
Abstract
Key message
A single-nucleotide insertion resulted in a premature stop codon that is responsible for white immature fruit color in cucumber.
Abstract
Despite our previous progress in the mapping of the gene controlling white color in immature cucumber fruit and the identification of candidate genes, the specific gene that governs chlorophyll metabolism and its regulatory mechanism remains unknown. Here, we generated a mapping population consisting of 9497 F2 plants to delimit the controlling gene to an 8.2-kb physical interval that defines a sole candidate gene, APRR2. Sequencing the full-length DNA and cDNA of APRR2 allowed for identification of an allele, aprr2, encoding a truncated 101-amino acid protein due to a frameshift mutation and a premature stop codon. Gene structure prediction indicated that these 101 residues are located in a domain necessary for the function of the protein. The expression patterns of APRR2 were entirely consistent with the visual changes in green color intensity during fruit development. A microscopic observation of the fruit pericarp revealed fewer chloroplasts and a lower chloroplast chlorophyll storage capacity in Q24 (white) than in Q30 (green). A single-base insertion in the white color gene w, which leads to a premature stop codon, is hypothesized to have disabled the function of this gene in chlorophyll accumulation and chloroplast development. These findings contribute to basic research and the genetic improvement of fruit color.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Cucumber (Cucumis sativus L.; 2n = 2x = 14), a member of the Cucurbitaceae family, is one of the most economically important crops in the world (Yundaeng et al. 2015). The cucumber is generally considered to have originated and domesticated in India (Yundaeng et al. 2015), and it is currently widely cultivated in China, Turkey, Iran, Russian, the Ukraine, Spain and the USA, where its yield exceeded 1 % of the global production (71.4 million tons) in 2013 (see Table S1 in the online supplementary files for details, data from http://faostat.fao.org/). China is the largest producer of cucumbers, with an annual production of 54.3 million tons (76.1 % of global production). The visual quality is a vital index that influences consumer choice. Two types of cucumber are available in China: the North China type, which has long and green fruit with numerous white spines and ribbed epicarp; and the South China type, which has short and mottled fruit with few spines and a relatively smooth surface. Therefore, an important goal of cultivation programs is the improvement of fruit texture.
The release of the cucumber draft genome has provided valuable insights into the genetic inheritance of traits in this plant (Huang et al. 2009), and many molecular markers and high density genetic maps have been developed in the areas of genetics and breeding to assist in the process of cultivar development. To date, 21 simply inherited cucumber genes have been mapped to chromosomes (Weng 2014). Aside from several genes, such as Bt (Bitter fruit), Bl (Bitter leaf), Tu (Warty fruit) and M (bisexual flower) (Li et al. 2012; Shang et al. 2014; Yang et al. 2014b), few have been map-based cloned, markedly hindering the progress of marker-assisted selection (MAS) breeding and transgenic breeding.
Five skin color-related genes, dark green, green, yellow-green (yg), light green and white (w) skin genes, are included in the cucumber gene list published in 2001 (Xie and Wehner 2001). Although white-skinned cucumbers are uncommon, they are being increasingly consumed in parts of the Chinese mainland, such as Gansu and Shandong provinces, and the desirability of these cultivars in China has grown over time. Our previous research (Liu et al. 2015) mapped the white immature fruit gene w to a 33.0-kb region, followed by annotation of four candidate genes using the online program FGENESH. However, BLASTP analyses indicated that no predicted gene was directly involved in the visual trait. Putative amino acid sequence alignment and qPCR (quantitative real-time PCR) analyses revealed differences between the two parental lines of two candidate genes, PSP and RRP (two-component Response Regulator-like Protein). Therefore, further research should be performed to identify the controlling gene and characterize its function.
Based on our previous research, the present study was conducted to identify the exact gene responsible for fruit color by enlarging the mapping populations and developing new molecular markers. Candidate gene DNA and cDNA were sequenced to investigate gene structure, and full-length cDNAs from ten different cucumber lines were used to detect mutated sites responsible for the diversity between green- and white-colored fruit. To assess gene expression patterns, mRNA was extracted from fruit and plant tissues during different developmental periods. In addition, the chlorophyll content and chloroplast abundance of green and white cucumber fruit were assessed. This work will facilitate transgenic MAS breeding as well as the exploration of gene interactions among skin color-related genes.
Materials and methods
Plant materials
The F2 generation was derived from the two parental lines, Q30 and Q24, which were also used for chloroplast microscopy. Q30 is green and produces long fruit, whereas Q24 produces white spherical fruit. Additional green (W33) and white (HY, M33, B-2-2, and L66) cucumber samples were used to analyze mutations in the target gene. Moreover, these seven lines and Chang Chun Mi Ci (CCMC) were applied to measure chlorophyll levels. All cucumber seeds were provided by the cucumber research team of the Horticulture College at Northwest A&F University in Yangling, China. All eight lines and the F2 generation were grown in plastic greenhouses at Northwest A&F University under natural sunlight from spring 2014 to summer 2015. The fruit samples used for qPCR were harvested in June 2015 in Yangling (34°16′N, 108°4′E). The field work was conducted as described in our previous work (Liu et al. 2015).
Mapping strategy
Based on our previous study (Liu et al. 2015), the mapping population was enlarged to 9497 plants. Two flanking SSR markers, C120-indel and GXH3913 (physically at 39282299 and 39131194 on chromosome 3, respectively, Chinese long V2 from Cucurbit Genomics Database, Fig. 1a), were used to screen the mapping populations to obtain recombinants. For both markers, the gel bands, which were same as those for Q30, F1 or Q24, were classified as a, h or b, respectively. In this case, each individual that differed in classification from C120-indel and GXH3913 was regarded as a recombinant and was then selected for further linkage analysis.

Definitive genetic mapping of the gene for white skin (w) in cucumber. a A previous fine genetic map that delimited the w gene to a 33-kb physical region based on 2971 F2 plants. b Definitive map of the w gene; a total 9497 F2 plants were used to narrow the gene to 8.2 kb on chromosome 3. Asterisks indicate the number of recombinant plants
The whole genomes of Q30 and Q24 were sequenced using an Illumina HiSeq™ 2000 (Biomarker Technologies, Beijing, China) at 24- and 27-fold sequencing depths to develop effective InDel and SNP markers for our research. High-throughput SNPs were applied to develop ASPCR (Liu et al. 2015), cleaved amplified polymorphic sequences (CAPS) (Neff et al. 1998) or derived CAPS (dCAPS) markers. Primers for the dCAPS markers were designed using a web-based program, dCAPS Finder 2.0 (http://helix.wustl.edu/dcaps/dcaps.html); other markers were designed with Primer Premier 5.0 (http://www.premierbiosoft.com/). The physical locations of linkage markers were determined by BLAST searches of the primer sequences against the 9930 genome (Cucurbit Genomics Database; Chinese Long V2; http://www.icugi.org/cgi-bin/ICuGI/index.cgi) (known as in silico PCR). All designed primer sequences were first used for specificity validation over the whole-genome sequence of 9930. Polymorphism in each newly synthesized marker was first assessed using parental DNA. The polymorphic markers were then utilized to determine the genotype of recombinant plants. The genotype and phenotype data were used for linkage analyses and map construction using Join-Map 4.0, with a LOD threshold score of 6.0.
Molecular cloning and sequencing
Genomic DNA was extracted following the CTAB method, as described previously (Clark 1997). Both the DNA and cDNA of the candidate gene were cloned in this study. Four primer pairs (APRRclone1-APRRclone4 in Table S2, online recourses) were designed to amplify the genome sequences of Q30 and Q24. The APRRclone1-forward/APRRclone4-reverse pair was used to amplify the cDNA of the target gene from cucumber lines. The PCR reactions were performed in a total volume of 20 μl according to the manual supplied with Phusion® High-Fidelity PCR Kit (NEB, #E0553S/L, Singapore). The products were electrophoresed through a 1 % agarose gels and extracted using a gel extraction kit (BioTeke, China). The purified PCR products were then used for “+A base” reactions with Taq DNA polymerase (#EP0402, Thermo). The 20-μl reaction volume included 1 U Taq polymerase, 1.5 mM MgCl2, 0.2 mM of each dNTP, 2 μl of 10× Buffer (KCl) and the purified PCR products to a final volume of 20 μl. The mixture was heated to 72 °C for 30 min, and the PCR products were subsequently used for TA cloning (Zhou and Gomez-Sanchez 2000). Single bacterial colonies were validated by PCR, and triplicate positive clones of each cucumber line were sequenced by Sangon Biotech Co., Ltd (Shanghai, China). All primers used in this study were synthesized by Sangon Biotech Co., Ltd. The complete coding sequences of the seven selected cucumber lines have been submitted to GenBank under the following accession numbers: Q24 (KU179198), B-2-2 (KU179199), HY (KU179200), L66 (KU179201), M33 (KU179202), Q30 (KU179203), and W33 (KU179204).
Sequence analyses
Gene prediction was performed using the online program FGENESH (http://linux1.softberry.com/berry.phtml) (Salamov and Solovyev 2000) and Cucumber Genome Browser (http://www.icugi.org/cgi-bin/ICuGI/index.cgi). ORF Finder (http://www.ncbi.nlm.nih.gov/gorf/orfig.cgi) was used to investigate open reading frame (ORFs). The gene structural profile was determined based on alignment analysis of DNA and cDNA with spidey (NCBI, http://www.ncbi.nlm.nih.gov/spidey/spideyweb.cgi) as well as structural prediction from the FGENESH website. cDNA and amino acid sequences were aligned with the DNAMAN program. All DNA sequences related to cucumber line 9930 and PI183967 were downloaded from the Cucurbit Genomics database. The sequences for GY14 were obtained from Phytozome (http://phytozome.jgi.doe.gov/pz/portal.html#).
Chlorophyll determination and chloroplast microscopy
Circular pieces were cut from the epicarp and extracted with 80 % acetone in the dark. The pigment extracts were immediately spectrophotometrically assayed at specific absorption coefficients as described in a previous study (Inskeep and Bloom 1985).
To observe the chloroplasts of epidermis cells, tissues from 9-day-old cucumber fruit (after pollination) were excised with a sterile razor blade and fixed immediately in 3.5 % (v/v) glutaraldehyde solution for 1 h in the dark. The fruit tissue was disrupted as described previously (Forth and Pyke 2006; Pan et al. 2013; Pyke and Leech 1992). In brief, the tissue was heat-treated at 65 °C in a solution of disodium EDTA-Na2 (ethylene diamine tetraacetate, 0.1 M, pH 9.0) for 20 min followed by fragmentation using a Tissuelyser (Shanghai Jingxin Industrial Development Co., Ltd, Shanghai, China). The tissue fragment solution was then filtered through degreasing cotton. The pure tissue was stored at 4 °C in EDTA-Na2 solution for up to 6 months. The samples were observed and photographed using an OLYMPUS BX51 microscope and an OLYMPUS DP72 digital camera (Olympus Corporation, Japan).
Gene expression assay
Quantitative real-time PCR (qPCR) was performed to elucidate the development-specific and tissue- specific expression of the candidate gene. Pericarp samples from 4- to 30-day fruit after pollination and other tissues (root, stem, flower, and leaves) were excised, and RNA was isolated using a Column Plant mRNAout kit (TIANDZ, China) according to the manufacturer’s instructions. First-strand cDNA was synthesized with a 5× All-In-One Master Mix kit (AccuRT Genomic DNA Removal Kit, ABM, Canada). The cucumber ubiquitin extension protein gene was used as the reference. The 20-μl qPCR volume included 3 μl of cDNA template (100 ng/μl), 2 μl of each primer (10 μM), 10 μl of EvaGreen qPCR Master Mix (ABM, Canada), and RNase-free water to a final volume of 20 μl. The PCR was performed using an iQTM 5 Multicolor Real-Time PCR Detection System (Bio-Rad, USA). The values from triplicate reactions were averaged, and the relative expression level was determined with the 2−ΔΔCt method (Livak and Schmittgen 2001).
Phylogenetic analyses
cDNA sequences were analyzed using ORF Finder (NCBI, http://www.ncbi.nlm.nih.gov/gorf/gorf.html), and the amino acid (aa) sequences were then compared with known sequences in the standard SmartBLAST databases (NCBI, http://blast.ncbi.nlm.nih.gov/smartblast/smartBlast.cgi?CMD=Web) (Wheeler et al. 2005). High-level homologous aa sequences were downloaded in FASTA format. The sequence accession numbers are shown in supplemental online Table S3. Phylogenetic trees were constructed using MEGA 6.0 (http://www.megasoftware.net/index.php) with the neighbor-joining method (Li et al. 2015). Bootstrapping was performed with 1000 replicates.
Results
Definitive mapping of the w gene
Our previous work mapped the w gene to a 33.0-kb physical region (Fig. 1a). To further delineate this gene, we selected 9497 F2 plants for detailed mapping analyses. By screening the population with two InDel markers, GXH3913 and C120-indel, 46 recombination events were detected. In addition, two InDel (LH39253, LH39258) and two SNP (ASPCR39259, ASPCR39250) markers residing within the physical interval that spans the target gene were developed (primer sequences are available in Table S2 online). We investigated the genotypes of the newly developed four markers and the adjacent molecular markers mapped in our previous study. Twenty-five recombination events delimited w to an 8.2-kb physical region on chromosome 3 flanked by two newly developed markers: InDel-type LH39258 and SNP-type ASPCR39250 (Fig. 1b). The full-length DNA sequences of 9930 were used for putative gene prediction via FGENESH and Cucumber Genome Browser, and only one gene structure was consistently identified by both methods. Thus, definitive genetic mapping, along with the identification of candidate genes, defined a single candidate gene for w. The physical position indicated that this single candidate gene corresponds to the RRP gene, as predicted in our previous study (Liu et al. 2015).
Molecular cloning of the candidate gene
We aligned the putative aa sequences with those in the SmartBLAST database of NCBI. A series of ‘two-component response regulator-like’ (APRR2) genes were identified and the mRNA of the homolog from Cucumis sativus showing the highest identity was downloaded and analyzed by ORF Finder to determine the start and stop codons. As the DNA sequence between the start and stop codons in the 9930 genome is 5878 bp long, we designed four primer pairs (shown in supplementary Table S2, online) to clone the full-length DNA from both parents, Q30 and Q24. Sequencing of the APRR2 indicated lengths of 5875 and 5907 nucleotides in each line that were 99.46 and 100 % similar to the whole-genome sequencing assembly data (see Fig. S1 in the online supplementary materials for additional details). Fourteen single-base substitutions and nine InDels (including seven single- and double-base InDels) were detected between Q30 and Q24. Gene prediction of both parental DNA sequences based on a comparison with the FGENESH database yielded a consistent gene structure containing 12 exons.
Insight analyses of diversified allelic genes
To better understand the amino acid variations that may affect function, the full-length cDNAs from seven lines (five white and two green) were sequenced and translated into the corresponding amino acids using ORF Finder. Additionally, the cDNAs from three green cucumbers, GY14, 9930 and PI183967 (PI17), were downloaded from Phytozome and Cucurbit Genomics Database. Alignments between the cDNA sequences and the genomic sequences of Q30 confirmed that APRR2 comprises 12 exons and 11 introns (Fig. 2a). Both the full-length cDNAs and deduced amino acid sequences were aligned, and the results are shown in online supplemental Fig. S2 and Fig. S3, respectively. Two SNPs, one SNP and one InDel were identified in the cDNAs from PI17, B-2-2 and W33, respectively. Specifically, a single-nucleotide insertion (G) was detected in all white cucumbers, as shown in Fig. 2b. This insertion consistent in white cucumbers occurs at the last nucleotide site of the ninth exon (Fig. 2a). We further analyzed the putative amino acid sequences and detected a premature stop codon due to a frameshift after the SNP (Fig. 2c) that results in a deletion of 101 amino acid residues in white cucumbers (Fig. S3). To evaluate the domain necessary for the function of the protein, we BLAST searched the amino acid sequences against the protein database of NCBI. Graphics of the highest identity in Cucumis sativus revealed that the truncated 101 residues are located within a functional domain (244 amino acids) of a ripening-related transcription factor (Fig. S4) (Pan et al. 2013). Taken together, these data indicated that this SNP variation is responsible for white/green immature fruit.

Gene structure and homologous analyses of the w gene. a Twelve exons and 11 introns comprise the coding region. A single-nucleotide insertion (G) occurs at the last nucleotide in exon 9. b Five white and five green lines were used for homologous analyses. The G insertion is consistent in white cucumbers. c The G insertion causes a premature stop codon due to a frameshift
Phylogenetic relationship
It has been reported that an APRR2 ortholog overexpressed in transgenic tomato lines increased the plastid number, area, and pigment content, enhancing the levels of chlorophyll in immature unripe fruit (Pan et al. 2013). In Arabidopsis, a pair of Golden2-like genes (GLK1 and GLK2) have similar biological functions in regulating chlorophyll biosynthesis (Waters et al. 2009) and chloroplast development (Fitter et al. 2002). In addition, GLK1 and GLK2 expression in tomato predicts their roles in leaves and fruit. GLK2 was demonstrated to determine chlorophyll accumulation and distribution in developing tomato fruit (Powell et al. 2012), and the gene exhibits high sequence similarity to tomato APRR2 (Pan et al. 2013). APRR1, which is related to APRR2, is suggested to be involved in plastid development and ripening (Makino et al. 2000; Pan et al. 2013). Moreover, w was demonstrated to control white skin color in cucumber, and its allele (W) in Q30 showed the highest identity (99 %) to the predicted Cucumis sativus APRR2 sequence available in the NCBI protein database. Therefore, to further understand the evolutionary relationship of APRR2 in diverse plant species and its relationship to GLK2/GLK1 and APRR1 in Arabidopsis, a phylogenetic analysis was performed using the neighbor-joining (NJ) method (Fig. 3). W/w genes in cucumber parents share the highest amino acid homology with those from 9930 and muskmelon, and they were placed into the same clade as other APRR2 proteins from the species examined. However, the GLK2/GLK1 and APRR1 protein sequences in Arabidopsis are distinct from this clade. These observations illustrated that W in cucumber is an APRR2 homolog that is distinct from APRR1 and GLK2/GLK1.

Phylogenetic relationship of APRR2 among selected species and its relationship with CKL1/CKL2 and APRR1 in Arabidopsis. Amino acid sequences were downloaded from NCBI. Accession numbers are presented in supplemental Table S3 on line
Developmental- and tissue-specific expression
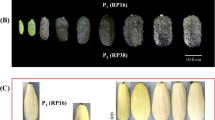
Fruit from both green and white lines were harvested 4–30 days after pollination and subjected to qPCR (Fig. 4). The mRNA levels in the roots, stems, leaves and flowers (mixture of female and male) were also detected to evaluate tissue-specific expression. As shown in Fig. 4, APRR2 expression was higher in the green compared to the white lines. Moreover, the expression levels in Q30 tended to be higher in immature stages (before 12 days) and gradually decreased as the plant matured (after 12 days). This tendency was entirely consistent with the observed visual changes in fruit color during development, i.e., the plant gradually turned green during the immature fruit stage and then became chlorotic in the ripening stage. APRR2 in 12-day-old fruit exhibited the highest expression level, a 12.7-fold increase in Q30 compared with Q24. Additionally, enhanced expression in the leaves of Q30 was also recorded and was consistent with the elevated green intensity in Q30 compared with Q24. The expression levels in the root, stem and flower were low in both the Q30 and Q24 lines.

Development- and tissue-specific expression in Q30 and Q24. a The developmental visual appearances of the Q30 fruit; the ten pictures of fruit correspond to the ten respective columns in the histogram. b Examined stem, leaf and flower tissues. Female and male flowers were pooled for mRNA extraction. c Transcript levels of the candidate gene in different tissues, as detected by real-time quantitative PCR. The data are presented as the average values of three replicates (±SD). The relative transcript level is presented using Q24 as the reference
Chlorophyll content and chloroplast number
Overexpression of an APRR2 ortholog in transgenic tomato lines increased the plastid number and levels of chlorophyll in immature unripe fruit (Pan et al. 2013). To further investigate the potential function of the APRR2 in cucumber, we evaluated the chloroplast number in both Q30 and Q24 as well as the chlorophyll content in four white cucumbers (HY,B-2-2,M33 and Q24) and four green cucumbers (9930, CCMC, W33 and Q30). As shown in Fig. 5, the levels of chlorophyll a and chlorophyll b were higher in the green compared to the white lines. High levels of chlorophyll were also observed in tissues rich in chloroplasts. Microscopy revealed that the chloroplasts predominantly accumulated as green foci in the epidermal cells of fruit. Moreover, the epicarp of the Q30 fruit contained more chloroplasts than that of Q24 fruit, and the chloroplasts in Q24 exhibited a much lower storage capacity for chlorophyll, as evidenced by the pale green color of the chloroplasts.

Chlorophyll content and chloroplast number in white and green cucumber fruit. Four white (HY, B-2-2, M33, and Q24) and four green (9930, CCMC, W33, and Q30) lines were used to measure the chlorophyll content. Fruits from Q30 and Q24 plants were used for chloroplast number examination under microscopy; FW fresh fruit
Discussion
Mapping of the w gene using an efficient strategy
Fruit color is a target of breeding. Color is determined by the proportions of endogenous pigments, such as carotenoids, chlorophylls, flavonoids, and anthocyanins (Wang et al. 2015), and the visual color of cucumber fruit ranges widely, from white to dark green. Nonetheless, there are a limited number of white cultivars. A clear understanding of the specific genetic patterns of the w gene will promote more effective plant breeding. Since the release of the cucumber genome sequence, 203 economically important genes/QTLs have been molecularly mapped or cloned (Weng 2014). A map-based cloning strategy, combined with recombinant inbred lines (RIL), near isogenic lines (NIL) or a F2 population is widely used for gene mapping in cucumber. F2 populations are easier and faster to generate but require a larger population size. Next-generation DNA sequencing technology now permits cost-effective genome sequencing at a population scale (Qi et al. 2013), promoting the progress of gene mapping. Within this context, we sequenced the genomes of the parents and enlarged the F2 mapping population to 9497 individuals. The target gene was efficiently delimited to a physical interval of 8.2 kb.
Subtle variation responses to the white trait
Comparative analyses suggest that fruit crops may have undergone narrower bottlenecks during domestication than grain crops and that their genetic diversity has been more strongly reduced within their cultivated gene pools (Qi et al. 2013). The genetic diversity of cultivated cucumbers is low (Knerr et al. 1989; Li et al. 2011; Weng et al. 2010; Zhang et al. 2010), and several trait-related genes are associated with a single copy in the genome (Li et al. 2009, 2011, 2013a, Liu et al. 2015; Pramnoi et al. 2013; Tan et al. 2015; Weng et al. 2010; Yang et al. 2014a). Our study demonstrated that the white immature fruit color in cucumber is controlled by a single simply inherited gene. One SNP close to the splice site that is reserved during transcription results in the translation of a premature stop codon in white-fruit cucumbers. This SNP appears to be responsible for the color trait. Further qPCR analysis confirmed APRR2 as a strong candidate regulator of fruit color, as evidenced by increased expression at the immature stage and decreased expression during gradual ripening.
The functional role of APRR2 is associated with chloroplast development
Color-related genes have been cloned in pear (Yang et al. 2013), pepper (Popovsky and Paran 2000; Li et al. 2013b), peach (Adami et al. 2013), cauliflower (Lu et al. 2006), apple (Han et al. 2011), tomato (Ma et al. 2014), and grape (Azuma et al. 2007). Previous studies have indicated that the R2R3-MYB transcription factor is a common regulator that controls fruit color in several species (Borevitz et al. 2000; Feller et al. 2011; Kayesh et al. 2013; Li et al. 2013a; Motamayor et al. 2013; Singh et al. 2014), primarily by regulating anthocyanin biosynthesis (Han et al. 2011; Kayesh et al. 2013). Carotenoids are orange, yellow, and red pigments that have a variety of critical functions in plants and accumulate at high levels in chloroplasts and chromoplasts (Lu et al. 2006). Chlorophylls are responsible for the green appearance of leaves and immature fruit in plants and chlorophyll metabolism is a highly coordinated process that consists of a series of cooperative reactions catalyzed by many enzymes (Beale 1999). Specifically, 27 genes encode these enzymes in Arabidopsis thaliana (Beale 2005); however, the candidate APRR2 gene does not appear to be involved in the chlorophyll biosynthetic pathway.
In the present study, we mapped a single candidate gene responsible for cucumber fruit color. A BLASTP search against the NCBI protein database indicated that the candidate gene has sequence similarity to Arabidopsis pseudo-response regulator 2 gene (APRR2) (accession number, NP_567548.1). However, the function of APRR2 in Arabidopsis remains unclear (Pan et al. 2013). APRR2 orthologs expressed in tomato and sweet pepper are associated with increased plastid numbers and pigment accumulation in the chloroplasts of fruit tissue (Pan et al. 2013). Moreover, a major quantitative trait locus (QTL) close to the APRR2 gene explained over 50 % of the variation in chlorophyll content and was responsible for increased plastid compartment size in pepper fruit (Brand et al. 2012; Pan et al. 2013). In our investigation, analyses of chlorophyll content and chloroplast number in both green and white cucumbers revealed a strong and similar association between the color trait and the function of the APRR2 gene in immature fruit. Overall, these findings suggest that APRR2, a gene mainly associated with a critical intermediate in chloroplast development and chlorophyll biosynthesis, regulates the appearance of fruit skin in cucumber.
Putative mechanism reveals in the green/white trait
At least 22 members of the response regulatory family (ARRs) are expressed in Arabidopsis, and these proteins can be classified into two subtypes: type A and type B (D’Agostino and Kieber 1999; Hosoda et al. 2002). In Arabidopsis, type B response regulators are transcription factors that act as positive regulators in the two-component cytokinin signaling pathway (Argyros et al. 2008). A-type members might function as negative regulators of the cytokinin response (Cheng and Kieber 2014). Histidine phosphotransfer proteins mediate phosphotransfer from receptors kinases to ARRs in the cytokinin signaling pathway (Cheng and Kieber 2014; Hutchison et al. 2006). Plant ARRs contain a canonical Asp residue that is necessary for phosphorylation, but the so-called “pseudo response regulators (APRRs)” proteins commonly lack this Asp (Heyl et al. 2013). However, it has been confirmed that APRR2 contains this Asp residue and exhibits phosphotransfer ability (Hwang et al. 2002; Satbhai et al. 2011). Moreover, the B motif, originally identified as a signature of type-B ARRs involved in His-to-Asp phosphorelay systems in Arabidopsis, is found in the sequence of the APRR2 protein from both Arabidopsis and cucumber (Fig. S4) (Fitter et al. 2002; Hwang et al. 2002). These analyses suggest that APRR2 functions as an ARR (Satbhai et al. 2011), which may act as a regulator in cytokinin signaling. The plant hormone cytokinin has been described as playing a role in regulating the development and activity of chloroplasts, especially during the etioplast-to-chloroplast transition (Cortleven and Schmülling 2015). Indeed, cytokinin promotes chlorophyll synthesis, chloroplast ultrastructure, and chloroplast number (Cortleven and Schmülling 2015). Mapping analyses have indicated that an almost complete form of APRR2 is required for the function of the protein in plants (Perochon et al. 2010).
To date, the mechanism of action of Arabidopsis, tomato or pepper APRR2 is not known (Pan et al. 2013). One possibility is that APRR2 expression in green cucumber participates in a ripening signal mechanism involving chlorophyll accumulation and chloroplast development (Barry et al. 2012; Pan et al. 2013). The single-nucleotide insertion in the coding region of APRR2 leads to a frameshift mutation that results in a premature stop codon and forms an allele, aprr2, in white cucumber lines. This lack of the C-terminal 101 amino acids prevents the biological function of the gene product during ripening signaling and results in low levels of chlorophyll and fewer chloroplasts. The mechanistic basis of APRR2 demands further investigation. The findings of this study contribute to basic research and the genetic improvement of fruit color and may provide a basis for designing diagnostic tools for practical cucumber breeding.
Author contribution statement
H. L. constructed the map, cloned the gene, conducted the qPCR analysis, measured the chlorophyll content, conducted the chloroplast microscopy, and wrote the paper. J. J. helped clone the gene. X. L. and J. L. performed some of the fieldwork. H. M., S. C. and Y. L. provided valuable research ideas. Z. C. designed and supervised the study.
References
Adami M, De Franceschi P, Brandi F et al (2013) Identifying a carotenoid cleavage dioxygenase (ccd4) gene controlling yellow/white fruit flesh color of peach. Plant Mol Biol Reporter 31:1166–1175
Argyros RD, Mathews DE, Chiang Y-H et al (2008) Type B response regulators of arabidopsis play key roles in cytokinin signaling and plant development. Plant Cell 20:2102–2116
Azuma A, Kobayashi S, Yakushiji H et al (2007) VvmybA1 genotype determines grape skin color. Vitis 46:154–155
Barry CS, Aldridge GM, Herzog G et al (2012) Altered chloroplast development and delayed fruit ripening caused by mutations in a zinc metalloprotease at the lutescent2 locus of tomato. Plant Physiol 159:1086–1098
Beale S (1999) Enzymes of chlorophyll biosynthesis. Photosynth Res 60:43–73
Beale SI (2005) Green genes gleaned. Trends Plant Sci 10:309–312
Borevitz JO, Xia Y, Blount J et al (2000) Activation tagging identifies a conserved MYB regulator of phenylpropanoid biosynthesis. Plant Cell 12:2383–2393
Brand A, Borovsky Y, Meir S et al (2012) pc8.1, a major QTL for pigment content in pepper fruit, is associated with variation in plastid compartment size. Planta 235:579–588
Cheng C-Y, Kieber J (2014) Signaling: cytokinin signaling. In: Howell SH (ed) Molecular biology. Springer New York, pp 1–19
Clark MS (1997) Plant molecular biology—a laboratory manual. In: Clark DMS (ed). Springer, Berlin Heidelberg
Cortleven A, Schmülling T (2015) Regulation of chloroplast development and function by cytokinin. J Exp Bot 66:4999–5013
D’Agostino IB, Kieber JJ (1999) Phosphorelay signal transduction: the emerging family of plant response regulators. Trends Biochem Sci 24:452–456
Feller A, Machemer K, Braun EL et al (2011) Evolutionary and comparative analysis of MYB and bHLH plant transcription factors. Plant J 66:94–116
Fitter DW, Martin DJ, Copley MJ et al (2002) GLK gene pairs regulate chloroplast development in diverse plant species. Plant J 31:713–727
Forth D, Pyke KA (2006) The suffulta mutation in tomato reveals a novel method of plastid replication during fruit ripening. J Exp Bot 57:1971–1979
Han SE, Lee HE, Heo S et al (2011) Isolation and characterization of genes expressed differently in mature fruits of ‘redfield’ and ‘greensleeves’ apples. Hortic Environ Biote 52:413–421
Heyl A, Brault M, Frugier F et al (2013) Nomenclature for members of the two-component signaling pathway of plants. Plant Physiol 161:1063–1065
Hosoda K, Imamura A, Katoh E et al (2002) Molecular structure of the GARP family of plant Myb-related DNA binding motifs of the arabidopsis response regulators. Plant Cell 14:2015–2029
Huang S, Li R, Zhang Z et al (2009) The genome of the cucumber, Cucumis sativus L. Nat Genet 41:1275–1281
Hutchison CE, Li J, Argueso C et al (2006) The arabidopsis histidine phosphotransfer proteins are redundant positive regulators of cytokinin signaling. Plant Cell 18:3073–3087
Hwang I, Chen H-C, Sheen J (2002) Two-component signal transduction pathways in arabidopsis. Plant Physiol 129:500–515
Inskeep WP, Bloom PR (1985) Extinction coefficients of chlorophyll a and b in N, N-dimethylformamide and 80 % acetone. Plant Physiol 77:483–485
Kayesh E, Shangguan LF, Korir NK et al (2013) Fruit skin color and the role of anthocyanin. Acta Physiologiae Plantarum 35:2879–2890
Knerr LD, Staub JE, Holder DJ et al (1989) Genetic diversity in Cucumis sativus L. assessed by variation at 18 allozyme coding loci. Theor Appl Genet 78:119–128
Li Z, Huang S, Liu S et al (2009) Molecular isolation of the M gene suggests that a conserved-residue conversion induces the formation of bisexual flowers in cucumber plants. Genetics 182:1381–1385
Li YH, Yang LM, Pathak M et al (2011) Fine genetic mapping of cp: a recessive gene for compact (dwarf) plant architecture in cucumber, Cucumis sativus L. Theor Appl Genet 123:973–983
Li Z, Wang S, Tao Q et al (2012) A putative positive feedback regulation mechanism in CsACS2 expression suggests a modified model for sex determination in cucumber (Cucumis sativus L.). J Exp Bot 63:4475–4484
Li Y, Wen C, Weng Y (2013a) Fine mapping of the pleiotropic locus B for black spine and orange mature fruit color in cucumber identifies a 50 kb region containing a R2R3-MYB transcription factor. Theor Appl Genet 126:2187–2196
Li Z, Wang S, Gui X-L, et al. (2013b) A further analysis of the relationship between yellow ripe-fruit color and the capsanthin-capsorubin synthase gene in pepper (Capsicum sp.) Indicated a new mutant variant in C. annuum and a tandem repeat structure in promoter region. PLoS One 8
Li C, Li D, Shao F et al (2015) Molecular cloning and expression analysis of WRKY transcription factor genes in Salvia miltiorrhiza. BMC Genom 16:1–21
Liu H, Meng H, Pan Y, et al (2015) Fine genetic mapping of the white immature fruit color gene w to a 33.0-kb region in cucumber (Cucumis sativus L.). Theor Appl Genet 128:2375–2385
Livak KJ, Schmittgen TD (2001) Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods 25:402–408
Lu S, Van Eck J, Zhou X et al (2006) The cauliflower Or gene encodes a DnaJ cysteine-rich domain-containing protein that mediates high levels of beta-carotene accumulation. Plant Cell 18:3594–3605
Ma N, Feng H, Meng X et al (2014) Overexpression of tomato SlNAC1 transcription factor alters fruit pigmentation and softening. BMC Plant Biol 14:351
Makino S, Kiba T, Imamura A et al (2000) Genes encoding pseudo-response regulators: insight into his-to-asp phosphorelay and circadian rhythm in Arabidopsis thaliana. Plant Cell Physiol 41:791–803
Motamayor JC, Mockaitis K, Schmutz J et al (2013) The genome sequence of the most widely cultivated cacao type and its use to identify candidate genes regulating pod color. Genome Biol 14:r53
Neff MM, Neff JD, Chory J et al (1998) dCAPS, a simple technique for the genetic analysis of single nucleotide polymorphisms: experimental applications in Arabidopsis thaliana genetics. Plant J 14:387–392
Pan Y, Bradley G, Pyke K et al (2013) Network inference analysis identifies an APRR2-Like gene linked to pigment accumulation in tomato and pepper fruits. Plant Physiol 161:1476–1485
Perochon A, Dieterle S, Pouzet C et al (2010) Interaction of a plant pseudo-response regulator with a calmodulin-like protein. Biochem Bioph Res Co 398:747–751
Popovsky S, Paran I (2000) Molecular genetics of the y locus in pepper: its relation to capsanthin-capsorubin synthase and to fruit color. Theor Appl Genet 101:86–89
Powell ALT, Nguyen CV, Hill T et al (2012) Uniform ripening encodes a Golden 2-like transcription factor regulating tomato fruit chloroplast development. Science 336:1711–1715
Pramnoi P, Somta P, Chankaew S et al (2013) A single recessive gene controls fragrance in cucumber (Cucumis sativus L.). J Genet 92:147–149
Pyke KA, Leech RM (1992) Chloroplast division and expansion is radically altered by nuclear mutations in Arabidopsis thaliana. Plant Physiol 99:1005–1008
Qi J, Liu X, Shen D et al (2013) A genomic variation map provides insights into the genetic basis of cucumber domestication and diversity. Nature Genet 45:1510–1515
Salamov AA, Solovyev VV (2000) Ab initio gene finding in Drosophila genomic DNA. Genome Res 10:516–522
Satbhai SB, Yamashino T, Okada R et al (2011) Pseudo-response regulator (PRR) homologues of the moss Physcomitrella patens: insights into the evolution of the PRR family in land plants. DNA Res 18:39–52
Shang Y, Ma Y, Zhou Y et al (2014) Biosynthesis, regulation, and domestication of bitterness in cucumber. Science 346:1084–1088
Singh R, Low E-TL, Ooi LC-L et al (2014) The oil palm VIRESCENS gene controls fruit colour and encodes a R2R3-MYB. Nature communications 5:4106
Tan J, Tao Q, Niu H, et al (2015) A novel allele of monoecious (m) locus is responsible for elongated fruit shape and perfect flowers in cucumber (Cucumis sativus L.). Theor Appl Genet 128:2483–2493
Wang L, Li J, Zhao J et al (2015) Evolutionary developmental genetics of fruit morphological variation within the Solanaceae. Front Plant Sci 6:248
Waters MT, Wang P, Korkaric M et al (2009) GLK transcription factors coordinate expression of the photosynthetic apparatus in Arabidopsis. Plant Cell 21:1109–1128
Weng Y (2014) Molecularly tagged genes and quantitative trait loci in cucumber. CUCURBITACEAE 2014, Bay Harbor, Michigan, USA, p 48
Weng Y, Johnson S, Staub JE et al (2010) An extended intervarietal microsatellite linkage map of cucumber, Cucumis sativus L. HortScience 45:882–886
Wheeler DL, Barrett T, Benson DA et al (2005) Database resources of the National Center for Biotechnology Information. Nucleic Acids Res 33:D39–D45
Xie J, Wehner TC (2001) Gene list 2001 for cucumber. Cucurbit Genet Coop. Report 24:110–136
Yang YN, Zhao G, Yue WQ et al (2013) Molecular cloning and gene expression differences of the anthocyanin biosynthesis-related genes in the red/green skin color mutant of pear (Pyrus communis L.). Tree Genet Genomes 9:1351–1360
Yang X, Li Y, Zhang W et al (2014a) Fine mapping of the uniform immature fruit color gene u in cucumber (Cucumis sativus L.). Euphytica 196:341–348
Yang X, Zhang W, He H et al (2014b) Tuberculate fruit gene Tu encodes a C2 H2 zinc finger protein that is required for the warty fruit phenotype in cucumber (Cucumis sativus L.). Plant J Cell Mol Biol 78:1034–1046
Yundaeng C, Somta P, Tangphatsornruang S, et al (2015) A single base substitution in BADH/AMADH is responsible for fragrance in cucumber (Cucumis sativus L.), and development of SNAP markers for the fragrance. Theor Appl Genet 128:1881–1892
Zhang SP, Miao H, Gu XF et al (2010) Genetic mapping of the scab resistance gene in cucumber. J Am Soc Hortic Sci 135:53–58
Zhou M-Y, Gomez-Sanchez CE (2000) Universal TA cloning. Curr Issues Mol Biol 2:1–7
Acknowledgments
We thank Dr. Husain Ahmad and Cuinan Wu for critical reading of the manuscript. We are grateful for the assistance provided by Yupeng Pan. This research was supported by a grant from the National Natural Science Foundation of China (Project #31372074).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Ethical standards
The authors declare that this study complies with the current laws of the countries in which the experiments were performed.
Additional information
Communicated by R. G. F. Visser.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Liu, H., Jiao, J., Liang, X. et al. Map-based cloning, identification and characterization of the w gene controlling white immature fruit color in cucumber (Cucumis sativus L.). Theor Appl Genet 129, 1247–1256 (2016). https://doi.org/10.1007/s00122-016-2700-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00122-016-2700-8