat mass (Fig. 1c, d). Moreover, blood glucose and plasma insulin levels also gradually decreased (Fig. 1e, f). At 4 weeks after TM injection, Ogt-KO mice exhibited hypothermia and severe malnutrition, although their food intake did not significantly decrease (Fig. 1g, h, ESM Table 2). Finally, Ogt-KO mice began to die, and only 20% of Ogt-KO mice survived until 6 weeks after TM injection (Fig. 1i). As a result, we were unable to analyse glucose metabolism in these mice. These data indicate that O-GlcNAcylation is essential not only for embryonic development but also for survival in adult mice.

Inducible Ogt deletion in adult mice resulted in a lethal phenotype. (a) Schematic diagram of Ogt-KO mouse generation. (b) Immunohistochemical analysis of O-GlcNAcylation using anti-O-GlcNAc (RL2) antibodies in multiple tissues (original magnification ×100). iWAT, inguinal white adipose tissue; N/D, not determined due to lean phenotype. (c) Body weight of Ogt-KO mice and control mice following TM injection (n = 6 for Ogt-flox and n = 10 for Ogt-KO). (d) Inguinal fat mass of Ogt-KO mice and control mice 3 weeks after TM injection (n = 3 each). (e) Blood glucose levels of Ogt-KO mice and control mice following TM injection (n = 6 for Ogt-flox and n = 10 for Ogt-KO). (f) Plasma insulin levels of Ogt-KO mice and control mice following TM injection (n = 6 for Ogt-flox and n = 10 for Ogt-KO). (g) Body temperature of Ogt-KO mice and control mice 3 weeks after TM injection (n = 5 for Ogt-flox and n = 4 for Ogt-KO). (h) Food intake of Ogt-KO mice and control mice 3 weeks after TM injection (n = 8 for Ogt-flox and n = 6 for Ogt-KO). (i) Survival rates following TM injection (n = 8 for Ogt-flox and n = 18 for Ogt-KO). White circles, Ogt-flox; black circles, Ogt-KO. In (c), (e) and (f) MANOVA Pillai’s trace p < 0.01, p < 0.05, p < 0.05, respectively. Data are means ± SEM. *p < 0.05, † p = 0.08
To examine the role of O-GlcNAcylation in glucose metabolism, we generated tissue-specific Ogt-KO mice and analysed the role of O-GlcNAcylation in each tissue.
Insulin-sensitive tissue-specific Ogt deletion had no major effects on glucose metabolism
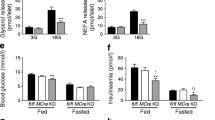
We generated tissue-specific Ogt-KO mice for insulin-sensitive tissues, including liver (Ogt-LKO), skeletal muscle (Ogt-MKO) and adipose tissue (Ogt-FKO), by crossbreeding Ogt-flox mice with Alb-Cre [17], Mlc1f-Cre [18] and Adipoq-Cre mice [19], respectively (ESM Fig. 2a). Western blot analysis indicated significant decreases in OGT protein levels and O-GlcNAcylation in these tissues (ESM Fig. 2b, c). Ogt-LKO, Ogt-MKO and Ogt-FKO mice grew normally and displayed no significant changes in fasting glucose levels compared with those of control littermates at 10 weeks of age (Fig. 2a–f). Furthermore, insulin-sensitive tissue-specific Ogt-KO mice exhibited normal glucose levels during an IPGTT. Additionally, they displayed little difference in insulin sensitivity by insulin tolerance test compared with control littermates (Fig. 2g–l). Since Ogt knockdown in insulin-sensitive tissues had little impact on glucose metabolism under physiological conditions, we evaluated the role of O-GlcNAcylation in pancreatic beta cells.

Ogt deletion in insulin-sensitive tissues had little impact on glucose metabolism. (a–c) Body weights and (d–f) blood glucose levels of insulin-sensitive tissue-specific Ogt-KO mice and littermate control mice at 10 weeks of age (n = 5–6 each). (g–i) Blood glucose concentrations during IPGTT (n = 4–5 each). MANOVA Pillai’s trace p = 0.964, p = 0.191, p = 0.926, respectively. (j–l) Glucose changes from baseline during i.p. insulin tolerance tests (n = 4–9 each). MANOVA Pillai’s trace p = 0.182, p = 0.879, p = 0.525, respectively. White circles, Ogt-flox; black circles, Ogt-LKO; black triangles, Ogt-MKO; black squares, Ogt-FKO. Data are means ± SEM
Pancreatic beta cell-specific Ogt-KO mice displayed biphasic changes in glucose metabolism
To evaluate the role of Ogt in glucose metabolism in pancreatic beta cells, we generated TM-inducible beta cell-specific Ogt-KO mice (Ogt-βKO) by crossbreeding female Ogt-flox mice with male Pdx1-CreER™ mice [20] (Fig. 3a). At 10 weeks of age, TM was injected i.p. in both groups of mice. Western blot and immunohistochemical analyses indicated that OGT protein levels and protein O-GlcNAcylation were markedly diminished in pancreatic beta cells from Ogt-βKO mice (Fig. 3b–e). Interestingly, in Ogt-βKO mice body weight was markedly increased 6 weeks after TM injection because of mild hyperphagia but subsequently decreased from 8 weeks post-injection (Fig. 3f, ESM Fig. 3a). In contrast, Ogt-βKO blood glucose levels transiently decreased at 5–6 weeks and then dramatically increased after 8 weeks post-injection (Fig. 3g). Finally, Ogt-βKO mice displayed severe hyperglycaemia at 10 weeks post-injection (Fig. 3g). Consistent with temporal changes in glucose levels, plasma insulin levels in Ogt-βKO mice transiently increased 5–7 weeks post-injection and were eventually depleted at 10 weeks (Fig. 3h). IPGTT were performed every week between 5 and 8 weeks post-injection. The results indicated a biphasic change in glucose tolerance, with a hypoglycaemic state occurring at 5–6 weeks post-injection (early phase) and a hyperglycaemic state occurring at 7–8 weeks post-injection (late phase) (Fig. 3i–m).

Ogt deletion in pancreatic beta cells resulted in biphasic changes in body weight and glucose metabolism. (a) Schematic diagram of pancreatic beta cell-specific Ogt-βKO mouse generation. (b) Representative western blot analysis for OGT and O-GlcNAcylation (RL2) levels in isolated islets. (c, d) Quantification of western blot analysis for OGT and RL2 (n = 4 each). (e) Representative immunohistochemical analysis for RL2 in the pancreas of Ogt-βKO and control mice at 4 weeks post-TM injection (original magnification ×100). (f–h) Temporal changes in body weight and blood glucose and plasma insulin levels in Ogt-βKO mice and control mice following TM injection (n = 7 for Ogt-flox and n = 8 for Ogt-βKO). (i–l) Glucose concentrations during IPGTT 5–8 weeks, respectively, after TM injection (n = 7 for Ogt-flox and n = 8 for Ogt-βKO). (m) Gradual changes in AUC during IPGTT 5–8 weeks after TM injection (n = 7 for Ogt-flox and n = 8 for Ogt-βKO). MANOVA Pillai’s trace: (f, h) p < 0.05; (g, i, j, l, m) p < 0.01; (k) p = 0.592. White circles, Ogt-flox; black circles, Ogt-βKO. Data are means ± SEM. *p < 0.05
Transient hyperinsulinaemia improved glucose tolerance and accelerated adiposity during the early phase
Ogt-βKO mice displayed lower glucose levels during the early phase but hyperglycaemia during the late phase (Fig. 3g). During the early phase, we observed significant increases in body weight in Ogt-βKO mice (Figs 3f, 4a). Furthermore, marked steatosis and increased adiposity in Ogt-βKO mice could be explained by hyperphagia resulting from enhanced insulin secretion followed by hypoglycaemia (Fig. 4b, c). In fact, Ogt-βKO mice displayed increased insulin secretion in response to a glucose challenge prior to weight gain (Fig. 4d, e). Histological analysis demonstrated that islet area, size and Ki67 positive cells were increased in Ogt-βKO mice (Fig. 4f–i), but no significant changes were detected in pancreatic islets by immunohistochemistry for insulin (Fig. 4j, k). Moreover, insulin secretion and content per isolated islet was increased in Ogt-βKO mice (Fig. 4l, m) and was accompanied by increased expression of the gene encoding insulin (Ins2) (Fig. 4n). These findings suggest that loss of O-GlcNAcylation in pancreatic beta cells may have lowered glucose levels during the early phase by increasing insulin secretion and enhancing islet growth. Taken together, in the early phase, deletion of Ogt-mediated O-GlcNAcylation resulted in accelerated insulin secretion in vivo, accompanied by increased insulin expression and islet growth, resulting in enhanced insulin-induced adiposity in vivo.

Loss of O-GlcNAcylation in pancreatic beta cells resulted in transient hyperinsulinaemia and subsequent adiposity. (a) Representative images of a beta cell-specific Ogt-βKO mouse and a control mouse 7 weeks after TM injection. (b) iWAT mass of Ogt-βKO and control mice (n = 6 each). BW, body weight. (c) Liver H&E staining of Ogt-βKO and control mice (original magnification ×100). (d, e) Insulin secretion and AUC during IPGTT in Ogt-βKO and control mice 5 weeks after TM injection (n = 7 for Ogt-flox and n = 8 for Ogt-βKO). In (d), MANOVA Pillai’s trace p < 0.05. (f–h) Representative H&E staining, islet/pancreas area and islet size in Ogt-βKO and control mice 5 weeks after TM injection (original magnification ×100). (i) Ki67 positive cells/islets in Ogt-βKO and control mice 5 weeks after TM injection. (j, k) Representative insulin immunohistochemical images and quantification (n = 6 each; original magnification ×100). (l) Insulin secretion in response to 2.8 mmol/l glucose, 25 mmol/l glucose and 25 mmol/l glucose +30 mmol/l KCl from isolated islets (ten islets; n = 4–5 each). (m) Insulin content in isolated islets from Ogt-βKO and control mice (ten islets; n = 12 for Ogt-flox and n = 15 for Ogt-βKO). (n) qPCR analysis of isolated islets from Ogt-βKO and control mice (n = 5 each). White circles and bars, Ogt-flox; black circles and bars, Ogt-βKO. Data are means ± SEM. *p < 0.05
Depletion of Ogt resulted in hypoinsulinaemia because of beta cell apoptosis
During the late phase, a significant increase in blood glucose levels was accompanied by depleted insulin secretion (Figs 3g, h, 5a, b). Histological analysis demonstrated decreased islet number and size (Fig. 5c–e). Furthermore, insulin-positive area was significantly decreased in Ogt-βKO mice compared with control mice (Fig. 5f, g). Transmission electron microscopic analysis revealed fewer insulin granules with abnormal morphology (Fig. 5h). Similarly, insulin and beta cell-related gene expression levels were significantly reduced (Fig. 5i). TUNEL staining indicated a significant increase in apoptotic cells in Ogt-βKO mouse islets (Fig. 5j, k). These findings suggest that Ogt-mediated O-GlcNAcylation is essential for beta cell survival.

Ogt-βKO mice displayed severe hyperglycaemia and insulin depletion associated with beta cell apoptosis. (a, b) Blood glucose and plasma insulin levels in Ogt-βKO and control mice 10 weeks after TM injection (n = 7 for Ogt-flox and n = 5 for Ogt-βKO). (c–e) Representative H&E staining, islet/pancreas area and islet size in Ogt-βKO and control mice 10 weeks after TM injection (original magnification ×100). (f, g) Insulin immunohistochemistry and quantification of insulin-positive area/islet (n = 6 each; original magnification ×100). (h) Transmission electron microscopy of Ogt-βKO and control mice 10 weeks after TM injection (original magnification ×2000). (i) qPCR analysis of isolated islets (n = 5 each). (j, k) TUNEL staining and TUNEL-positive cells/islet (n = 6 each; original magnification ×100). White bars, Ogt-flox; black bars, Ogt-βKO. Data are means ± SEM. *p < 0.05
Discussion
In this study, we comprehensively evaluated the role of O-GlcNAcylation in glucose metabolism in multiple tissues using tissue-specific Ogt-KO mice under physiological conditions. Because congenital Ogt gene deletion in mice caused embryonic lethality [15], little was known about the role of O-GlcNAcylation in systemic glucose metabolism. In this study, for the first time, we generated TM-inducible systemic Ogt-KO mice, which allowed us to analyse the role of O-GlcNAcylation in glucose metabolism in adult mammals. Ogt-KO mice displayed gradual body weight loss (Fig. 1c). Furthermore, Ogt-KO mouse mortality rates increased dramatically 4 weeks after TM injection (Fig. 1i). These results suggest that O-GlcNAcylation is essential not only for organ development in embryos but also for survival in adult mice.
Next we created insulin-sensitive tissue-specific Ogt-KO mice. Ogt knockdown in liver, skeletal muscle and adipose tissue had minimal impact on glucose homeostasis under physiological conditions (Fig. 2). In contrast to our results, previous studies have reported that excess O-GlcNAcylation in mice led to insulin resistance [22,23,24]. Collectively, these results suggest that gain-of-function of O-GlcNAcylation in insulin-sensitive organs is associated with the pathogenesis of insulin resistance, whereas loss-of-function does not impact glucose metabolism under physiological conditions. Further examination is necessary to investigate the loss of O-GlcNAcylation during various pathological conditions (e.g. diabetes), starvation and exercise.
A prominent phenotype was observed in Ogt-βKO mice. Acquired Ogt deletion revealed a unique phenotype. Specifically, mice displayed reduced glucose levels accompanied by enhanced insulin secretion during the early phase and hyperglycaemia accompanied by insulin depletion during the late phase (Fig. 3h–k). A recent study reported that loss of Ogt in pancreatic beta cells induced severe hyperglycaemia and insulin depletion, potentially because of endoplasmic reticulum (ER) stress, using a similar mouse model [25]. Their results were consistent with the late phase phenotype of our study, and increased levels of ER stress markers, such as Chop and Bip, were also observed in our model. Additionally, our study revealed that Ogt deletion transiently promoted insulin secretion and beta cell growth and subsequently increased adiposity during the early phase (Fig. 3). These results suggest that O-GlcNAcylation regulates not only beta cell survival but also insulin secretion and beta cell capacity under physiological conditions.
Two unresolved issues remain. The first issue is a technical limitation. A recent study reported that Pdx1 PB-CreER™ mice also express Cre in pancreatic alpha cells and the hypothalamus [26]. In our model, there were no significant differences in plasma glucagon levels between Ogt-βKO mice and control littermates, suggesting a limited effect of Ogt deletion on glucagon secretion (ESM Fig. 3b). In the hypothalamus, a recent report demonstrated that loss of O-GlcNAcylation in paraventricular nucleus neurons resulted in a rapid obese phenotype in which food intake more than doubled within 2 weeks following Cre induction [27]. Our mouse model also displayed transient obesity with mild hyperphagia. However, the increase in food intake was modest, and body weight gain was delayed compared with mice lacking O-GlcNAcylation in paraventricular nucleus neurons (ESM Fig. 3a). Furthermore, we observed hyperinsulinaemia and hypoglycaemia in the mice 5–6 weeks after Ogt deletion. Hypoglycaemia induced by hyperinsulinaemia is often accompanied by hyperphagia to alleviate hypoglycaemia. Therefore, it induces weight gain, which is often observed in people with type 2 diabetes receiving insulin treatment [28, 29]. Thus, we speculate that body weight gain between 5 and 8 weeks post-deletion is a secondary effect resulting from hypoglycaemia rather than an effect in the hypothalamus. Further investigation will be necessary to confirm the direct effects in pancreatic beta cells.
Second, the cause of death in the global Ogt-KO mouse is unclear. Although Ogt-βKO mice exhibited severe hyperglycaemia, none of the tissue-specific KO mice displayed the mortality rates observed in the global Ogt-KO mouse. One recent study demonstrated that heart-specific Ogt deletion was also lethal because of heart failure [30]. However, we did not detect elevated Bnp (also known as Nppb) mRNA expression in Ogt-KO mice (ESM Fig. 1b). Interestingly, although food intake did not decrease, severe malnutrition and hypometabolism were observed before death (ESM Table 2). Thus, we speculate that heart failure was not responsible for increased mortality rates in this model; however, malabsorption of nutrients could be the cause of the increased mortality rates. In addition, our results from global and beta cell-specific Ogt-KO mice clearly indicate that O-GlcNAcylation inhibition is inappropriate for the treatment of insulin resistance and diabetes complications because of its harmful effects. A recent proteomics approach revealed over 1000 OGT targets in mammalian cells [31]. Identification of the specific O-GlcNAcylated protein may provide a novel therapeutic strategy for the treatment of diabetes mellitus.
In conclusion, O-GlcNAcylation plays a pivotal role in glucose and cellular homeostasis in adult mammals. In addition, O-GlcNAcylation in insulin-sensitive tissues does not substantially impact glucose metabolism under physiological conditions, whereas O-GlcNAcylation in pancreatic beta cells has multiple effects on insulin secretion and beta cell homeostasis.
Abbreviations
- KO:
-
Knockout
- O-GlcNAc:
-
O-linked N-acetylglucosamine
- OGT:
-
O-GlcNAc transferase
- Ogt-βKO:
-
Beta cell-specific Ogt-knockout mice
- Ogt-FKO:
-
Adipose tissue-specific Ogt-knockout mice
- Ogt-LKO:
-
Liver-specific Ogt-knockout mice
- Ogt-MKO:
-
Skeletal muscle-specific Ogt-knockout miceTM Tamoxifen
- UDP-GlcNAc:
-
Uridine diphosphate N-acetylglucosamine
References
Kahn BB, Alquier T, Carling D, Hardie DG (2005) AMP-activated protein kinase: ancient energy gauge provides clues to modern understanding of metabolism. Cell Metab 1:15–25
Popovic D, Vucic D, Dikic I (2014) Ubiquitination in disease pathogenesis and treatment. Nat Med 20:1242–1253
Menzies KJ, Zhang H, Katsyuba E, Auwerx J (2016) Protein acetylation in metabolism—metabolites and cofactors. Nat Rev Endocrinol 12:43–60
Hanover JA, Krause MW, Love DC (2012) Bittersweet memories: linking metabolism to epigenetics through O-GlcNAcylation. Nat Rev Mol Cell Biol 13:312–321
Hardiville S, Hart GW (2014) Nutrient regulation of signaling, transcription, and cell physiology by O-GlcNAcylation. Cell Metab 20:208–213
Torres CR, Hart GW (1984) Topography and polypeptide distribution of terminal N-acetylglucosamine residues on the surfaces of intact lymphocytes. Evidence for O-linked GlcNAc. J Biol Chem 259:3308–3317
Janetzko J, Walker S (2014) The making of a sweet modification: structure and function of O-GlcNAc transferase. J Biol Chem 289:34424–34432
Zachara NE, Hart GW (2004) O-GlcNAc a sensor of cellular state: the role of nucleocytoplasmic glycosylation in modulating cellular function in response to nutrition and stress. Biochim Biophys Acta 1673:13–28
Love DC, Ghosh S, Mondoux MA et al (2010) Dynamic O-GlcNAc cycling at promoters of Caenorhabditis elegans genes regulating longevity, stress, and immunity. Proc Natl Acad Sci USA 107:7413–7418
Sekine O, Love DC, Rubenstein DS, Hanover JA (2010) Blocking O-linked GlcNAc cycling in drosophila insulin-producing cells perturbs glucose-insulin homeostasis. J Biol Chem 285:38684–38691
Ruan HB, Han X, Li MD et al (2012) O-GlcNAc transferase/host cell factor C1 complex regulates gluconeogenesis by modulating PGC-1alpha stability. Cell Metab 16:226–237
Wang X, Feng Z, Wang X et al (2016) O-GlcNAcase deficiency suppresses skeletal myogenesis and insulin sensitivity in mice through the modulation of mitochondrial homeostasis. Diabetologia 59:1287–1296
Vosseller K, Wells L, Lane MD, Hart GW (2002) Elevated nucleocytoplasmic glycosylation by O-GlcNAc results in insulin resistance associated with defects in Akt activation in 3T3-L1 adipocytes. Proc Natl Acad Sci USA 99:5313–5318
Gao Y, Miyazaki J, Hart GW (2003) The transcription factor PDX-1 is post-translationally modified by O-linked N-acetylglucosamine and this modification is correlated with its DNA binding activity and insulin secretion in min6 beta cells. Arch Biochem Biophys 415:155–163
Shafi R, Iyer SP, Ellies LG et al (2000) The O-GlcNAc transferase gene resides on the X chromosome and is essential for embryonic stem cell viability and mouse ontogeny. Proc Natl Acad Sci USA 97:5735–5739
Ventura A, Kirsch DG, McLaughlin ME et al (2007) Restoration of p53 function leads to tumour regression in vivo. Nature 445:661–665
Weisend CM, Kundert JA, Suvorova ES, Prigge JR, Schmidt EE (2009) Cre activity in fetal albCre mouse hepatocytes: utility for developmental studies. Genesis 47:789–792
Bothe GW, Haspel JA, Smith CL, Wiener HH, Burden SJ (2000) Selective expression of Cre recombinase in skeletal muscle fibers. Genesis 26:165–166
Eguchi J, Wang X, Yu S et al (2011) Transcriptional control of adipose lipid handling by IRF4. Cell Metab 13:249–259
Zhang H, Fujitani Y, Wright CV, Gannon M (2005) Efficient recombination in pancreatic islets by a tamoxifen-inducible Cre-recombinase. Genesis 42:210–217
Fujimoto S, Ishida H, Kato S et al (1998) The novel insulinotropic mechanism of pimobendan: direct enhancement of the exocytotic process of insulin secretory granules by increased Ca2+ sensitivity in beta cells. Endocrinology 139:1133–1140
Virkamaki A, Daniels MC, Hamalainen S, Utriainen T, McClain D, Yki-Jarvinen H (1997) Activation of the hexosamine pathway by glucosamine in vivo induces insulin resistance in multiple insulin sensitive tissues. Endocrinology 138:2501–2507
McClain DA, Lubas WA, Cooksey RC et al (2002) Altered glycan-dependent signaling induces insulin resistance and hyperleptinemia. Proc Natl Acad Sci U S A 99:10695–10699
Yang X, Ongusaha PP, Miles PD et al (2008) Phosphoinositide signalling links O-GlcNAc transferase to insulin resistance. Nature 451:964–969
Alejandro EU, Bozadjieva N, Kumusoglu D et al (2015) Disruption of O-linked N-acetylglucosamine signaling induces ER stress and beta cell failure. Cell Rep 13:2527–2538
Wicksteed B, Brissova M, Yan W et al (2010) Conditional gene targeting in mouse pancreatic β-cells: analysis of ectopic Cre transgene expression in the brain. Diabetes 59:3090–3098
Lagerlof O, Slocomb JE, Hong I et al (2016) The nutrient sensor OGT in PVN neurons regulates feeding. Science 351:1293–1296
The Diabetes Control and Complications Trial Research Group (1993) The effect of intensive treatment of diabetes on the development and progression of long-term complications in insulin-dependent diabetes mellitus. N Engl J Med 329:977–986
Dewan S, Gillett A, Mugarza JA, Dovey TM, Halford JC, Wilding JP (2004) Effects of insulin-induced hypoglycaemia on energy intake and food choice at a subsequent test meal. Diabetes Metab Res Rev 20:405–410
Watson LJ, Long BW, DeMartino AM et al (2014) Cardiomyocyte Ogt is essential for postnatal viability. Am J Physiol Heart Circ Physiol 306:H142–H153
Nagel AK, Ball LE (2014) O-GlcNAc transferase and O-GlcNAcase: achieving target substrate specificity. Amino Acids 46:2305–2316
Acknowledgements
We are indebted to K. Kosaka, Y. Asano, T. Yamamoto and the Central Research Laboratory of Shiga University of Medical Science for their expert technical assistance in this study. We thank S.J. Burden (New York University Medical Center) and K. Ueki (Tokyo University) for providing Mlc1f-Cre mice. We also thank E. Rosen (Harvard University), W. Ogawa and T. Hosooka (Kobe University) for providing Adipoq-Cre mice, and M. Gannon (Vanderbilt University Medical Center) for providing Pdx1 PB-CreER™ mice. Some of the data were presented as abstracts at the 75th Scientific Sessions of the ADA in 2015 and Keystone Symposia in 2015, and have been submitted to the third Korea–Japan Diabetes Forum in 2017.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Data availability
The data that support the findings of this study are available from the corresponding author upon reasonable request.
Funding
This study was supported by Grants-in-Aid for Scientific Research (KAKENHI) from the Japan Society for the Promotion of Science (No. 15K09383 to OS, No. 16K09743 to SU, No. 16K09744 to KM). This study was partially supported by the Shiga University of Medical Science President’s Grant for the Encouragement of Young Researchers. The study was funded by the Shiga University of Medical Science. The Department of Medicine, Shiga University of Medical Science reports receiving research promotion grants (Shogaku Kifukin) from Astellas, Boehringer-Ingelheim, Daiichi-Sankyo, Kowa Pharmaceuticals, Kyowa Hakko Kirin, Mitsubishi Tanabe, MSD, Ono Pharmaceutical, Sanofi, Sanwa Kagaku Kenkyusho, Shionogi, Taisho-Toyama, Takeda and Teijin Pharma. However, the research topics of these donation grants are not restricted.
Duality of interest
The authors declare that there is no duality of interest associated with this manuscript.
Contribution statement
SI, OS, SK, SU and HM designed the study. SI, OS, NO, KI and NH conducted the research. SI, KM, TC, NH and NI analysed and interpreted the data. SI, KM and HM wrote the manuscript. OS, NO, KM, SU, SK, TC, KI, NH, NI and HM reviewed and edited the manuscript. KM had primary responsibility for the final content. All authors read and approved the final manuscript.
Electronic supplementary material
ESM
(PDF 565 kb)
Rights and permissions
About this article

Cite this article
Ida, S., Morino, K., Sekine, O. et al. Diverse metabolic effects of O-GlcNAcylation in the pancreas but limited effects in insulin-sensitive organs in mice. Diabetologia 60, 1761–1769 (2017). https://doi.org/10.1007/s00125-017-4327-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00125-017-4327-y