
Abstract
Aims/hypothesis
Pancreatic islet microendothelium exhibits unique features in interdependent relationship with beta cells. Gastrointestinal products of the ghrelin gene, acylated ghrelin (AG), unacylated ghrelin (UAG) and obestatin (Ob), and the incretin, glucagon-like peptide-1 (GLP-1), prevent apoptosis of pancreatic beta cells. We investigated whether the ghrelin gene products and the GLP-1 receptor agonist exendin-4 (Ex-4) display survival effects in human pancreatic islet microendothelial cells (MECs) exposed to chronic hyperglycaemia.
Methods
Islet MECs were cultured in high glucose concentration and treated with AG, UAG, Ob or Ex-4. Apoptosis was assessed by DNA fragmentation, Hoechst staining of the nuclei and caspase-3 activity. Western blot analyses and pharmacological inhibition of protein kinase B (Akt) and extracellular signal-related kinase (ERK)1/2 pathways, detection of intracellular cAMP levels and blockade of adenylyl cyclase (AC)/cAMP/protein kinase A (PKA) signalling were performed. Levels of NO, IL-1β and vascular endothelial growth factor (VEGF)-A in cell culture supernatant fractions were measured.
Results
Islet MECs express the ghrelin receptor GHS-R1A as well as GLP-1R. Treatment with AG, UAG, Ob and Ex-4 promoted cell survival and significantly inhibited glucose-induced apoptosis, through activation of PI3K/Akt, ERK1/2 phosphorylation and intracellular cAMP increase. Moreover, peptides upregulated B cell lymphoma 2 (BCL-2) and downregulated BCL-2-associated X protein (BAX) and CD40 ligand (CD40L) production, and significantly reduced the secretion of NO, IL-1β and VEGF-A.
Conclusions/interpretation
The ghrelin gene-derived peptides and Ex-4 exert cytoprotective effects in islet MECs. The anti-apoptotic effects involve phosphoinositide 3-kinase (PI3K)/Akt, ERK1/2 and cAMP/PKA pathways. These peptides could therefore represent a potential tool to improve islet vascularisation and, indirectly, islet cell function.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Despite the evidence of some turnover in adult humans and the apparent capacity of endocrine pancreatic beta cells to regenerate throughout life, patients with type 1, as well as type 2, diabetes have a substantial deficit in beta cell mass. Concomitant with the immune-mediated beta cell destruction, additional impairments in insulin secretion and beta cell regeneration have been proposed in type 1 diabetes. Likewise, type 2 diabetes is accompanied by beta cell apoptosis and failure of beta cell mass to expand adequately in response to rising secretory demand [1]. Therefore, identification of molecules promoting both beta cell survival and function, or regulating pancreatic gene expression, would help in understanding the pathophysiology of diabetic disease and to develop new therapeutic strategies to maintain or restore beta cell mass.
Within this context, recent studies indicate that newly described gastrointestinal peptides may stimulate proliferation and prevent apoptosis of pancreatic beta cells. These peptides include the ghrelin gene products, acylated ghrelin (AG), unacylated ghrelin (UAG) and the recently identified peptide, obestatin (Ob). The ghrelin system appears to have a paracrine/autocrine role in the regulation of hormonal secretion and islet growth and differentiation in fetal life [2, 3]. A reciprocal relationship exists between AG and insulin, suggesting that AG negatively regulates glucose homeostasis, whereas UAG counteracts the metabolic activities of AG [4]. Further, the ghrelin isoforms may have a role in regulating beta cell fate and function, in both physiological and pathological conditions [5]. The actions of the 23-amino-acid amidated peptide Ob, initially described as a physiological opponent of ghrelin, are still mainly unknown [6]. Ob has been shown to promote beta cell survival, activate signalling pathways and induce expression of genes that regulate beta cell growth, differentiation, insulin synthesis and glucose metabolism [7]. The incretin glucagon-like peptide-1 (GLP-1), the major gastrointestinal product of proglucagon processing, has been shown to exert trophic effects on beta cells [8]. GLP-1 stimulates beta cell proliferation, enhances differentiation of new beta cells from progenitors and inhibits beta cell apoptosis. GLP-1 receptors are abundantly expressed in many cell types other than pancreatic islet cells, indicating a role wider than expected [9].
In their broad range of biological actions, the ghrelin gene products, as well as GLP-1, feature a variety of cardiovascular activities, and share protective effects on cardiomyocytes and endothelial cells [9–13]. This also renders these peptides of interest at the microvascular level, which is endowed with a remarkable heterogeneity of endothelial phenotype and function [14, 15]. Within the pancreatic islets, the existence of an endothelial–endocrine axis from fetal to adult life is well established, and the dense islet capillary network plays an integral role in a number of islet functions [16–19]. These functions include insulin gene expression, optimised beta cell secretory function and blood glucose regulation, mediated by growth factors, collagen IV or other basement membrane components [14, 20]. The islet microcirculation appears to participate in sensing the environment and generating signals to induce the growth of the islets in response to physiological stimuli, such as insulin resistance or pregnancy [21]. Indeed, increased islet vasculature has been observed in association with expanded islet mass, and disrupted islet vascular architecture and perfusion are pathological hallmarks in animal models of diabetes [22–24]. Moreover, a diabetic milieu induces relevant alterations in islet endothelial cells, leading to apoptosis and NO and cytokine production [25–27]. At present, no data are available on the effects of gastrointestinal peptides in islet endothelium.
The aim of the present study was to evaluate whether AG, UAG, Ob and the GLP-1R agonist exendin-4 (Ex-4) exert cytoprotective effects in human islet endothelium. We investigated the potential anti-apoptotic and survival actions of these peptides in human islet endothelial cells exposed to hyperglycaemia and the underlying signalling pathways involved.
Methods
Cell culture conditions
Primary islet microendothelial cells (MECs) were isolated from human islets discarded from transplant use and prepared using a modification of Ricordi’s technique [28]. The immortalised cell line was prepared using human islets obtained from an organ donor and prepared in the Cell Isolation Unit at King’s College Hospital [29]; these studies were approved by the local Ethical Review Committee. Primary islet MECs were positively selected using anti-CD105 immunomagnetic beads, purified, cultured onto endothelial cell attachment factor (Sigma Aldrich, Milan, Italy) in complete endothelial basal medium (EBM; Clonetics, San Diego, CA, USA) and characterised [30] (see the electronic supplementary material [ESM]). In high glucose conditions, the medium was adjusted to 28 mmol/l glucose (Sigma) [25]. Experiments were conducted in parallel with physiological (5.6 mmol/l) concentration of glucose. Because of the need for a long-term culture in high glucose conditions, the SV40-immortalised human cell line, established from purified islet MECs, was used [29].
Cell survival and proliferation were assessed by trypan blue exclusion cell count and by measuring DNA synthesis by the 5-bromo-2′-deoxyuridine (BrdU) incorporation immunoassay (Cell Proliferation ELISA, BrdU assay; Roche Diagnostics, Mannheim, Germany). Cell viability was assessed by the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay [31]. Five separate experiments in triplicate, at different time points and with different conditions, were performed.
In experimental conditions evaluating the effects of AG, UAG, Ob and Ex-4, cells were incubated with 100 μg/ml oxidised LDL (oxLDL) for 24 h at 37°C, as a known pro-apoptotic stimulus [25]. To evaluate the effects of AG, UAG, Ob and Ex-4 on cell proliferation, survival and apoptosis, induced by high glucose or oxLDL, as well as the effects on protein kinase B (Akt), extracellular signal-related kinase (ERK)1/2 and cAMP/protein kinase A (PKA) pathways, islet MECs were incubated with increasing doses of AG, UAG, Ob or Ex-4 (10–100 nmol/l; Phoenix Pharmaceuticals, CA, USA), on the basis of previous reports [7, 31]. Two different conditions were assayed: peptides were given daily or added overnight before cell collection. Peptide 14-1 (GSSFLSPEHQRVQQ), the inverse sequence of UAG fragment 1–14 (Tib MolBiol, Genoa, Italy) was used as the control peptide. The effects of peptides on cell survival and proliferation were also assessed in human umbilical vein-derived endothelial cells (HUVECs).
To assess the secretion of vascular endothelial growth factor (VEGF)-A and to dissect the distinct contribution to its secretion of islet endothelium and beta cells, human islet MECs and a rat glucose-sensitive clonal beta cell line, INS-1E, were used and cultured as described [31].
Receptor expression
Total RNA was extracted using RNeasy Mini spin columns and reverse transcribed (2 μg) according to standard protocols using avian myeloblastosis virus reverse transcriptase and oligo(dT). GHSR1A, GLP1R and GPR39 mRNA expression were assessed by standard PCR, as described previously [7, 31]. Quantitative real-time PCR was also performed (see the ESM).
To assess whether the effects of AG were mediated by the GHS-R1A receptor, cells were incubated with AG in the presence of 10 nmol/l of d-[Lys3]-GHRP-6 (Phoenix Pharmaceuticals), selective antagonist of the cognate AG receptor.
At the protein level, cell surface expression of GHS-R1A and GPL-1R was assessed by immunofluorescence (IF) and confocal analysis; IF studies were also performed on cryostatic sections of pancreas tissue (ESM).
Carboxytetramethylrhodamine (TAMRA)-Ob, a fluorescent Ob derivative (Inbios, Naples, Italy) was synthesised and used as a probe for Ob binding sites; binding was performed as described [13] at 4°C and 37°C. 5(6)-TAMRA was used as negative control.
Detection of apoptosis
Apoptosis was evaluated in time course experiments, at 3–6 days intervals, by a photometric enzyme immunoassay measuring mono- and oligonucleosomes in the cytoplasmic fraction of cell lysates, as an index of DNA fragmentation (Cell Death Detection ELISAPLUS, Roche). Further, islet MECs were also subjected to Hoechst 33258 assay analysis, as described previously [31], and activation of the caspase family was assessed using the caspase-3 Colorimetric Activity Assay kit (Chemicon International, Temecula, CA, USA). Three separate experiments, in triplicate, at different time points during different culture conditions, were performed.
Western blot analyses and intracellular cAMP levels
Islet MECs, subjected to different experimental conditions, were lysed at 4°C for 30 min in lysis buffer [20]. Samples were centrifuged, normalised to 50 μg/sample in 20 μl, resolved by 8% SDS-polyacrylamide gel electrophoresis under reducing conditions and transferred to nitrocellulose. Membranes were blocked and incubated with one of the specific antibodies (mouse monoclonal anti-phosphorylated Akt (p-Akt) or anti-Akt, or anti-phosphorylated ERK (p-ERK) or anti-ERK, Cell Signaling Technology, Beverly, MA, USA) overnight at 4°C [25, 31]. Further, MECs were incubated with 100 μmol/l 3-isobutyl-1-methylxanthine and cAMP was measured from lysates using the Direct Cyclic AMP EIA kit (Assay Designs, Milan, Italy), according to the manufacturer’s instructions.
In experiments on the pharmacological inhibition of phosphoinositide 3-kinase (PI3K), ERK1/2, adenylyl cyclase (AC) or PKA, islet MECs were treated 1 h before AG, UAG or Ob treatment with two unrelated PI3K pharmacological inhibitors, wortmannin (0.1 μmol/l) and LY294002 (10 μmol/l), or an ERK inhibitor, PD98059 (50 μmol/l), an AC inhibitor, MDL12330A (100 nmol/l), or a PKA inhibitor, KT5720 (5 μmol/l). Three experiments were performed for each condition.
Membranes were incubated overnight at 4°C with anti-CD40, anti-CD40L, anti-BAX and anti-BCL-2 antibodies (Santa Cruz Biotechnology, Heidelberg, Germany; 1:200). Blots were probed with peroxidase-conjugated goat anti-mouse IgG (1:5,000; Pierce, Rockford, IL, USA) or peroxidase-conjugated goat anti-rabbit IgG (Amersham, Little Chalfont, UK) for 1 h at room temperature and developed with chemiluminescence reagents (ECL; Amersham, Milan, Italy). Three separate experiments, in duplicate, for each condition, were performed. Equal protein loading was confirmed by β-actin production.
The level of CD40L was also evaluated by flow cytometric analysis using CellQuest software (BD Biosciences, Milan, Italy) [32], in the presence of interleukin (IL)-1β (10 ng/ml) and TNF-α (50 ng/ml) for 24 h.
IL-1β, NO and VEGF-A detection
Cell culture supernatant fractions were collected before each subculture and medium exchange and stored at −80°C. IL-1β was measured by quantitative sandwich enzyme immunoassay (R&D Systems, Abingdon, UK), according to the manufacturer’s instructions. NO was determined as nitrite concentration in supernatant fractions by diazotisation reaction, using NaNO2 as standard [25]. VEGF-A levels in supernatant fractions of islet MECs and INS-1E beta cells were detected by human and rat (respectively) quantitative sandwich enzyme immunoassay (R&D, Milan, Italy).
Statistical analysis
BrdU incorporation, MTT assay, levels of apoptosis (i.e. optical density for DNA fragmentation), levels of p-Akt/Akt, p-ERK1/2/ERK1/2, cAMP, IL-1β, NO and VEGF-A in different culture conditions were compared using the Mann–Whitney U test. Data were analysed using SPSS statistical package (SPSS, Chicago, IL, USA), and p values less than 0.05 were considered significant. Because of the batch-to-batch and interassay variations, in some experiments data are represented as percentages of variation (mean ± SD) of the results obtained in 5.6 mmol/l glucose conditions within each experiment, unless otherwise stated.
Results
Expression of receptors for AG, Ob and Ex-4 on islet MECs
PCR analyses showed that islet MECs express the genes encoding the AG receptor, GHSR1A, and the GLP1 receptor, GLP1R, but not GPR39, which encodes one of the potential Ob receptors (Fig. 1a). The expression was quantified by real-time PCR (Fig. 1b).

Expression of GLP1R, GHSR1A and GPR39 and Ob binding. a Conventional PCR analysis of GLP1R, GHSR1A and GPR39 and of ACTB mRNA expression in islet MECs (lanes 2, 5, 8 and 11). M, 100-bp ladder; omission of cDNA in the PCR mixture was used as a negative control (lanes 1, 4, 7 and 10) and pituitary cells were used as a positive control (3, 6, 9, 12). Omission in the PCR mixture of reverse transcriptase enzyme during retrotranscription was also used as a negative control, providing negative results. b Islet MEC mRNA levels of GHSR1A and GLP1R determined by real-time PCR. Results were normalised to an 18S rRNA transcript, and expressed as fold variation compared with brain gene expression (\( {2^{{ - \Delta \Delta {{\text{C}}_{\text{t}}}}}} \)cycle threshold [Ct]) (means ± SD of three independent experiments). Brain and adipose tissues were used as positive controls. Omission of RNA in the retrotranscription to cDNA provided background signals below the threshold line used to calculate the Ct value in the real-time PCR reaction. GPR39 was not expressed in islet MECs (data not shown). c Representative confocal immunofluorescence micrographs of islet MECs, stained with anti-GHS-R1A and anti-GLP-1R antibodies, showing a diffuse receptor expression in islet MECs (original magnification 630×, nuclei stained in blue with DAPI). Immunofluorescence analysis of TAMRA-Ob binding to islet MECs, which is reduced by incubation with unlabelled Ob (competitor). Ctrl, 5(6)-TAMRA. Ob appears to be localised mainly on the cell surface at 4°C and internalised in the cytoplasm at 37°C. d Representative immunofluorescence micrographs of pancreatic islets stained with anti-GHS-R1A and anti-GLP-1R antibodies (detected with Alexa Fluor 488 conjugated secondary antibody) (original magnification 400×), and anti-CD31 (detected with Alexa Fluor 594 conjugated secondary antibody). The merge indicates the association of GHS-R1A and GLP-1R with vessel endothelial cells. GLP-1R is also present on endocrine cells. Three different pancreases were examined with similar results
Immunofluorescence studies detected GHS-R1A and GLP-1R protein expression on islet MECs. These studies detected labelled Ob binding to islet MECs, which was reduced by unlabelled Ob. Ob appeared localised mainly on the cell surface at 4°C, while the majority was internalised at 37°C (Fig. 1c). GHS-R1A and GLP-1R proteins were also expressed on sections of human pancreatic islets. Co-localisation with CD31 indicated the expression of GHS-R1A and GLP-1R on endothelial cells lining islet vessels (Fig. 1d).
AG, UAG, Ob and Ex-4 inhibit the effects of high glucose on islet MEC survival and apoptosis
Under conditions of chronic hyperglycaemia, survival and proliferation of islet MECs progressively decreased and apoptosis increased, compared with normoglycaemia (p < 0.05; Figs 2a,b, 3a). Under this condition, the exposure of islet MECs to 10 nmol/l AG, UAG, Ob or Ex-4 promoted cell survival and proliferation. After a 1 week culture in high glucose, the number of viable cells, by trypan blue exclusion, was reduced by 33 ± 11% (n = 5) compared with cells cultured in physiological glucose concentration. Peptide treatment increased the viable cell number by 36 ± 15% (AG), 31 ± 25% (UAG), 38 ± 16% (Ob) and 58 ± 15% (Ex-4; n = 5) compared with cells cultured in high glucose without peptides (p < 0.05 for AG, Ob and Ex-4, p = 0.1 for UAG).

Ghrelin peptides and Ex-4 promote cell survival and proliferation of islet MECs cultured in high glucose. a Cell survival, assessed by MTT assay, of islet MECs cultured for 2 or 14 days in 28 mmol/l glucose without peptides (1, black bars) or treated with oxLDL (1, black bar), compared with parallel cultures in normal glucose (NG, white bar) or high glucose (HG) cultures of cells treated with 10 nmol/l AG (2, grey bars), UAG (3, grey bars), Ob (4, grey bars) or Ex-4 (5, crosshatched bars). Scrambled peptide 14-1 (6, white bar) in the 14 day culture represents the control peptide. Culture with 10 nmol/l AG and selective antagonist of the GHS-R1A receptor, d-[Lys3]-GHRP-6 (7, white bar). b Proliferation, assessed as BrdU incorporation, of islet MECs cultured for 2 or 14 days in 28 mmol/l glucose without peptides (1, black bars) or treated with oxLDL (1, black bar), compared with parallel cultures in normal glucose (white bar) or high glucose cultures of cells treated with 10 nmol/l AG (2, grey bars), UAG (3, grey bars), Ob (4, grey bars) or Ex-4 (5, crosshatched bars). Scrambled peptide 14-1 (6, white bar) at 14 day culture represents the control peptide. Culture with 10 nmol/l AG and selective antagonist of the GHS-R1A receptor, d-[Lys3]-GHRP-6 (7, white bar). OD, optical density. Data are expressed as mean ± SD of three to five different experiments for each time or treatment point.* p < 0.05 compared with parallel high glucose cultures without peptides. † p < 0.05 compared with normal glucose cultures

Ghrelin gene peptides and Ex-4 inhibit the effects of high glucose and oxLDL on apoptosis in islet MECs. a Apoptosis, assessed as DNA fragmentation of islet MECs, cultured for 2 or 14 days in 28 mmol/l glucose (HG) without peptides (1, black bars) or treated with oxLDL (1, black bar), compared with parallel cultures in normal glucose (NG, white bar) or high glucose cultures of cells treated with 10 nmol/l AG (2, grey bars), UAG (3, grey bars), Ob (4, grey bars) or Ex-4 (5, crosshatched bars). Scrambled peptide 14-1 (6, white bar) in the 14 day culture represents the control peptide. Culture with 10 nmol/l AG and a selective antagonist of the GHS-R1A receptor, d-[Lys3]-GHRP-6 (7, white bar). b Assessment of caspase-3 activity in islet MECs incubated for up to 7 days in normal (white bar) or 28 mmol/l glucose without peptides (1, black bars) compared with parallel high glucose cultures of cells treated with 10 nmol/l AG (2, grey bars), UAG (3, grey bars), Ob (4, grey bars), or Ex-4 (5, crosshatched bars). OD, optical density. Data are expressed as mean ± SD of three different experiments for each time or treatment point. * p < 0.05 compared with parallel high glucose cultures without peptides. † p < 0.05 compared with normal glucose cultures. c Representative micrographs of Hoechst 33258 nuclear immunofluorescence staining (original magnification ×100) of MECs cultured in normal glucose, or in high glucose, treated with 10 nmol/l AG, UAG, Ob or Ex-4
Cell survival by MTT assay was significantly enhanced as early as 2 days following treatment, reaching an increase of 80% (AG), 68% (UAG), 80% (Ob) and 84% (Ex-4) after 14 days of high glucose culture (p < 0.05 compared with parallel cultures in high glucose without peptides) (Fig. 2a, Table 1). Concomitantly, under similar conditions, peptide treatment increased cell proliferation, reaching a significant mean increase of 86% (AG), 64% (UAG), 87% (Ob) and 145% (Ex-4) after 14 days of high glucose culture (p < 0.05 compared with cells cultured in high glucose without peptides) (Fig. 2b, Table 1).
Peptide treatment also substantially increased HUVEC survival by 62% (AG), 51% (UAG), 70% (Ob) and 48% (Ex-4) and proliferation by 19% (AG), 22% (UAG), 28% (Ob) and 32% (Ex-4) after 14 days of high glucose culture, although without reaching statistical significance.
Exposure to 10 nmol/l AG, UAG, Ob and Ex-4 significantly inhibited apoptosis induced by high glucose and by 100 μg/ml oxLDL. In time course experiments, this effect was evident from the first 2 days of high glucose culture in the presence of peptides, progressively reaching a mean decrease in apoptosis of 84% (AG), 84% (UAG), 82% (Ob) and 68% (Ex-4) after 14 days of culture (p < 0.05 compared with parallel high glucose cultures without peptides; Fig. 3a, Table 1). These data were confirmed by the significant inhibition of caspase-3 activation (Fig. 3b) and by Hoechst staining of apoptotic nuclei (Fig. 3c). The cytoprotective activities of AG were abrogated by coincubation with d-[Lys3]-GHRP-6 (Figs. 2a, b, 3a), and incubation with peptide 14-1 did not show any protective effect.
AG, UAG, Ob and Ex-4 promote islet MEC survival through activation of PI3K/Akt, ERK1/2 and AC/cAMP/PKA signalling pathways
Figure 4 shows that protection against glucose-induced cell death was accompanied by increased phosphorylation of Akt and ERK1/2. The effect was evident both for overnight and daily peptide treatment, in 7 day high glucose culture. The ratio of p-Akt/Akt increased by 62% (AG), 73% (UAG), 155% (Ob) and 188% (Ex-4) in an overnight treatment regime, and by 56% (AG), 28% (UAG), 140% (Ob) and 162% (Ex-4) in a daily regime. The increase was statistically significant only for Ob and Ex-4 (Fig. 4a). The ratio of p-ERK1/2/ERK1/2 increased by 244% (AG), 279% (UAG), 311% (Ob) and 305% (Ex-4) in an overnight treatment regime, and by 77% (AG), 81% (UAG), 63% (Ob) and 49% (Ex-4) in a daily regime, with statistical significance for all peptides (Fig. 4b).

Ghrelin gene peptides and Ex-4 prevent the reduction of Akt and ERK phosphorylation and intracellular cAMP levels. a Phosphorylation of Akt and the p-Akt/Akt ratio evaluated as densitometric analysis of western blot bands in islet MECs cultured in normal (NG) or 28 mmol/l glucose (HG) without peptides for 7 days (1, black bars), or treated with 10 nmol/l AG (2, grey bars), UAG (3, grey bars), Ob (4, grey bars) or Ex-4 (5, grey bars) in two regime treatments, 7 days or 12 h. b Phosphorylation of ERK1/2 and the p-ERK/ERK ratio evaluated as densitometric analysis of western blot bands in islet MECs cultured in normal or 28 mmol/l glucose without peptides for 7 days (1, black bars) or treated with 10 nmol/l AG (2, grey bars), UAG (3, grey bars), Ob (4, grey bars) or Ex-4 (5, cross hatched bars) in two regime treatments, 7 days or 12 h. c Intracellular cAMP levels in islet MECs cultured in normal glucose or 28 mmol/l glucose without peptides for 7 days (1, black bar) compared with parallel cultures of cells treated with 10 nmol/l AG (2, grey bar), UAG (3, grey bar), Ob (4, grey bar) or Ex-4 (5, crosshatched bar). Data are expressed as mean ± SD of three different experiments for each time or treatment point.* p < 0.05 compared with parallel high glucose cultures without peptides. † p < 0.05 compared with normal glucose cultures
Cultures with specific inhibitors of either PI3K/Akt (LY294002 and wortmannin) or ERK1/2 (PD98059) significantly prevented the cytoprotective effects observed with peptide treatment (Fig. 5a–c).

Ghrelin gene peptides and Ex-4 effects are prevented by pharmacological inhibition of PI3K/Akt, ERK1/2, AC or PKA on cell survival and apoptosis of islet MECs cultured in high glucose. a A reduction in cell survival, assessed by MTT assay, was induced by simultaneous treatment of islet MECs cultured in 28 mmol/l glucose (HG) for 7 days, with AG, UAG, Ob, or Ex-4 (vehicle) and inhibitors of PI3K (LY294002, 10 μmol/l [Ly] and wortmannin, 0.1 μmol/l [Wort]) or ERK1/2 (PD98059, 50 μmol/l [PD]) or AC (MDL12330A, 100 nmol/l [MDL]) or PKA (KT5720, 5 μmol/l [KT]). The black bar shows high glucose cultures without peptides. b An increase in apoptosis, assessed as DNA fragmentation, was induced by simultaneous treatment of islet MECs cultured in 28 mmol/l glucose for 7 days, with AG, UAG, Ob, or Ex-4 (vehicle) and inhibitors of PI3K (LY294002, 10 μmol/l and wortmannin, 0.1 μmol/l) or ERK1/2 (PD98059, 50 μmol/l) or AC (MDL12330A, 100 nmol/l) or PKA (KT5720, 5 μmol/l). The bar shows high glucose cultures without peptides. c An increase in caspase-3 activity was induced by simultaneous treatment of islet MECs cultured in 28 mmol/l glucose for 7 days, with AG, UAG, Ob or Ex-4 (vehicle) and inhibitors of PI3K (LY294002, 10 μmol/l and wortmannin, 0.1 μmol/l), ERK1/2 (PD98059, 50 μmol/l), AC (MDL12330A, 100 nmol/l) or PKA (KT5720, 5 μmol/l). The black bar shows high glucose cultures without peptides. OD, optical density. Data are expressed as mean ± SD of three different experiments for each treatment point. *p < 0.05 compared with parallel normal glucose and high glucose cultures with peptides alone (vehicle). Incubation with pathway inhibitors did not significantly modify cell survival, apoptosis, or caspase-3 activity in high glucose cultures without peptides (data not shown)
Chronic high glucose culture significantly decreased the islet MEC intracellular levels of cAMP, but this was significantly restored by daily treatment with AG, UAG, Ob or Ex-4 (Fig. 4c). Furthermore, cultures with specific inhibitors of AC (MDL12330A) or PKA (KT5720) prevented the cytoprotective effects of all three ghrelin gene products and of Ex-4 (Fig. 5a–c), suggesting that their effects were at least in part dependent on activation of the AC/cAMP/PKA signalling pathway.
Consistent with data indicating Ob binding to GLP-1R [7], competition experiments with exendin-9 (Ex-9), a specific GLP-1R antagonist, prevented the Ob-induced effects on cell survival, proliferation and apoptosis in high glucose conditions (Fig. 6a–c). Ex-9 also significantly abrogated the effects of Ex-4 (Fig. 6a–c), while coincubation of Ob and Ex-4 did not significantly modify the cytoprotective effects exerted by the single peptides.

Effects of the specific GLP-1 antagonist Ex-9 (500 nmol/l). Effects of Ex-9 treatment on apoptosis (a), assessed as DNA fragmentation, survival (b), assessed by MTT assay, and proliferation (c), assessed as BrdU incorporation of islet MECs cultured for 7 days in 28 mmol/l glucose (HG) and treated with 10 nmol/l Ob or Ex-4 (grey bars). Normal glucose (NG, white bars), 28 mmol/l glucose (black bars). Data are expressed as mean ± SD of three different experiments for each treatment point. *p < 0.05 compared with parallel high glucose cultures without peptides and cultures in the presence of Ex-9
Effects of AG, UAG, Ob and Ex-4 on BCL-2, BAX, and CD40-CD40L expression
Western blot analysis of BCL-2 and BAX in hyperglycaemia-induced apoptosis showed that a 7 day culture in high glucose decreased the production of the anti-apoptotic BCL-2 protein and enhanced the production of the pro-apoptotic BAX protein in islet MECs (Fig. 7a, b). These changes were reversed by exposure to ghrelin gene products or Ex-4, with an increase in BCL-2 levels of 115% (AG), 92% (UAG), 109% (Ob) and 94% (Ex-4) (Fig. 7a), and a decrease of pro-apoptotic BAX levels to 46% (AG), 58% (UAG), 43% (Ob) and 45% (Ex-4) (Fig. 7b), compared with the production in parallel high glucose cultures without peptides.

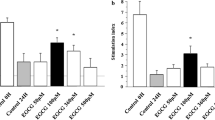
Production of BCL-2, BAX and CD40L. a Production of BCL-2 evaluated as densitometric analysis of western blot bands in islet MECs cultured in normal (1, white bar) or 28 mmol/l glucose (2, black bar) for 7 days or treated with 10 nmol/l of AG (3, grey bar), UAG (4, grey bar), Ob (5, grey bar) or Ex-4 (6, crosshatched bar). b Production of BAX evaluated as densitometric analysis of western blot bands in islet MECs cultured in normal (1, white bar) or 28 mmol/l glucose (2, black bar) for 7 days or treated with 10 nmol/l of AG (3, grey bar), UAG (4, grey bar), Ob (5, grey bar) or Ex-4 (6, crosshatched bar). c Production of CD40L evaluated as densitometric analysis of western blot bands in islet MECs cultured in normal (1, white bar) or 28 mmol/l glucose (2, black bar) for 7 days or treated with 10 nmol/l of AG (3, grey bar), UAG (4, grey bar), Ob (5, grey bar) or Ex-4 (6, crosshatched bar). OD, optical density. Data are expressed as mean ± SD of three different experiments. *p < 0.05 compared with parallel normal glucose and high glucose cultures with peptides. d Representative flow-cytometric analysis of human islet MECs cultured in normal glucose (NG) or in 28 mmol/l glucose (HG) (7 days) with IL-1β (10 ng/ml) and TNF-α (50 mg/ml) for 24 h, stained for CD40L (dark line histograms) and corresponding isotype control antibodies (dashed line histograms). Thin line histogram represents CD40L production in cells treated with 10 nmol/l Ob in high glucose. Three separate experiments showed similar results
Densitometric western blot analysis of the co-stimulatory CD40–CD40L pathway showed that high glucose upregulates the production of CD40L only. This upregulation was counteracted by exposure to ghrelin gene products or Ex-4 (Fig. 7c). Changes in CD40L were confirmed by flow-cytometric analysis of islet MECs in hyperglycaemic and inflammatory milieu without (mean fluorescence intensity [MFI] 22.6 ± 2.2) or with 10 mmol/l Ob (mean MFI 13.3 ± 1.5; Fig. 7d).
Effects of AG, UAG, Ob and Ex-4 on levels of NO, IL-1β and VEGF-A in cell supernatant fractions
Mean values for repeated measurements of IL-1β and of the stable NO oxidation product, nitrite, were significantly higher in supernatant fractions of high glucose cultures than normal glucose cultures, and were significantly reduced by treatment with AG, UAG, Ob or Ex-4 (Fig. 8a, b; p < 0.01 for IL-1β and p < 0.05 for NO).

Endothelial IL-1β and NO production, and endothelial and INS-1E rat beta cell VEGF-A production. a Mean value ± SD of IL-1β in cell free supernatant fractions of islet MECs in normal glucose (NG, white bar), in 28 mmol/l glucose (HG) (1, black bar), or treated with 10 nmol/l AG (2, grey bar), UAG (3, grey bar), Ob (4, grey bar) or Ex-4 (5, crosshatched bar). b Mean value ± SD of NO in cell free supernatant fractions of islet MECs in normal (white bar), in 28 mmol/l glucose (1, black bar) or treated with 10 nmol/l AG (2, grey bar), UAG (3, grey bar), Ob (4, grey bar) or Ex-4 (5, crosshatched bar). c Mean value ± SD of VEGF-A in cell free supernatant fractions of islet MECs in normal (white bar), in 28 mmol/l (1, black bars) glucose, or treated with 10 nmol/l AG (2, grey bars), UAG (3, grey bars), Ob (4, grey bars) or Ex-4 (5, crosshatched bars). d Mean value ± SD of VEGF-A in cell free supernatant fraction of INS-1E rat beta cells in normal (white bar), in 28 mmol/l glucose (1, black bars), or treated with 10 nmol/l AG (2, grey bars), UAG (3, grey bars), Ob (4, grey bars) or Ex-4 (5, crosshatched bars). Data are expressed as mean ± SD of three different experiments for each treatment or time point. *p < 0.05 compared with parallel normal glucose cultures and high glucose cultures with peptides
In time course experiments, levels of VEGF-A progressively increased in supernatant fractions of both islet endothelial and beta cells in high glucose (p < 0.05 compared with normal glucose for both cell types). The increased secretion was abrogated by the treatment with AG, UAG, Ob or Ex-4 in both cell cultures (Fig. 8c, d).
Discussion
The present study shows the ability of the ghrelin gene products and of the GLP-1 receptor agonist exendin-4 to inhibit apoptosis of human pancreatic endothelial cells induced by glucose toxicity. Islet MECs constitutively expressed GHS-R1A and GLP-1R, which may explain the biological effect of AG and Ex-4. UAG was also shown to be an active peptide despite acting on an unknown receptor [5]. Although AG desacylation and binding to the hypothetical UAG receptor may contribute to AG biological activity, receptor antagonist experiments suggest a direct AG effect, through GHS-R1A binding. The orphan receptor G-protein coupled receptor 39 (GPR39), a potential receptor of Ob [33], was not detected in islet MECs. However, immunofluorescence and GLP-1R competition studies indicated that Ob binds to GLP-1R expressed by islet MECs [7]. Expression of GHS-R1A in islet endothelial cells, together with the established production of ghrelin within pancreatic islets [3, 34], may sustain the autocrine/paracrine circuits active between endocrine and endothelial counterparts within the islet of Langerhans [14].
Sustained and intermittent hyperglycaemia affects endothelial cellular survival and proliferation, including the islet microendothelium [25]. Several metabolic mechanisms are involved, including oxidative stress, increased intracellular Ca2+, mitochondrial dysfunction, changes in fatty-acid metabolism and impaired phosphorylation of the protein kinase Akt [35]. The Akt signalling cascade plays a pivotal role in preventing apoptosis in a variety of settings, and has a role in insulin-mediated glucose transport and pancreatic beta cell mass and function [36]. In the present study, sustained high glucose levels impaired ERK1/2 tyrosine phosphorylation as well, and reduced intracellular cAMP and its targets, the cAMP-dependent PKA pathways, known to promote cell growth and to delay apoptosis [37]. The ghrelin gene products, independently of their acylation, promptly promoted cell survival and halted the cell proliferation impairment induced by the metabolic condition. The anti-apoptotic activity was mediated by tyrosine phosphorylation and activation of PI3K/Akt and ERK1/2 pathways, and by activation the cAMP/PKA signalling pathway, as indicated by cAMP intracellular levels and by inhibition of AC and of the downstream pathway of cAMP involving PKA. These multifunctional pathways transmit signals that result in prevention of apoptosis or induction of cell cycle progression, depending on the cell type, and can cross-regulate one another [37].
The mechanisms underlying apoptosis are regulated by defined biochemical pathways and involve members of the BCL-2 family [38]. In the present study, pro-survival BCL-2 protein, which stabilises the mitochondrial membrane and prevents the release of cytochrome C from the mitochondria and the activation of caspases, was found to be downregulated by high glucose. In contrast, the pro-apoptotic member, BAX, which antagonises BCL-2, was upregulated. These alterations were counteracted by treatment with AG, UAG and Ob, providing further insight into the mechanisms of their cytoprotective effects. Cytoprotective activities, mediated by similar mechanisms, were shown to be shared by the GLP-1R agonist Ex-4. GLP-1R is abundantly produced in extra-pancreatic tissues, including vascular endothelial cells and immune cells. Beyond its metabolic effects, GLP-1 has been demonstrated to ameliorate endothelial dysfunction, as well as to be involved in immunoregulatory processes [9, 39]. Our findings extend its regenerative actions from beta cells to the islet endothelium and add further interest to GLP-1R agonists, which not only improve the pathophysiology of the diabetic state, but may be of interest as an adjunctive therapy in the setting of immunomodulation/beta cell regeneration and islet transplantation [40].
High glucose conditions upregulated CD40L production in islet MECs. Functional CD40L is produced on the vascular endothelium [41] and contributes to B cell activation, isotype switching, co-stimulation in T cell mediated immunity and activation of extravasating monocytes [42], with an impact in atherosclerosis and in chronic inflammatory and autoimmune diseases. Blockers of CD40L have been strikingly effective in animal models of autoimmune diseases, such as systemic lupus erythematosus and type 1 diabetes [43]. Ghrelin gene products and Ex-4 were able to downregulate CD40L production in islet MECs. Together with early and intensive insulin therapy or combined with immunotherapies, these peptides might decelerate the targeting and loss of the remaining beta cell capacity during autoimmune insulitis. Human and murine studies indicate that, during insulitis, the endothelial cells surrounding the islets adopt an activated phenotype and are involved in the regulation of mononuclear cell transmigration and homing in the islets [44]. In line with our findings, Ex-4 has been recently shown to inhibit monocyte adhesion to the endothelium and to regulate inflammation in macrophages via the cAMP/PKA pathway [9]. It is noteworthy that overproduction of BCL-2 in endothelial cells decreases T cell cytotoxicity, suggesting that this protein may also protect endothelial cells from apoptosis resulting from an immunological insult [45].
Increasing evidence points to an interdependent physical and functional relationship between islet endothelium and beta cells, from organogenesis to adult life [14]. Post-natal beta cell mass is dynamic and can increase in function and mass for added demand, possibly by microendothelial inductive signals [20, 21, 46]. Diabetes, in all forms, is characterised by progressive reduction of beta cell function and mass together with impaired insulin secretion and beta cell regeneration. Several mechanisms have been proposed. These include increased vulnerability to apoptosis induced by inflammatory cytokines, induction of NO synthase (NOS)-dependent mechanisms and production of the vasoactive mediator, NO [1, 25], endothelial cell loss, and vasculature disruption [18, 23, 24]. NO, which is increased in hyperglycaemic conditions, has an established direct cytotoxicity in islets and potentially impairs insulin release [47]. Islet MECs are also a source of the proinflammatory cytokine IL-1β under hyperglycaemic conditions, independently of any viral or immune-mediated process. IL-1β impairs insulin release in human islets, induces TNF receptor superfamily, member 6 (FAS) expression enabling FAS-mediated apoptosis, and acts as a mediator of glucotoxicity [27]. In the present study, ghrelin gene products and Ex-4 significantly reduced the production of NO and IL-1β, providing an additional mechanism by which the peptides can directly protect beta cells, beyond their effects on islet vascularisation.
The major vascular endothelial growth factor, VEGF-A, is responsible for islet vascularisation [16, 17]. Persistent high glucose enhanced VEGF-A secretion by islet endothelial and beta cells. Glucose is reported to enhance synthesis of VEGF-A in cultured islets of Zucker diabetes fatty (ZDF) rats, a model of type 2 diabetes [24], which show progressive disruption of vascular endothelium and islet failure. Indeed, in murine models, hyperglycaemia in the short-term increases islet capillary blood pressure and perfusion in a glucose-dependent and reversible pathway [23]. When persistent, hyperglycaemia, by increasing islet flow and pressure, contributes to the disruption of islet endothelium. These changes lead to islet hypoperfusion and failure [18, 24]. In our report, the cytoprotective effects of ghrelin gene peptides and of Ex-4 were accompanied by abrogation of the glucose-induced VEGF-A increase. Such an effect has no immediate explanation. Studies on the angiogenic properties of ghrelin and GLP-1 are scanty and contradictory [48, 49], reflecting the heterogeneity of the microendothelium derived from different organs. However, it is noteworthy that thiazolidinedione pioglitazone prevented diabetes in the ZDF rat model [24], by preventing both the decrease in endothelial cell number and the endothelial hypertrophy, concomitant with a reduction in the VEGF-A level. Furthermore, renal production of VEGF-A is consistently upregulated by hyperglycaemia and in experimental and human diabetes. Inhibition of VEGF-A has beneficial effects on diabetes-induced glomerular functional and structural alterations, indicating a deleterious role for VEGF-A in the pathophysiology of diabetic nephropathy [50]. Pancreatic islets and renal glomeruli share ultrastructural features [14].
In conclusion, we provide evidence that metabolic stress is detrimental to islet endothelial cells, which are targets and effectors of the hyperglycaemic condition. The cytoprotective effect exerted by ghrelin gene peptides and incretin Ex-4, in association with their reported regenerative effect on residual beta cell capacity, renders these peptides an appealing therapeutic tool to regulate islet fate in vivo.
Abbreviations
- AC:
-
Adenylyl cyclase
- AG:
-
Acylated ghrelin
- Akt:
-
Protein kinase B
- BAX:
-
BCL-2 receptor-associated X protein
- BCL-2:
-
B cell lymphoma 2
- BrdU:
-
5-Bromo-2′-deoxyuridine
- CD40L:
-
CD40 ligand
- Ct :
-
Cycle threshold
- ERK:
-
Extracellular signal-related kinase
- Ex-4:
-
Exendin-4
- Ex-9:
-
Exendin-9
- GHS-R:
-
GH secretagogue receptor/ghrelin receptor
- GLP-1:
-
Glucagon-like peptide-1
- GLP-1R:
-
Glucagon-like peptide-1 receptor
- GPR39:
-
G-protein coupled receptor 39
- HUVEC:
-
Human umbilical vein-derived endothelial cell
- IF:
-
Immunofluorescence
- IL-1β:
-
Interleukin-1β
- MEC:
-
Microendothelial cell
- MFI:
-
Mean fluorescence intensity
- MTT:
-
3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide
- NOS:
-
Nitric oxide synthase
- Ob:
-
Obestatin
- oxLDL:
-
Oxidised LDL
- PI3K:
-
Phosphoinositide 3-kinase
- PKA:
-
Protein kinase A
- TAMRA:
-
Carboxytetramethylrhodamine
- UAG:
-
Unacylated ghrelin
- VEGF:
-
Vascular endothelial growth factor
- ZDF:
-
Zucker diabetes fatty
References
Meier JJ (2008) Beta cell mass in diabetes: a realistic therapeutic target? Diabetologia 51:703–713
Kojima M, Hosoda H, Date Y, Nakazato M, Matsuo H, Kangawa K (1999) Ghrelin is a growth-hormone-releasing acylated peptide from stomach. Nature 402:656–660
Prado CL, Pugh-Bernard AE, Elghazi L, Sosa-Pineda B, Sussel L (2004) Ghrelin cells replace insulin-producing beta cells in two mouse models of pancreas development. Proc Natl Acad Sci USA 101:2924–2929
Broglio F, Gottero C, Prodam F et al (2004) Non-acylated ghrelin counteracts the metabolic but not the neuroendocrine response to acylated ghrelin in humans. J Clin Endocrinol Metab 89:3062–3065
Granata R, Baragli A, Settanni F, Scarlatti F, Ghigo E (2010) Unraveling the role of the ghrelin gene peptides in the endocrine pancreas. J Mol Endocrinol 45:107–118
Zhang JV, Ren PG, Avsian-Kretchmer O et al (2005) Obestatin, a peptide encoded by the ghrelin gene, opposes ghrelin’s effects on food intake. Science 310:996–999
Granata R, Settanni F, Gallo D et al (2008) Obestatin promotes survival of pancreatic beta-cells and human islets and induces expression of genes involved in the regulation of beta-cell mass and function. Diabetes 57:967–979
Farilla L, Bulotta A, Hirshberg B et al (2003) Glucagon-like peptide 1 inhibits cell apoptosis and improves glucose responsiveness of freshly isolated human islets. Endocrinology 144:5149–5158
Arakawa M, Mita T, Azuma K et al (2010) Inhibition of monocyte adhesion to endothelial cells and attenuation of atherosclerotic lesion by a glucagon-like peptide-1 receptor agonist, exendin-4. Diabetes 59:1030–1037
Liu FQ, Zhang XL, Gong L et al (2011) Glucagon-like peptide 1 protects microvascular endothelial cells by inactivating the PARP-1/iNOS/NO pathway. Mol Cell Endocrinol 339:25–33
Baldanzi G, Filigheddu N, Cutrupi S et al (2002) Ghrelin and des-acyl ghrelin inhibit cell death in cardiomyocytes and endothelial cells through ERK1/2 and PI 3-kinase/Akt. J Cell Biol 159:1029–1037
Togliatto G, Trombetta A, Dentelli P et al (2010) Unacylated ghrelin rescues endothelial progenitor cell function in individuals with type 2 diabetes. Diabetes 59:1016–10125
Li WG, Gavrila D, Liu X et al (2004) Ghrelin inhibits proinflammatory responses and nuclear factor-kB activation in human endothelial cells. Circulation 109:2221–2226
Zanone MM, Favaro E, Camussi G (2008) From endothelial to beta cells: insights into pancreatic islet microendothelium. Curr Diabetes Rev 4:1–9
Swerlick RA, Lee KH, Li LJ, Sepp NT, Caughman SW, Lawley TJ (1992) Regulation of vascular cell adhesion molecule 1 on human dermal microvascular endothelial cells. J Immunol 149:698–705
Lammert E, Cleaver O, Melton D (2001) Induction of pancreatic differentiation by signals from blood vessels. Science 294:564–557
Lammert E, Gu G, McLaughlin M et al (2003) Role of VEGF-A in vascularization of pancreatic islets. Curr Biol 13:1070–1074
Brissova M, Shostak A, Shiota M et al (2006) Pancreatic islet production of vascular endothelial growth factor-a is essential for islet vascularization, revascularization, and function. Diabetes 55:2974–2985
Johansson A, Lau J, Sandberg M, Borg LA, Magnusson PU, Carlsson PO (2009) Endothelial signalling supports pancreatic beta cell function in the rat. Diabetologia 52:2384–2394
Nikolova G, Jabs N, Konstantinova I et al (2006) The vascular basement membrane: a niche for insulin gene expression and beta cell proliferation. Dev Cell 10:397–405
Johansson M, Mattsson G, Andersson A, Jansson L, Carlsson PO (2006) Islet endothelial cells and pancreatic beta-cell proliferation: studies in vitro and during pregnancy in adult rats. Endocrinology 147:2315–2324
Mizuno A, Noma Y, Kuwajima M, Murakami T, Zhu M, Shima K (1999) Changes in islet capillary angioarchitecture coincide with impaired B cell function but not with insulin resistance in male Otsuka–Long–Evans–Tokushima fatty rats: dimorphism of the diabetic phenotype at an advance age. Metabolism 48:477–483
Carlsson PO, Flodstrom M, Sandler S (2000) Islet blood flow in multiple low dose streptozotocin-treated wild-type and inducible nitric oxide synthase-deficient mice. Endocrinology 141:2752–2757
Li X, Zhang L, Meshinchi S et al (2006) Islet microvasculature in islet hyperplasia and failure in a model of type 2 diabetes. Diabetes 55:2965–2973
Favaro E, Miceli I, Bussolati B et al (2008) Hyperglycaemia induces apoptosis of human pancreatic islet endothelial cells: effects of pravastatin on the Akt survival pathway. Am J Pathol 173:442–450
Suschek C, Fehsel K, Kroncke KD, Sommer A, Kolb-Bachofen V (1994) Primary cultures of rat islet capillary endothelial cells. Constitutive and cytokine-inducible macrophagelike nitric oxide synthases are expressed and activities regulated by glucose concentration. Am J Pathol 145:685–695
Maedler K, Sergeev P, Ris F et al (2002) Glucose-induced beta cell production of IL-1beta contributes to glucotoxicity in human pancreatic islets. J Clin Invest 110:851–860
Huang GC, Zhao M, Jones P et al (2004) The development of new density gradient media for purifying human islets and islets-quality assessments. Transplantation 77:143–145
Favaro E, Bottelli A, Lozanoska-Ochser B et al (2005) Primary and immortalised human pancreatic islet endothelial cells: phenotypic and immunological characterisation. Diabetologia 48:2552–2562
Zanone MM, Favaro E, Doublier S et al (2005) Expression of nephrin by human pancreatic islet endothelial cells. Diabetologia 48:1789–1797
Granata R, Settanni F, Biancone L et al (2007) Acylated and unacylated ghrelin promote proliferation and inhibit apoptosis of pancreatic beta-cells and human islets: involvement of 3′,5′-cyclic adenosine monophosphate/protein kinase A, extracellular signal-regulated kinase 1/2, and phosphatidyl inositol 3-kinase/Akt signalling. Endocrinology 148:512–529
Zanone MM, Favaro E, Conaldi PG et al (2003) Persistent infection of human microvascular endothelial cells by coxsackie B viruses induces increased expression of adhesion molecules. J Immunol 171:438–446
Tremblay F, Perreault M, Klaman LD, Tobin JF, Smith E, Gimeno RE (2007) Normal food intake and body weight in mice lacking the G protein-coupled receptor GPR39. Endocrinology 148:501–506
Volante M, Allia E, Gugliotta P et al (2002) Expression of ghrelin and of the GH secretagogue receptor by pancreatic islet cells and related endocrine tumors. J Clin Endocrinol Metab 87:1300–1308
Datta SR, Brunet A, Greenberg ME (1999) Cellular survival: a play in three Akts. Genes Dev 13:2905–2927
Elghazi L, Balcazar N, Bernal-Mizrachi E (2006) Emerging role of protein kinase B/Akt signaling in pancreatic beta-cell mass and function. Int J Biochem Cell Biol 38:157–163
Stork PJ, Schmitt JM (2002) Crosstalk between cAMP and MAP kinase signaling in the regulation of cell proliferation. Trends Cell Biol 12:258–266
Choy JC, Granville DJ, Hunt DW, McManus BM (2001) Endothelial cell apoptosis: biochemical characteristics and potential implications for atherosclerosis. J Mol Cell Cardiol 33:1673–1690
Hattori Y, Jojima T, Tomizawa A et al (2010) A glucagon-like peptide-1 (GLP-1) analogue, liraglutide, upregulates nitric oxide production and exerts anti-inflammatory action in endothelial cells. Diabetologia 53:2256–2263
Rickels MR, Mueller R, Markmann JF, Naji A (2009) Effect of glucagon-like peptide-1 on beta- and alpha-cell function in isolated islet and whole pancreas transplant recipients. J Clin Endocrinol Metab 94:181–189
Mach F, Schönbeck U, Sukhova GK et al (1997) Functional CD40 ligand is expressed on human vascular endothelial cells, smooth muscle cells, and macrophages: implications for CD40-CD40 ligand signaling in atherosclerosis. Proc Natl Acad Sci U S A 94:1931–1936
Wagner AH, Güldenzoph B, Lienenlüke B, Hecker M (2004) CD154/CD40-mediated expression of CD154 in endothelial cells: consequences for endothelial cell-monocyte interaction. Arterioscler Thromb Vasc Biol 24:715–720
Homann D, Jahreis A, Wolfe T et al (2002) CD40L blockade prevents autoimmune diabetes by induction of bitypic NK/DC regulatory cells. Immunity 16:403–415
Hänninen A, Taylor C, Streeter PR (1993) Vascular addressins are induced on islet vessels during insulitis in nonobese diabetic mice and are involved in lymphoid cell binding to islet endothelium. J Clin Invest 92:2509–2515
Zheng L, Dengler TJ, Kluger MS et al (2000) Cytoprotection of human umbilical vein endothelial cells against apoptosis and CTL-mediated lysis provided by caspase-resistant Bcl-2 without alterations in growth or activation responses. J Immunol 164:4665–4671
Dor Y, Brown J, Martinez OI, Melton DA (2004) Adult pancreatic beta-cells are formed by self-duplication rather than stem-cell differentiation. Nature 429(6987):41–46
Corbett JA, Sweetland MA, Wang JL, Lancaster JR Jr, McDaniel ML (1993) Nitric oxide mediates cytokine-induced inhibition of insulin secretion by human islets of Langerhans. Proc Natl Acad Sci U S A 90:1731–1735
Ahluwalia A, Li A, Cheng G, Deng X, Tarnawski AS (2009) Reduced ghrelin in endothelial cells plays important mechanistic role in aging-related impairment of angiogenesis. J Physiol Pharmacol 60:29–34
Erdogdu O, Nathanson D, Sjöholm A, Nyström T, Zhang Q (2010) Exendin-4 stimulates proliferation of human coronary artery endothelial cells through eNOS-, PKA- and PI3K/Akt-dependent pathways and requires GLP-1 receptor. Mol Cell Endocrinol 325:26–35
Schrijver BF, Flyvbjerg A, Dr Vriese AS (2004) The role of vascular endothelial growth factor (VEGF) in renal pathophysiology. Kidney Int 65:2003–2017
Funding
This work was supported by Regione Piemonte (I), Ricerca Sanitaria Finalizzata 2008Bis.
Duality of interest
The authors declare that there is no duality of interest associated with the manuscript.
Contribution statement
EF and MMZ designed and performed the present study, analysed the data and wrote the manuscript. RG, IM, AB and FS contributed to the study design and critical revision of the manuscript. PCP, EG and GC contributed to analysis and interpretation of data and critical revision of the manuscript. All authors approved the version to be published.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM
PDF 731 kb
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Favaro, E., Granata, R., Miceli, I. et al. The ghrelin gene products and exendin-4 promote survival of human pancreatic islet endothelial cells in hyperglycaemic conditions, through phosphoinositide 3-kinase/Akt, extracellular signal-related kinase (ERK)1/2 and cAMP/protein kinase A (PKA) signalling pathways. Diabetologia 55, 1058–1070 (2012). https://doi.org/10.1007/s00125-011-2423-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00125-011-2423-y