
Abstract
Citrus tristeza virus (CTV) has caused the death of millions of trees grafted on sour orange (Citrus aurantium). However, this rootstock is very well adapted to the Mediterranean, semi-arid conditions. The aim of the present research is to genetically analyze the accumulation of CTV in a progeny derived from the cross between C. aurantium and Poncirus trifoliata, both resistant to CTV isolate T-346. Graft propagation of 104 hybrids was done on healthy sweet orange as a rootstock. Three months later, each rootstock was graft inoculated with two patches of infected tissue (isolate T-346). One, 2, and sometimes, 3 and 4 years after inoculation, hybrids and infected patches were tested for CTV by tissue-blot immuno-assay. Additionally, CTV multiplication was evaluated every year as the optical density of double-antibody sandwich enzyme-linked immuno-sorbent assay reactions. Linkage maps for P. trifoliata based on 63 markers, and for C. aurantium based on 157 markers, were used. Most molecular markers were microsatellites and IRAP (inter-retrotransposon amplified polymorphisms). Some analogues of resistance and expressed sequences were also included for candidate gene analysis.
Resistance against CTV was analyzed as a quantitative trait (CTV accumulation) by QTL (quantitative trait loci) analysis to avoid the assumption of monogenic control. Three major resistance QTLs were detected where the P. trifoliata resistance gene, Ctv-R, had been previously located in other progenies. Up to five minor QTLs were detected (Ctv-A 1 to Ctv-A 5 ). A significant epistatic interaction involving Ctv-R 1 and Ctv-A 1 was also found. An analogue of a resistance gene is a candidate for Ctv-A 3 , and two expressed sequences are candidates for Ctv-A 1 and Ctv-A 5 . Single-strand conformational polymorphism analysis of CTV genes QTL P20 and P25 (coat protein) in susceptible hybrids, was carried out to test whether or not any QTL accumulation was a defeated resistance gene. Since the same haplotype of the virus was visualized independently on the CTV titer, differences in the amount of virions are not explained through the selection of CTV genotypes by the host, but through differences among citradias in CTV replication and/or movement.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Citrus tristeza virus (CTV) is the causal agent of one of the most important diseases of citrus (Bar-Joseph and Lee 1989). This phloem-associated closterovirus exists in a large variety of isolates that differ in biological properties, such as symptoms in the field, reaction on indicator plants and aphid transmissibility. Since its outbreak in the early 1930s, tristeza has caused the death of millions of trees grafted on sour orange all around the world. The use of only tristeza-tolerant rootstock cultivars, worse adapted to semi-arid conditions than sour orange, has greatly narrowed the genetic diversity of rootstock cultivars increasing the vulnerability of citriculture in some areas. A sad example is ‘Citrus Sudden Death’; a new tristeza-like disease (Bassannezi et al. 2003) that affects sweet orange grafted on Rangpur lime. It has recently appeared in Brazil, in huge areas where only this rootstock is used for sweet orange production. The ability of viruses to expand their host range is an important practical effect of virus evolution (Roossnick 1997). Virus emergence is caused by several factors, including expanded host and vector range, human population increases, and rapid global movement of humans and their domesticated plants and animals. On the other hand, there are several factors contributing to the great variability of CTV: RNA polymerase errors in replication building up large viral populations, recombination and the fact of being hosted by a perennial crop where multiple CTV genotypes and other viruses may evolve together for long time increasing the probabilities of reassortment.
Resistance or tolerance to CTV is a major component of most citrus breeding programmes. Several studies have reported markers linked to a putative resistance gene against citrus tristeza virus (CTV) in Poncirus trifoliata, named Ctv-R (Gmitter et al. 1996; Mestre et al. 1997a; Fang et al. 1998). Nowadays it seems apparent that the genomic region where Ctv-R is located corresponds to a disease resistance gene cluster that includes other resistance genes, such as a putative QTL for nematode resistance (Ling et al. 2000; Deng et al. 2001). Chromosome walking along this region (Yang et al. 2001) delimited the Ctv-R gene location in a region between two resistance gene analogues (pY65 and pY28), spanning 322 kb approximately. However, all previous linkage studies trying to locate Ctv-R assume the hypothesis of a monogenic inheritance (resistant versus susceptible), and resistance was evaluated, at the most, a year after inoculation and not after a prolonged challenge. In a companion paper, Bernet et al. (2003) mapped Ctv-R in a different position within the Pa4 linkage group, than previously reported using a population of citradias (derived from the cross between sour orange and P. trifoliata) after a prolonged challenge. The change in positioning was interpreted as a deviation from the hypothesis of monogenic control for CTV resistance. In fact, Mestre et al. (1997b) had previously presented evidence supporting the involvement of more than one gene after the prolonged challenge.
Dawson and Hilf (1992) reviewed numerous results supporting the quantitative nature of susceptibility and resistance. Moreover, at the molecular level, the defense response triggered by many gene-for-gene systems also presents a quantitative nature due to the number of genes involved (Bent 1996). QTL analysis has provided geneticists with an opportunity to genetically analyze the plant-pathogen interaction; not only allowing the study of factors related to the resistance response but also factors involved in susceptibility, which are much more scarcely reported. Some aspects of plant-pathogen interaction complicate the QTL dissection of disease resistance. For example, disease incidence or severity may be rated on an ordinal scale rather than a truly quantitative evaluation, that has the normal distribution required for most statistical approaches. Genetic differences between strains or isolates may also result in different QTL profiles. Furthermore, the timing and method of inoculation may alter the QTL detected. In spite of all these complications, QTL analysis avoids the need to assume monogenic inheritance and provides information on the number, position and relative contribution of putative genes involved, which is very relevant for the success of future marker-assisted selection and genetic transformation experiments. Of course, it is impossible to unveil all genes just using a single segregating population because not all QTLs are segregating, or segregating alleles may not be different enough at all QTLs to allow their detection. Therefore, the larger the number of independent progenies the better the estimation for the number of QTLs or genes involved.
In this paper we describe the detection of QTLs involved in CTV accumulation in a family of citradias (104 C. aurantium×P. trifoliata hybrids), for more than 2 years after inoculation with a common CTV isolate. Epistatic interacions were also studied. A moderately dense linkage map for each parent was used that contains not only anonymous markers but also expressed sequences and analogues of resistance (Bernet et al. 2003).
Materials and methods
A segregating population (A×Pa) consisting of 104 hybrids was used for QTL analysis of CTV accumulation. It was derived from the cross between sour orange (C. aurantium L. var ‘Afin Verna’) and the trifoliate orange ‘Flying Dragon’ (FD).
JOINMAP 3.0 (Van Ooijen and Voorrips 2001) with the Kosambi mapping function was used for linkage. The A×Pa population was analyzed as a “cross pollinator” population type with no previous knowledge of the linkage phase of the markers. Two separate maps were developed for each parent by using the pseudotestcross coding form at each “loc” file. The nomenclature used for linkage groups is described in Ruiz and Asins (2003). These linkage maps were updated by increasing the family size from 63 to 104 hybrids, and increasing the number of molecular markers up to 157 and 63 in the sour orange and FD segregation analysis, respectively. New markers include some inter-retrotranposon amplified polymorphisms (IRAP) derived from gypsy-like retrotransposons sequences (C1, C2, C8 and C11, in Bernet and Asins 2003) and expressed sequences reported by Bernet et al. 2003.
The presence of Ctv-R was inferred from the presence of FD alleles at pY65 and pY28 marker loci (Yang et al. 2001). To do this, specific primers were designed to develop the corresponding SCARs (Bernet et al. 2003).
Evaluation of CTV resistance and accumulation
A×Pa hybrids and parents (sour orange and FD) were propagated on sweet orange rootstocks. After this propagation, the rootstocks were inoculated by grafting patches of infected sweet orange with CTV isolate T-346, a common Spanish isolate kept at the bank of CTV isolates at IVIA.
A first lot of 66 A×Pa hybrids was inoculated at the beginning of 1999 and the rest, 2 years later. The presence of the virus was checked every 6 months at five shoots (or petioles when shoots were not available) from different branches and at the inocula, as a positive control, by direct tissue-blot immuno-assay (DTBIA) following the procedure described in Garnsey et al. (1993). A plant was declared resistant when CTV was detected by this procedure at the inoculum but not at any branch. The CTV amount was also evaluated by DAS-ELISA (Double Antibody Sandwich Enzyme-Linked Immuno Sorbent Assay) using monoclonal antibodies 3CA5 and 3DFI together, as described in Cambra et al. (1993).
In spring of both 2001 and 2002, at least four branches of each inoculated plant were analyzed by DTBIA and DAS-ELISA. Those hybrids, where the virus was detected in all branches were considered susceptible (with uniform spatial distribution of the virus) and those where the virus was detected in at least one branch, but not in others, were declared susceptible with an irregular distribution of the virus.
QTL analysis
The mean optical density of DAS-ELISA reactions through years for the whole tree was used as a variable (CTV titre or CTV accumulation) for the QTL analysis.
Putative QTLs were identified using several statistical methodologies. Since CTV accumulation heavily departs from normality, a non-parametric test based on the Kruskal-Wallis (KW) methodology was considered for both C. aurantium and P. trifoliata linkage maps, separately, using the MapQTL 3.0 computer program (Van Ooijen and Maliepaard 1996). These linkage maps were first reported by Ruiz and Asins (2003), and enriched with new markers, analogues or resistance, expressed sequence tag (EST) and expressed sequences, by Bernet et al. (2003). For markers segregating as a backcross [as in the pseudo-test-cross design described by Grattapaglia et al. (1996); MiMj×MiMi for the C. aurantium genetic map, or MiMi×MiMj for the P. trifoliata one], log-transformed ELISA means were used for QTL analysis by interval mapping (IM) and composite interval mapping (CIM) using the QTL Cartographer computer program (Basten et al. 2002). All these analysis were repeated omitting resistant hybrids; i.e. using data on CTV accumulation only from susceptible individuals.
Epistatic interactions affecting CTV accumulation were analyzed by two-way ANOVA for pairwise combinations of significant marker loci. Only epistatic interactions between pY65 and candidate resistance genes (strategies 4 and 5, in Bernet et al. 2003) or P. trifoliata markers where a QTL had been detected, were tested. The same study was carried out in sour orange but using as reference marker pY28. The Bonferroni adjustment was employed to obtain an overall protection level of 0.05.
Results
The updated genetic maps of sour orange and FD (nine linkage groups each) are presented in Fig. 1. Segregation distortion was shown by 28% and 25.4% of sour orange and FD markers, respectively. Six and seven markers did not join any linkage group of the sour orange and FD maps, respectively. In both cases, four of theses unlinked markers presented distorted segregation ratios.


Sour orange and P. trifoliata “Flying Dragon” linkage maps based on the segregation analysis of 157 and 63 makers, respectively, within the A×Pa progeny (Bernet et al. 2003). Asterisks at markers indicate deviation from the expected segregation ratio (1:1). Nomenclature is described in Ruiz and Asins (2003)
The distribution of the mean DAS-ELISA values, although continuous, is not normally distributed. A little improvement was obtained by using the logarithm-transformed values (Fig. 2). Mean DAS-ELISA values for the resistant parents were 0.375 and 0.379 for C. aurantium and trifoliate orange, respectively. Some hybrids showed important changes in DAS-ELISA values through time (time-irregular hybrids). In some of these hybrids the distribution of the virus was also irregular among branches. In general, all hybrids showing high levels (over 1.5 O.D.) of CTV, presented also a uniform distribution among branches. In a hybrid with a very low amount of CTV (similar to the titre of resistant genotypes), the distribution of the virus was highly irregular among branches. This hybrid is one of those three susceptible ones carrying the putative Ctv-R gene from P. trifoliata (Bernet et al. 2003). Given that this result and others previously reported (Mestre et al. 1997b) disagree with the hypothesis of one single gene controlling the abundance of CTV in inoculated citrus plants; the involvement of other genomic regions was investigated by QTL analysis.

Histogram of the distribution of ELISA means (a) and ELISA log-transformed means (b) from data recorded in Spring from 0.5 to 3 years after inoculation
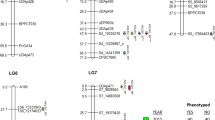
Although some differences in QTL detection were obtained depending on the statistical method, a general agreement was observed between QTLs detected by the Kruskal-Wallis (KW) and the Interval Mapping (IM) or Composite Interval Mapping (CIM) methodologies. The nearby marker given by the KW test, its linkage group, the position from the leftmost marker given by CIM and the statistical significance of each QTL, is presented in Table 1. QTLs detected by KW for DAS-ELISA means in the whole population or only for the susceptible hybrids, are located on linkage groups Pa4, A(7+3+4), A(8+6) and A(10+5b). They are presented in Fig. 3.

Value of the Kruskal-Wallis statistic for ELISA at markers of relevant linkage groups. Linkage groups as in Bernet et al. 2003. W: Whole population; S: susceptible subpopulation. Horizontal lines correspond to a 1% significant level
QTLs with the greatest contributions, Ctv-R 1 , -R 2 and -R a , are located on linkage group 4 where Ctv-R has been previously mapped (Gmitter et al. 1996; Mestre et al. 1997a; Fang et al. 1998), and in disagreement with our analysis of these data in which the monogenic inheritance of CTV resistance was assumed (Bernet et al. 2003). Two maxima were observed; therefore, two closely linked QTLs at pY65 and Z16 (Ctv-R1 and Ctv-R2, respectively) are hypothesized. Both KW (Fig. 3) and CIM (data not shown) presented those maxima, although CIM gave it at a position between markers “520” and “cO7-AB_850” (see Table 1) with a clear change in the likelihood ratio (LR). However, if the size of the window to adjust for adjacent markers is set to 2 cM instead of 5 cM, the highest peak is on marker Z16 with a LR of 46.78, then decreases in pY28 (LR=15.06) and then another peak on pY65 (LR=29.85). Markers pY65 and Z16 map about 2 cM apart and the population size is not too large; hence the resolution could not be enough to ensure the presence of the two QTLs. Nevertheless, there are some indications that should be investigated further. Interestingly, another QTL is also detected within this small region at sour orange linkage group 4. It has been named Ctv-R a .
A QTL with smaller gene effect (Ctv-A 3 ) is also involved in CTV accumulation, at the position of a putative analogue of resistance in the sour orange linkage map (S2-AS4_1700).
Some QTLs are detected using KW, but not with CIM in the whole population. However, when analyzing only susceptible hybrids some of these QTLs, and Ctv-A 5 , are also detected by CIM. In susceptible hybrids, an important QTL, Ctv-A 1 , seems to be involved in CTV accumulation. Its position (Fig. 4a) coincides with a cDNA clone obtained by differential-display analysis (Bernet et al. 2003), and it is specific to (always) susceptible (rr) Pp×Pp plants (derived from the self-pollination of P. trifoliata).

Composite Interval Mapping of log ELISA in the whole population (c) and in the susceptible subpopulation (a and b) for linkage groups Pa4 (a) and A(7+3+4) (b and c). LR: Likelihood ratio test value
Some QTLs were detected using CIM but not KW; for instance, a peak at the ‘520’ marker (an Rps2 analogue) in Fig. 4, and another between markers CL140_300 and CR12_1400 in linkage group AIII (data not shown). Given that CIM assumes a normal distribution of the trait, we tend to believe they might be artifacts of the analysis and rely more on the results using KW.
The possible existence of epistatic interactions involved in the genetic control of CTV accumulation was investigated. Only epistatic interactions between pY65 and P. trifoliata markers, where a QTL had been detected or candidate resistance genes (strategies 4 and 5 in Bernet et al. 2003) were tested. The same study was carried out in sour orange but using as the reference marker pY28. Two significant epistatic interactions were detected involving P. trifoliata markers VIC (p<0.01) and TAA 15 (p<0.02) using the whole population (Fig. 5). For sour orange, a significant epistatic interaction was found involving the differential cDNA clone 28R (p<0.02). Other interactions, near significance, involved 1R (p<0.06) and SCAB (p<0.08).

Means and standard errors of log ELISA in the whole population (a and b) and in the susceptible subpopulation (c) for pairs of interacting markers. 1: allele b (low-molecular-weight band); 2: allele a (high-molecular-weight-band)
Discussion
As expected, QTL analysis of CTV accumulation gives information that is more complex, but also more complete than that obtained under the assumption of monogenic inheritance. One (or several) major closely linked QTLs (Ctv-R 1 , Ctv-R 2 ) are involved in the resistance coming from P. trifoliata. Their contribution is very large (67–47%) and their position coincides with that reported by previous studies for Ctv-R using other progenies, after less prolonged challenges (Gmitter et al. 1996; Mestre et al. 1997a; Fang et al. 1998). Sour orange also presents a major QTL (Ctv-R a ), contributing almost a 60% to the total variance. Its position on A(7+3+4) seems to coincide with that of Ctv-R 1 and it would explain the CTV resistance of sour orange. Therefore, Ctv-R 1 and Ctv-R a might be allelic, and their most likely position would be between pY28 and ck16. In addition to the resistant QTLs, two CTV accumulation QTLs are also located at linkage group 4, Ctv-A 1 in P. trifoliata and Ctv-A 3 in C. aurantium; but they do not correspond to the same locus, because VIC (the closest marker to Ctv-A 1 ) is in another linkage group (AI+AVII) in the C. aurantium map. An analogue of resistance, S2-AS4-1700, is the candidate gene for Ctv-A 3 , on linkage group A(7+3+4).
Sequencing data on chromosome walking throughout the region containing Ctv-R 1 and Ctv-R 2 has shown how rich in resistance-gene analogues this region is (Deng et al. 2001; Yang et al. 2001). As with any QTL analyses, demonstration of a significant association between resistance and a chromosomal region does not distinguish between a single QTL of large effect or multiple QTLs of smaller effects. No resistance QTL has yet been dissected by recombination to distinguish between these possibilities. However, some resistance genes have already been cloned (Hammond-Kosack and Jones 1997), and one of them, the Pto gene that confers resistance to races of Pseudomonas syringe pv tomato that carry the AvrPto gene, was cloned using the map-based approach (Martin et al. 1993). Even in this case, where the disease resistance gene has a very large effect, further mutagenesis experiments have revealed an additional gene, Prf, that is required for Pto function and is located 24 kb from Pto (Salmeron et al. 1996). Clusters of disease-resistance genes are quite common, and would explain their evolution (Hammond-Kosack and Jones 1997; Takken and Joosten 2000) and involvement in the resistance response to very different pathogens (Van der Vossen et al. 2000). In Arabidopsis, five of these R gene-rich regions are now recognized and have been designed as “major resistance complexes”. Linkage group 4 of P. trifoliata and Citrus, definitively contains at least one major resistance complex. How many resistance genes of this complex are necessary to confer resistance to CTV? Will it depend on other loci in which P. trifoliata is homozygous and C. aurantium is heterozygous? A genetic approach based on mutagenesis to find interacting partners of R proteins (Required for Disease Resistance or RDR loci) has been fruitfully followed in Arabidopsis, tomato and barley (reviewed by Takken and Jooste 2000). In species, like citrus, where this approach is not affordable, an alternative one might be QTL analysis of the virus-plant interaction using progenies derived from distantly related resistant parents which constitutes the objective of the present paper.
QTL analysis presents several limitations (Asins 2002) and most of them affect this study. CTV accumulation presents a skewed distribution, even after log-transforming data (Fig. 2), and we do not know how this deviation from normality affects type I and II errors of IM and CIM methodologies. Consequently, we assume KW is the proper methodology for the QTL analysis of the trait. Of course, QTLs detected by all methodologies are expected to be the most reliable ones. QTLs in linkage groups A(8+6), and mainly A(10-5b), are only detected by using the KW methodology, while some QTLs were only detected by IM and CIM. Although CIM is expected to provide more accurate estimates of the QTL position than any other methodology (Zeng 1994) we think that, in the present situation, its % of false positives (type I error) might be greatly affected. Thus, the QTL detected between Ctv-A 1 and Ctv-R 1 (Fig. 4a) is considered a likely artifact or a ghost QTL.
Since Ctv-R 1 , Ctv-R 2 and Ctv-R a have large effects, another QTL analysis of CTV accumulation has been carried out using only susceptible hybrids (those where the virus was detected at least once along the experiment). Besides, it could consider a way to unveil factors involved in susceptibility; i.e. host factors needed by CTV for replication, movement and systemic infection. The obvious limitation of this second study is the reduced progeny size, of 43 hybrids. Additionally, the elimination of resistant progeny will also eliminate segregation in the regions adjacent to major R genes. This means that QTLs in these regions will be more difficult to detect. Although the distribution in these regions might be more skewed, it might improve in other regions, as our data have shown; but in any case, the use of a non-parametric approach may take into account the skewed segregation. This analysis has two advantages: in addition to improvement towards normality, other minor QTLs, localized at group Pa4 or A(3+7+4), can now be detected by reducing the variance associated with their phenotypic effects. This is the case of Ctv-A 1 in Pa4 and Ctv-A 4 , which is closely linked to Ctv-R a . However, why does Ctv-A 3 disappear? We think this is due to its relatively large distance from Ctv-R a , its low contribution (14%) and its reduction in progeny size.
The position of an expressed sequence (VIC) coincides with the position of the maximum LOD for QTL Ctv-A 1 (Fig. 4a). VIC also presents epistatic interactions with Ctv-R 1 using data on CTV accumulation from the whole progeny (Fig. 5). Thus, in those hybrids that inherited Ctv-r 1 (pY65-1) the amount of CTV will be significantly different depending on the inherited allele at VIC. VIC is a good candidate gene for Ctv-A 1 . Unfortunately, the search for homologous sequences, resulted in a gene of A. thaliana that codes for a hypothetical protein of unknown function. These results support the need to deeply study the expression and function of the VIC protein. Another gene involved in cell-to-cell movement, Ctm, different from Ctv-R, was reported to affect resistance of P. trifoliata against CTV (Mestre et al. 1997b). Because of its positioning, Ctv-A 1 could be it.
A QTL of moderate effect (28%) has been detected in susceptible hybrids (Ctv-A5). This QTL is also detected by using the KW methodology with the whole progeny. The maximum LOD score of this QTL coincides with the position of a microsatellite marker (CR16-780) that was derived from an EST (Ruiz and Asíns 2003). This EST corresponds to a NADP-isocitrate dehydrogenase. Another microsatellite (CR13-515), derived from an EST (Miyagawa-wase maturation stage pcMA1MA1M0910-38) at linkage group A(10+5b), is associated with CTV accumulation. The in silico translated protein coded by this EST contains an RNA recognition motif characteristic of splicing factors. The expression profile of all these candidate genes need to be studied to assess their putative involvement in CTV accumulation, as a previous step before confirmation by genetic transformation experiments.
The presence of two major types of disease resistance to plant pathogens, vertical resistance and horizontal resistance, has long been recognized in interactions between plant hosts and their pathogens. Vertical resistance in many plant host-pathogen relationships is hypersensitive, race-specific, and governed by interactions between avirulence genes in pathogens and resistance genes in plant hosts. In contrast, horizontal resistance is quantitative, presumably non-race specific, and controlled by polygenes, though these assumptions have not been actively tested. In this context, a hypothesis is that resistance genes would correspond to a QTL of large effect that would be responsible for vertical resistance, while a QTL with moderate or small effect would be responsible for horizontal resistance. Going further, Li et al al. (1999) have shown that a “defeated” resistance gene against a virulent strain of Xanthomonas oryzae acts as a QTL. If any QTL involved in the accumulation of CTV was a defeated resistance gene, then we should observe a change in the genetic composition of the CTV population hosted by citradias with a low CTV titre, in comparison to that of citradias with a high CTV titer. The haplotype composition for two CTV genes (P20 and P25, coding for the coat protein) was studied by single-strand conformation polymorphism analysis (Ayllon et al. 1999) in all susceptible citradias from the first inoculation lot at the 4th year after inoculation, and the original T-346 isolate was maintained in a sweet orange tree. All samples, including sweet orange, presented the same haplotype for CTV. Therefore the QTLs of moderate and small effect we have detected do not seem to have defeated resistance genes, although different results may be found if other CTV isolates are used.
Dawson and Hilf (1992) reviewed mounting evidence that systemic infection is an additive effect of replication, cell-to-cell movement and long-distance movement. Before a virus can invade a plant systemically it must accomplish a series of tasks, including infection, replication, cell-to-cell movement and long-distance movement. The virus must be able to perform each of these functions above some threshold rate. Systemic infection appears to be a balance of these functions, which must occur in concert. Due to their parasitic nature, viral replication and movement throughout the plant is controlled by a combination of viral and plant genes. We do not know which citrus gene products are used by CTV for replication and movement, but they have to exist since citrus is its natural host. Even P. trifoliata, that is a “nonhost” species, seems to be a cellular host since CTV can replicate within its protoplasts (Albiach-Martí et al. 1999). Therefore, it is possible that some accumulation QTLs, at least, correspond to loci where different alleles correspond to proteins used by CTV with a varying degree of efficiency in replication and/or movement. Additionally, the resistance response of the gene-for-gene system seems to require more than one single-gene product; not only the products of RDR genes but also a pathogenicity target, as in the ‘guard’ model proposed by Van der Biezen and Jones (1999). If the progenies used segregate for more than one of those genes, and gene effects of the different alleles can be detected, then the genetic analysis of the virus-plant interaction provides a method to detect and locate them simultaneously.
This paper is the first time that the CTV-citrus interaction has been studied by QTL analysis of virus accumulation. This allowed the study of its genetic control without assuming the restrictive hypothesis of monogenic inheritance. At least one major QTL has been detected at the position where Ctv-R had been previously located in other progenies. The detection of epistatic interactions with Ctv-R 1 and Ctv-R a suggests that the response of the citrus plant to infection with CTV involves interaction between the resistance gene and other polymorphic genes of the plant genome. Candidate genes have been selected that map where three QTLs locate (Ctv-A 1 , Ctv-A 3 and Ctv-A 5 ), and will be the objective of future experiments on expression profiling.
References
Albiach-Martí MR, Grosser JW, Hilf ME, Gowda S, Mawassi M, Satyanarayana T, Garnsey SM, Dawson WO (1999) CTV resistant plants are not immune to CTV at the cellular level. Proc 18th Annual Meeting of the American Society for Virology 192
Asíns MJ (2002) Present and future of quantitative trait locus analysis in plant breeding. Plant Breed 121:281–291
Ayllon MA, Rubio L, Moya A, Guerri J, Moreno P (1999) The haplotype distribution of two genes of citrus tristeza virus altered after host change of aphid transmission. Virology 255:32–39
Bassanezi RB, Bergamin-Filho A, Amorim L, Gimenes-Fernandes N, Gottwald TR, Bové JM (2003) Spatial and temporal analyses of citrus sudden death as a tool to generate hypothesis concerning its etiology. Phytopathology (in press)
Bar-Joseph M, Lee RF (1989) Citrus tristeza virus: descriptions of plant viruses no. 353. Assoc App Biol, Wellsbourne, UK
Basten CJ, Weir BS, Zeng Z-B (2002) QTL Cartographer, Version 1.17. Department of Statistics, North Carolina State University, Raleigh, USA
Bent AF (1996) Plant disease resistance genes: function meets structure. Plant Cell 8:1757–1771
Bernet GP, Asins MJ (2003) Identification and genomic distribution of gypsy like retrotransposons in Citrus and Poncirus. Theor Appl Genet (in press)
Bernet GP, Bretó MP, Asins MJ (2003) Expressed sequence enrichment of linkage group 4 for candidate gene analysis of citrus tristeza virus resistance. Theor Appl Genet: DOI 10.1007/s00122-003-1479-6
Cambra M, Camarasa E, Gorris MT, Garnsey SM, Gumpf DJ, Tsai MC (1993) Epitope diversity of citrus tristeza virus isolates in Spain. In: Proc 12th Conf IOCV, Riverside, California, USA, pp 33–38
Dawson WO, Hilf ME (1992) Host-range determinants of plant viruses. Annu Rev Plant Physiol Plant Mol Biol 43:527–555
Deng Z, Tao Q, Chang YL, Huang S, Ling P, Yu C, Chen C, Gmitter FG, Zhang HB (2001) Construction of a bacterial artificial chromosome (BAC) library for citrus and identification of BAC contigs containing resistance-gene candidates. Theor Appl Genet 102:1177–1184
Fang DQ, Federici CT, Roose ML (1998) A high-resolution linkage map of the citrus tristeza virus resistance gene-region in Poncirus trifoliata (L.) Raf. Genetics 150:883–890
Garnsey SM, Pemar TA, Cambra M, Henderson CT (1993) Direct tissue-blot immunoassay (DTBIA) for detection of citrus tristeza virus (CTV). In: Moreno P, Da Graça JV, Timmer LW (eds) Proc 12th Conf Int Organiz Citrus Virol, IOCV, Riverside, California, pp 39–50
Gmitter FG, Xiao SY, Huang S, Hu XL, Garnsey SM, Deng Z (1996) A localized linkage map of the citrus tristeza virus resistance-gene region. Theor Appl Genet 92:688–695
Grattapaglia D, Bertolucci FLG, Penchel R, Sederoff R (1996) Genetic mapping of quantitative trait loci controlling growth and wood quality traits in Eucalyptus grandis using a maternal half-sib family and RAPD markers. Genetics 144:1205–1214
Hammond-Kosack K, Jones JDG (1997) Plant disease resistance genes. Annu Rev Plant Physiol Mol Biol 48:575–607
Li ZK Luo LJ, Mei H, Paterson AH, Zhao XH, Zhoung DB, Wang YP, Yu XQ, Zhu L, Tabien R, Stansel JW, Ying CS (1999) A defeated rice resistance gene acts as a QTL against a virulent strain of Xanthomonas oryzae pv oryzae. Mol Gen Genet 261:58–63
Ling P, Duncan LW, Deng Z, Dunn D, Xu X, Huang S, Gmitter FG (2000) Inheritance of citrus nematode resistance and its linkage with molecular markers. Theor Appl Genet 100:1010–1017
Martin GB, Brommonschenkel SH, Chunwongse J, Frary A, Ganal MW, Spivey R, Wu T, Earle ED, Tanksley SD (1993) Map-based cloning of a protein kinase gene conferring disease resistance in tomato. Science 262:1432–1436
Mestre PF, Asíns MJ, Pina JA, Carbonell EA, Navarro L (1997a) Molecular markers flanking the citrus tristeza virus resistance gene from Poncirus trifoliata (L.) Raf. Theor Appl Genet 94:458–464
Mestre PF, Asíns MJ, Carbonell EA, Navarro L (1997b) New gene(s) involved in the resistance of Poncirus trifoliata (L.) Raf. to citrus tristeza virus. Theor Appl Genet 95:691–695
Roossinck MJ (1997) Mechanisms of plant virus evolution. Annu Rev Phytopathol 35:191–209
Ruiz C, Asíns MJ (2003) Comparison between Poncirus and Citrus genetic linkage maps. Theor Appl Genet 106:826–836
Salmeron JM, Oldroyd GED, Rommens CMT, Scofield SR, Kim HS (1996) Tomato PRF is a member of the leucine-rich repeat class of plant disease resistance genes and lies embedded within the Pto kinase gene cluster. Cell 86:123–33
Takken FLW, Joosten MHAJ (2000) Plant resistance genes: their structure, function and evolution. Eur J Plant Pathol 106:699–713
Van der Biezen EA, Jones JDG (1999) Plant disease-resistance proteins and the gene-for-gene concept. Trends Biochem Sci 23:454–456
Van der Vossen EG, van der Voort JR, Kanyuka K, Bendahmane A, Sandbrink H, Baulcombe DC, Bakker J, Stiekema WJ, Klein-Lankhorst RM (2000) Homologues of a single resistance-gene cluster in potato confer resistance to distinct pathogens: a virus and a nematode. Plant J 23:567–576
Van Ooijen JW, Maliepaard C (1996) MapQTL version 3: software for the calculation of QTL positions on genetic maps. CPRO-DLO, Wageningen
Van Ooijen JW, Voorrips RE (2001) JoinMap 3.0, Software for the calculation of genetic linkage maps. Plant Research International, Wageningen, The Netherlands
Yang Z-N, Ye X-R, Choi S, Molina J, Moonan F, Wing RA, Roose ML, Mirkov TE (2001) Construction of a 1.2-Mb contig including the citrus tristeza virus resistance gene locus using a bacterial artificial chromosome library of Poncirus trifoliata (L.) Raf. Genome 44:382–393
Zeng ZB (1994) The precision mapping of quantitative trait loci. Genetics 136:1457–1468
Acknowledgments
This work was supported in part by grants from Conselleria de Cultura, Educació i Ciència (GPB), INIA (SC99-047) and MCYT (AGL2002-02395). Authors thank J. Puchades, M. Boil and J. Piquer for technical assistance.
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by C. Möllers
Rights and permissions
About this article
Cite this article
Asins, M.J., Bernet, G.P., Ruiz, C. et al. QTL analysis of citrus tristeza virus-citradia interaction. Theor Appl Genet 108, 603–611 (2004). https://doi.org/10.1007/s00122-003-1486-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00122-003-1486-7