
Abstract
Watermelon [Citrullus lanatus (Thunb.) Matsum & Nakai] is an important cucurbit crop cultivated worldwide. In an effort to map WBNV resistance, the current study consisted of two populations derived from a WBNV resistant accession, IIHR-82 belonging to Citrullus lanatus var. citroides (Syn. Citrullus amarus). The first population is a backcross inbred population (BC1F6) of 141 families (Pop I) that was developed by crossing IIHR-82 to an elite cultivar Arka Manik. A resistant family, BIL-53 belonging to Pop I was crossed to IIHR-140 to develop F3 population, consisting of 112 families (Pop II). Each of these populations was evaluated for WBNV incidence for two years under natural epiphytotic conditions. A significant correlation was observed for various resistance traits across years and populations. The frequency distribution graphs of Area Under Disease Progress Curve for WBNV incidence in both the populations exhibited a continuous distribution, indicating that trait is quantitative in nature. A total of 163 markers were mapped for the Pop I which spread onto 15 linkage groups (LGs) spanning a total map length of 3310.17 cM with a mean marker interval of 20.31 cM. The linkage map for the Pop II was constructed with 135 markers spread over 12 linkage groups spanning a total length of 1965.53 cM with an average interval between markers of 14.56 cM.The QTL analysis for WBNV resistance related traits revealed 14 major QTL's for Pop I and 19 for Pop II with a maximum PVE upto 21.20%. Multi-trait QTL regions were observed on LG 3b for Pop I and LG 2, LG 4, LG 7 and LG 8 for Pop II. Importantly, we observed common QTL regions for plant survival on LG 2 and PDI on LG 3 and LG 8 for both the populations. As this is the first attempt to map QTL's for WBNV resistance, the results obtained in the present study may provide a guide for fine mapping multi-trait QTL regions.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Watermelon [Citrullus lanatus (Thunb.) Matsum & Nakai] is an important cucurbit crop cultivated throughout the world in an area of 3.50 million hectares with a production of 117 million tonnes (FAO 2016). In India, it is cultivated in an area of 1.01 lakh hectares with a production of 24.8 lakh tonnes (MAFW 2017). Important among the several constraints limiting its cultivation in India is watermelon bud necrosis orthotospovirus (WBNV), (Family: Tospoviridae, Bunyavirales) belonging to the watermelon silver mottle orthotospovirus (WSMoV) serogroup (Jain et al. 1998, 2015; Adams et al. 2017). It was first reported during 1991 infecting watermelon at Indian Council of Agricultural Research-Indian Institute of Horticultural Research (IIHR), Bangalore, India (Singh and Krishnareddy 1996) and later found to affect several other cucurbits, such as cucumber, ridge gourd and muskmelon (Jain et al. 1998, 2007; Mandal et al. 2003; Kumar et al. 2010). Recently, WBNV was also detected in non-cucurbitaceous crops like chilli pepper, tomato (Kunkalikar et al. 2011) and chrysanthemum (Holkar et al. 2017) in northern India. WBNV is widely distributed and endemic in majority of the watermelon growing states of India (Mandal et al. 2012). The virus is reported to be transmitted by melon thrips, Thrips palmi Karny (Rajasekharam 2010; Rebijith et al. 2012, 2016). Several studies have reported up to 100% yield loss due to this disease (Singh and Krishnareddy1996; Jain et al. 1998, 2007; Kunkalikar et al. 2011).The field symptoms of WBNV appear as leaf chlorosis, silver mottling of leaves, shortened internodes, brittle and upright growth of younger shoots, necrosis on apical bud, stem, petiole, and fruit stalk. Infected plants produce unmarketable, small, deformed fruits with uneven surface and necrotic or chlorotic rings, depending on the cultivar (Mandal et al. 2012 and Nagesh et al. 2018).
Currently, vector management (cultural, physical, chemical and biological) has been the focus for controlling this disease, but with limited success (Kumar et al. 2006 and Mandal et al. 2012). Hence, host plant resistance has been suggested as an economically viable and environment friendly option for managing this disease (Riley and Pappu 2000). In this context, breeding efforts are underway at IIHR, Bengaluru, India, to deploy resistant varieties to WBNV. Recent work of our group has focused on identifying source of resistance in IIHR-82, an accession belonging to Citrullus lanatus var. citroides and understanding its genetics in a prebred line derived from this source (Nagesh et al. 2018).
Several genomic resources including SSR markers (Ren et al. 2012), SNP markers (Sandlin et al. 2012; Nimmakayala et al. 2014 and Lambel et al. 2014), genome sequence (Guo et al. 2013), comparative map (Sandlin et al. 2012), high resolution maps (Ren et al. 2012, 2015a, b; Reddy et al. 2014; Shang et al. 2016, Branham et al. 2019), integrated genetic maps (Nimmakayala et al. 2014 and Ren et al. 2014), resequencing of diverse watermelon accessions of related species (Guo et al. 2013), cultivated and wild watermelon accessions (Guo et al. 2019), draft genome assemblies for two diploid watermelon cultivars (Wu et al. 2019) became available for watermelon in the public domain dawning a genomic era for this crop. This aided in QTL analyses of important horticultural traits (Prothro et al. 2012; Sandlin et al. 2012; Ren et al. 2014; Lambel et al. 2014; Kim et al. 2015; Liu et al. 2015; Ren et al. 2015a, b; Cheng et al. 2016; Park et al. 2016; Zhang et al. 2018) including recent identification of candidate genes for flesh color, lycopene (Wang et al. 2019), flowering time (Gimode et al. 2020) dwarfness (Dong et al. 2018) and fruit shape (Dou et al. 2018) in watermelon. Recently, linkage mapping has been carried out for resistance to tomato spotted wilt orthotospovirus (TSWV) in peanut (Khera et al. 2016) and melon yellow spot orthotospovirus (MYSV) in cucumber (Sugiyama et al. 2015). Dominant genes Sw-5, Tsw and Sws conferring resistance to TSWV in tomato (Stevens et al. 1995), chilli (Jahn et al. 2000) and MYSV in cucumber (Sugiyama et al. 2015) respectively have been mapped. As no such reports are available for WBNV resistance in watermelon, the current experiment was designed to identify QTL’s conferring resistance to this disease in the accession IIHR-82.
Materials and methods
Mapping populations
IIHR, Bengaluru maintains a collection of 347 accessions of cultivated watermelon and related species, which were screened for WBNV resistance in earlier studies (unpublished results). An accession, IIHR-82 belonging to Citrullus lanatus var. citroides was identified as possessing resistance to this disease. A backcross inbred population (BC1F6) of 141 families was developed by crossing IIHR-82 to Arka Manik (recurrent parent, which is a mega variety in Indian subcontinent) hereafter called Pop I. A resistant family, BIL-53 belonging to this population was crossed to a red fleshed icebox segment inbred, IIHR-140 to develop F3 population, consisting of 112 families, hereafter referred as Pop II. Genotyping was performed in BC1F5 and F2 families while, phenotyping was carried out in BC1F6 and F2:3 families in Pop I and Pop II respectively.
Phenotyping for WBNV resistance
Natural epiphytotic screening of the mapping populations for reaction to WBNV was carried out during summer season, when natural vector population is high favouring natural disease occurrence. Paired row spot planting technique under unmulched condition along with infector genotypes and yellow ribbons were used to attract thrips (Vector of WBNV). Popular commercial varieties viz., NS-295 and Arka Manik that are susceptible to WBNV were used as infector lines. These were planted 10 days prior to planting of test progenies to build up the field inoculum.
The disease severity was scored visually for symptoms on a scale of 0–3 as suggested by Sugiyama et al. (2009) with slight modifications: where, 0 = no symptom, 1 = slight crinkling of leaves, 2 = crinkling with yellowing or silver mottling of leaves and 3 = die back or severe bud necrosis (Fig. 1). The disease severity scores of individual plants thus recorded were used to calculate percent disease index (PDI) using the following formula:

Disease severity scale (0–3) for watermelon bud necrosis orthotospovirus under natural epiphytotic conditions
The PDI recorded over four intervals of 10 days from 35 to 65 DAS was used to calculate Area under disease progress curve (AUDPC) (Campbell and Madden, 1990) using the formula:
where Xi = disease index at ith observation, Xi+1 = disease index at i + 1st observation ti+1 − ti = number of days between two observations, and n = total number of observations.
Further, plant survival (%) and vine length (cm) were recorded from 35 to 65 DAS at 10 days interval in both the populations. Vine length is important criteria for WBNV resistance, as apical growth is arrested due to meristem necrosis in WBNV affected plants. These four traits viz., PDI, AUDPC, plant survival and vine length representing component traits of WBNV resistance were used for QTL analysis.
The broad sense heritability was calculated for each trait by using the formula:
Experimental design
The populations along with their parents and F1′s were evaluated for WBNV reaction in randomized complete block design (RCBD) with two replications and five plants per replication. The phenotyping was carried out during summer seasons of 2016 and 2018 for Pop I and during 2017 and 2018 for Pop II.
Diagnosis of WBNV
Direct antigen-coated enzyme-linked immunosorbent assay (DAC-ELISA) and reverse transcription-polymerase chain reaction (RT-PCR) techniques were used to confirm WBNV infection in the test entries during screening. DAC-ELISA was performed using general orthotospovirus antiserum as per the protocol described by Hobbs et al. (1987). To specifically identify WBNV infection, RT-PCR was performed using primers specific to the nucleocapsid (N) gene of WBNV as described by Holkar et al. (2017). The diseased samples recorded an amplification of ~ 750 bp size, while it was absent in healthy samples.
DNA isolation and marker genotyping
Genomic DNA was isolated from the true leaves of three-week old plants of parents, F1's and mapping populations employing modified CTAB method (Keb-Llanes et al. 2002). Genome wide markers including (SSRs, Indels & SV’s) were assembled from earlier reports in watermelon viz., Lee et al. (2007), Zhang et al. (2012), Ren et al. (2012) and cucurbit genomics database (https://www.icugi.org/). DNA amplification was carried out in a 15.4 µl of master mix containing 2.5 µl of 10 × Taq assay buffer, 2.0 µl of 1 mM dNTP’s, 2.0 µl of 5 pM of forward and reverse primer each, 0.5 µl of 3B 1U/µl of Taq polymerase, 3.0 µl of 30 ng template DNA and 3.4 µl of nuclease free water (Sanbiomed Pvt. Ltd. Bengaluru). The polymerase chain reaction (PCR) conditions were followed as: 94 °C for 5 min initial denaturation; 35 cycles of amplification at 94 °C for 20 s, 65 °C for 20 s and 72 °C for 90 s, and 72 °C for 8 min of final extension followed by hold at 4 °C. The PCR was performed in eppendorf thermal cycler machine (Effendorf, AG 22331, Hamburg, Germany). The PCR amplicons were resolved by agarose gel electrophoresis (3%) in 0.5 × Tris acetic acid buffer (TAE) and documented on a UV gel doc (UVTEC, Cambridge, Firereader V10).
Construction of linkage map
The genotypic data for each of the mapping populations was used to construct linkage map using default parameters in software package, QTL IciMapping v. 4.1, released and updated in January, 2016 (Wang et al. 2016). The marker order was determined with a generally accepted minimum threshold logarithm of odds (LOD) score of 3.0 and recombination fraction (θ) of 0.3. Map distances between markers were estimated using Kosambi mapping function (Kosambi 1944).
QTL analysis
QTL analysis of WBNV resistance traits was conducted using composite interval mapping (CIM) as implemented in QGene version 4.4.0 (Joehanes and Nelson 2008). The threshold values for each trait-marker linkage were determined using permutation tests (1000 permutations at p = 0.05). The default parameters of interval mapping at LOD threshold of 3.0 were used to carry out the QTL analysis.
Results
Phenotyping of populations for WBNV resistance
The infector rows of susceptible checks were planted 10 days in advance of the test entries to build up inoculum and avoid escapes. The check varieties served as a reference to decide the level of disease incidence. The checks used in our experiments viz., Arka Manik and NS-295 recorded a PDI of 95.87% and 98.28% during 2016 (Table 1), 87.04% and 96.67% during 2017 (Table 2) and 90.00% and 96.67% during 2018 (Table 2) respectively by 65 DAS confirming that the escapes were less than 5–10%. Affected plants were severely stunted, had shortened internodes and became very brittle with upright growth of younger branches. Other predominant symptoms were the presence of longitudinal brown necrotic streaks on vines, tendrils and petioles. As the disease progressed, the necrotic streaks on stem and the growing branches started drying from the tip leading to bud necrosis and die-back. All these symptoms observed during the experiment tested to be positive for WBNV infection employing DAC-ELISA and RT-PCR techniques.
Analysis of variance revealed significant differences for all the WBNV resistance related traits (PDI, AUDPC, plant survival and vine length) during 2016 and 2018 except AUDPC in 2018 for Pop I (Table 1). Significant differences were also observed for all the WBNV resistance related traits for Pop II during 2017, where as significant differences were observed only for PDI and vine length in 2018 (Table 2). The resistant parents viz., IIHR-82 for Pop I and BIL-53 for Pop II performed better than their corresponding susceptible parents for all resistance traits during all evaluations.
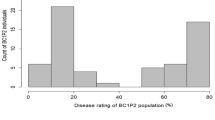
Frequency distribution of AUDPC for both populations are shown in Fig. 2. The AUDPC for Pop I ranged from 350 to 2306 during 2016, while it ranged from 187 to 1729 during 2018. The parents recorded AUDPC values of 1283 for IIHR-82 and 2013 for Arka Manik during 2016, while 742 for IIHR-82 and 1236 for Arka Manik during 2018. The AUDPC for Pop II ranged from 611 to 1792 during 2017 and 367 to 1632 during 2018. The parents BIL-53 and IIHR-140 recorded an AUDPC of 1250 and 1833 respectively during 2017; 1229 and 1354 respectively during 2018.

Frequency distribution of Area under disease progress curve (AUDPC) values for watermelon bud necrosis orthotospovirus infection under natural epiphytotic conditions for a BC1F6 families of the cross IIHR-82 × Arka Manik (Pop I) during the years 2016 and 2018 and b F2:3 families of the cross BIL-53 × IIHR-140 (Pop II) during the years 2017 and 2018
Results of correlation analysis for the Pop I and Pop II during the years 2016, 2017 and 2018 are shown in the Table S3. A significant positive correlation was observed among PDI recorded at different intervals in both populations. Vine length at 45, 55 and 65 days was negatively correlated with PDI at different intervals in both populations during 2016 and 2017. Similarly, plant survival at 45, 55 and 65 days was also negatively correlated with PDI at different intervals in both populations and all years except for Pop I during 2018. Likewise, significant positive correlation was also observed between AUDPC and PDI recorded at different intervals in both populations. Further, vine length and plant survival at 45, 55 and 65 days was negatively correlated with AUDPC at different intervals in both populations during 2016 and 2017. Similarly, a significant correlation was observed for all the four traits between the two years of evaluation in both populations (Tables S4 and S5).
A moderate level of heritability ranging from 15.98% to 35.55% was observed for the end of season PDI across populations (Table S6). Similarly, AUDPC also recorded low to moderate levels (11.04 to 21.40%) of heritability. Vine length (14.91 to 34.76%) and plant survival (11.38 to 29.73%) at the end of season recorded a moderate level of heritability.
Construction of linkage map
A total of 477 PCR based markers including SSR’s, Indel’s and SV’s were screened to assess the parental polymorphism between IIHR-82 and Arka Manik for Pop I. Among them, 238 (49.89%) markers were found to be polymorphic. Of these, a total of 163 markers (109 SSR’s, 49 Indel’s and 5 SV’s) could be mapped based on 141 BC1F5 families of Pop I (Fig. 3). The 163 markers spanned onto 15 linkage groups (LGs) with a total map length of 3310.17 cM and a mean marker interval of 20.31 cM. The marker loci per linkage group varied from 3 (LG 11b) to 23 (LG 10) with a mean of 10 loci per LG. The longest interval in the map is 51.21 c (LG 6) and the shortest interval is 0.83 cM (LG 5) (Table 3).


Linkage map and QTL’s for watermelon bud necrosis orthotospovirus resistance traits identified in the BC1F6 population of the cross IIHR-82 × Arka Manik (Pop I). PDI: Percent Disease Index, VL: Vine length, SUR: Plant Survival, AUDPC: Area Under Disease Progress Curve are indicated in pink, red, brown and green colors respectively. In the QTL naming format q: QTL, 35, 45, 55 and 65: Observations taken at different days after sowing, Iihr-82: Resistant parent in Pop I and 16 and 18: Summer 2016 and 2018
A total of 593 markers were used to detect parental polymorphism between BIL-53 and IIHR-140 for Pop II. Of these, 156 (26.30%) markers were found to be polymorphic and were used to genotype Pop II. Among them, 21 markers were eliminated from the map, because they recorded > 50 cM distance from their nearest markers. Finally, linkage map was constructed with 135 markers (93 SSR’s, 40 Indels and 2 SV’s) based on 112 F2 individuals of Pop II (Fig. 4). The map consisted of 12 linkage groups spanning a total length of 1965.53 cM with an average interval between markers of 14.56 cM.The mapped marker loci per linkage group varied from 4 (LG 6a) to 24 (LG 10) with a mean of 11 loci per LG. The largest interval in the map is 50.76 cM (LG 6b) and the smallest interval is 0.94 c (LG 10) (Table 4).


Linkage map and QTL’s for watermelon bud necrosis orthotospovirus resistance traits identified in the F2:3 population of the cross BIL-53 × IIHR-140. PDI: Percent Disease Index, VL: Vine length, SUR: Plant Survival, AUDPC: Area Under Disease Progress Curve are indicated in pink, red, brown and green colors respectively. In the QTL naming format q: QTL, 35, 45, 55 and 65: Observations taken at different days after sowing, BIL-53: Resistant parent in Pop II and 17 and 18: Summer 2016 and 2018
Among the two populations, the common mapped markers are 61. Out of this the highest number of common markers were noticed on LG10 (13) followed by LG (7). In our study, we have noticed that 77.91% of markers exhibited segregation distortion (SD) in Pop I and 28.24% in Pop II (P ≤ 0.05). Of these SD markers, 90.55% were skewed towards Arka Manik in Pop I where as 56.76% were skewed towards IIHR-140 in Pop II.
QTL analysis
QTL analysis was carried out for four WBNV resistance traits in both populations and loci exhibiting more than 10.0% phenotypic variance explained (PVE) were considered as major QTL’s. A total of 14 major QTL’s were identified for WBNV resistance related traits in Pop I with a LOD score of > 3.0. The PVE for these QTL’s ranged from 10.0% (q_Pdi45_Iihr82_16_3.1, & q _Audpc_Iihr82_18_3.2) to 21.20% (q _Sur45_Iihr82_18_2.1). The 14 QTL’s were distributed on to different linkage groups viz., one QTL each on LG 1b (q_VL45_Iihr82_16_1.1), LG 2b (q _Sur45_Iihr82_18_2.1), LG 5 (q _VL45_Iihr82_18_5.1), LG 6 (q_Pdi55_Iihr82_16_6.3), LG 7 (q_Pdi55_Iihr82_16_7.1), LG 8 (q_Pdi65_Iihr82_18_8.1), LG 10 (q_VL35_Iihr82_18_10.1) and LG 11b (q_Pdi65_Iihr82_18_11.1); six QTL’s on LG 3 (q_Pdi45_Iihr82_16_3.1, q_Pdi45_Iihr82_18_3.1, q_Pdi55_Iihr82_18_3.1, q_Pdi65_Iihr82_18_3.1, q_Audpc_Iihr82_18_3.1 & q_Audpc_Iihr82_18_3.2) (Table 5).
A total of 19 major QTL’s for WBNV resistance related traits were identified in Pop II with PVE ranging from 12.00% (q _VL55_Bil53_18_4.1 & q _VL65_Bil53_18_4.1) to 17.00% (q _Sur65_ Bil53_17_7.1) at LOD > 3.0.The QTL’s were distributed on different linkage groups viz., one QTL on LG 3 (q_Pdi55_Bil53_18_3.1); two each on LG 4 (q_VL55_Bil53_18_4.1, q_VL65_18_4.1), and LG 8 (q_Pdi55_Bil53_18_8.1 and q_Audpc_Bil53_18_8.1); three on LG2 (q _Sur65_Bil53_17_2.1, q _Sur65_Bil53_17_2.2 and q _Sur65_Bil53_17_2.3) and eleven on LG 7 (q _Pdi65_Bil53_17_7.1, q _ Pdi65_Bil53_17_7.2, q _Pdi65_Bil53_17_7.3, q_Sur65_Bil53_17_7.1, q_Sur65_Bil53_17_7.2, q_Sur65_Bil53_17_7.3, q_Sur65_Bil53_17_7.4, q_Sur65_Bil53_17_7.5, q _Sur65_Bil53_17_7.6, q_Sur65_Bil53_17_7.7 and q _ Sur65_Bil53_17_7.8) (Table 6).
Among the 14 major QTL’s identified in Pop I, IIHR-82 contributed favourable alleles for five QTL’s while Arka Manik contributed for nine QTL’s. Similarly, BIL-53 contributed favourable alleles for 8 QTL’s and IIHR-140 for 11 QTL’s in Pop II.
Discussion
Narrow genetic base of cultivated watermelon makes it vulnerable to several biotic stresses. Wild species of Citrullus have the potential to provide useful diversity and broaden the genetic base. Citrullus lanatus var. citroides is considered a reservoir of elite genes for several agronomic traits (Hawkins et al. 2001; Levi et al. 2001a, b; Hashizume et al. 2003; Zhang et al. 2004; Guo et al. 2010; Levi et al. 2011; Ren et al. 2012, 2014; Nimmakayala et al. 2014). In the current experiment, an attempt was made to map resistance loci for WBNV from this species.
The frequency distribution graphs of AUDPC (Fig. 2) in both Pop I and Pop II showed that there was a continuous distribution, indicating that this trait is quantitative in nature. Our recent study also reported that resistance to WBNV is inherited dominantly with involvement of major gene along with several background genes with minor effect in BIL-53 (Nagesh et al. 2018). The segregation pattern of WBNV resistance in the current study also suggests dominant inheritance. Similarly, dominant genes mapped for other orthotospovirus resistance include Sw-5 in tomato (Stevens et al. 1995), Tsw in chilli (Jahn et al. 2000) for TSWV and Sws for MYSV in cucumber (Sugiyama et al. 2015). Most of these dominant virus resistant genes encode for proteins with nucleotide binding site leucine rich repeat (NBS-LRR) domains (Maule et al. 2007).
The highly significant correlation observed among the four traits (PDI, AUDPC, vine length and plant survival) across populations and years suggests that these are related component traits contributing towards resistance. In general, the PDI and AUDPC were found to be negatively correlated with vine length and plant survival of populations under WBNV infection. However, vine length and survival did not correlate negatively with PDI and AUDPC during 2018 in both populations (Table S3). This might be attributed to the slow rate of disease progress observed due to intermittent rain during the screening season.
There is no artificial screening protocol with high through put and efficiency currently available for this disease. Therefore, we have adopted a natural epiphytotic screening protocol in our experiments, which we assume resulted in higher GxE interaction leading to underestimation of heritability of the resistance traits. Further, moderate levels of heritability for these four disease resistance traits observed in the current experiment is of typical polygenic nature of resistance and indicates that a combined selection for all these traits seems to be more effective (Table S6). This further justifies the development and use of markers for transferring resistance into a desirable background.
Linkage map
Construction of useful genetic linkage maps and identifying QTL’s associated with disease or pest resistance has been challenging in watermelon due to low polymorphism (Levi et al. 2011). The majority of watermelon genetic maps have been from inter-varietal populations (C. lanatus var. lanatus × C. lanatus var. citroides) because citroides is a valuable source of disease resistance alleles (Levi et al. 2013), while lanatus harbors the ideal horticultural traits such as high sugar and lycopene content (Ren et al. 2014). In the current study, we developed linkage maps from two populations derived from this species of watermelon (Citrullus lanatus var. citroides) using PCR based markers (SSR’s, Indels and SV’s) available in the public domain. Although the current maps are not highly saturated, with mean marker interval of ~ 15-20 cM, they can be definitely useful for broadly locating the causal loci.
Reddy et al. (2013) reported the presence of wide chromosomal structural differences between lanatus and citroides, such groups resulting in distorted segregation in the mapping populations derived from them. Several researchers also opined that populations may exhibit quasi-linkage of molecular markers (Levi et al. 2001a, b, 2002, 2006, 2011). In the current study also, we have noticed greater SD in Pop I (77.91%) than Pop II (28.24%). This might be due to the reason that Pop I is derived directly from wild species where as Pop II is obtained from one of the advanced line of Pop I which might have increased chromosomal synteny reducing the SD in Pop II. Hence, using advanced breeding lines derived from the wild species as one of the parent in mapping studies instead of directly using the wild species seems appropriate where SD is severe. Highly skewed markers result in the over estimation of recombination frequency leading to larger map length as observed in Pop I (3310.17 cM) compared to that of Pop II (1965.53 cM) which was well within the expected range. Earlier reports of inter-varietal genetic maps range from 800 to 2162 cM (Levi et al. 2001a, 2002, 2006, 2011; Zhang et al. 2004; Ren et al. 2012, 2015a, b; Sandlin et al. 2012).
QTL analysis
The additive effect observed for a QTL is an estimate of the change in the average phenotype that would be produced by substituting a single allele of one type with that of another type (e.g., a replaced by A) in a population and is calculated as half of the difference between the mean of all cases that are homozygous for one parental allele (aa) compared to the mean of all cases that are homozygous for the other parental allele (AA). Therefore, the range of absolute values for a trait would determine the magnitude of additive effect, which would explain the reason for observing higher values for QTLs related to AUDPC.
The ± sign of the additive effect would determine the parent contributing the favourable allele (Sun et al. 2016). The negative additive effects for various QTLs identified indicate that the male parent contributed alleles for higher phenotypic value, while positive additive effects indicate alleles for higher trait value from the female parent (Tables 5, 6). Presence of both positive and negative additive effects for various resistance traits reveals that both parents are contributing favorable alleles for resistance justifying the possibility of transgressive segregation. We observed several families performing better than the resistant parent which might be attributed to transgressive segregation. Such transgressive segregants were also observed in several other studies including Ren et al. (2015a, b) in watermelon.
Out of several QTL’s detected in Pop I, LG 3b recorded the highest number of QTL’s contributing for PDI and AUDPC with common QTL region (6 cM & 66–68 cM) and the favorable alleles for majority of these QTL’s were contributed by Arka Manik. We also identified QTLs for PDI on LG 6 (q _Pdi55_Iihr82_16_6.3) and LG 7 (q _ Pdi55_Iihr82_16_7.1). Multi trait QTL regions with more than one QTL in a region were observed on LG 3b (51.28–68.49 cM). The LG 3b recorded common QTLs for PDI/AUDPC during both the years of evaluation in this population (Table 5).
Among the various QTL’s detected in Pop II, LG 7 recorded the highest number of QTL’s contributing for plant survival and PDI. Among them, PDI and plant survival recorded three common QTL regions (90 cM, 158–160 cM & 166–174 c) and the favourable allele for all these QTL regions were contributed by the susceptible parent, IIHR-140.The sign of additive effects for these QTL’s revealed that resistance allele was contributed by BIL-53 in Pop II, while BIL-53 itself received this allele from Arka Manik in Pop I. This proves that seemingly susceptible varieties may also carry resistance alleles. Multi trait QTL regions with more than one QTL in a region were observed in Pop II on LG 7 (72-180 c), LG 2 (26–44 cM), LG 8 (8 cM) and LG 4 (52 cM). Importantly, we observed common QTL regions for plant survival on LG 2 and PDI on LG 3 and LG 8 for both the populations.
Finding the right combination of highly effective alleles at flanking loci of the QTL region that forms a tolerant haplotype would be more relevant than the individual allele itself at a single marker locus and is expected to be more effective in selection/ introgression (Collard and Mackill 2008). Therefore, haplotypes for alleles of flanking markers of the multi-trait QTLs observed in the current study were manually derived and were studied for their influence on WBNV resistance phenotype (Table 7). A clear gradation in phenotypes was observed between the resistant and susceptible alleles for all the loci studied.
In our analysis, the highest effect of selection for favourable alleles at flanking markers of QTL’s was observed for q _ Pdi45_Iihr82_16_3.1 (reduction in PDI by 21.88%) followed by q_Audpc_Iihr82_18_3.2 (reduction in AUDPC by 15.93%) in Pop I and q_Pdi55_Bil53_18_8.1 (reduction in PDI by 22.64%), followed by q_Audpc_Bil53_18_8.1 (reduction in AUDPC by 18.71%), q_Pdi65_Bil53_17_7.3 (reduction in PDI by 13.45%), q_Sur65_Bil53_17_7.1 (increase in plant survival by 29.85%) and q_Sur65_Bil53_17_7.5 (increase in plant survival by 25.78%) in Pop II (Table 7). This analysis is only indicative of the improvement that can be achieved in terms of the trait value because of a particular QTL region. However, fine mapping shall be a prerequisite for conduct of selection using flanking markers.
To conclude, these multi-trait QTL regions observed on LG 3b in Pop I and LG 2, LG 4, LG 7 and LG 8 in Pop II are potential targets for further analysis. As this is the first attempt to map QTL’s for WBNV resistance, the results obtained in the present study may provide a guide for further validation and fine mapping multi-trait QTL regions.
References
Adams MJ, Lefkowitz EJ, King AM, Harrach B, Harrison RL, Knowles NJ, Kropinski AM, Krupovic M, Kuhn JH, Mushegian AR, Nibert M (2017) Changes to taxonomy and the international code of virus classification and nomenclature ratified by the international committee on taxonomy of viruses. Arch Virol 162:2505–2538
Anonymous (2012) Cucurbit Genomics Database. Available at https://www.icugi. org
Branham SE, Wechter WP, Ling KS, Chanda B, Massey L, Zhao G, Guner N, Bello M, Kabelka E, Fei Z, Levi A (2019) QTL mapping of resistance to Fusarium oxysporum f. sp. niveum race 2 and Papaya ringspot virus in Citrullus amarus.Theor Appl Genet. https://doi.org/10.1007/s00122-019-03500-3
Campbell CL, Madden LV (1990) Introduction to plant disease epidemiology. Wiley, New York
Cheng Y, Luan F, Wang X, Gao P, Zhu Z, Liu S, Baloch AW, Zhang Y (2016) Construction of a genetic linkage map of watermelon (Citrullus lanatus) using CAPS and SSR markers and QTL analysis for fruit quality traits. Sci Hort 202:25–31. https://doi.org/10.1016/j.scienta.2016.01.004
Collard BCY, Mackill DJ (2008) Marker-assisted selection: an approach for precision plant breeding in the twenty-first century. Phil Trans R Soc B 363:557–572. https://doi.org/10.1098/rstb.2007.2170
Dong W, Wu D, Li G, Wu D, Wang Z (2018) Next-generation sequencing from bulked segregant analysis identifies a dwarfism gene in watermelon. Sci Rep 8:2908. https://doi.org/10.1038/s41598-018-21293-1
Dou J, Zhao S, Lu X, He N, Zhang L, Ali A, Kuang H, Liu W (2018) Genetic mapping reveals a candidate gene (ClFS1) for fruit shape in watermelon (Citrullus lanatus L.). Theor Appl Genet. https://doi.org/10.1007/s00122-018-3050-5
FAO (2016) Food and Agricultural organization (https://www.faostat.fao.org), Rome, Italy. Retrived 10 Febraury 2018
Gimode W, Clevenger J, McGregor C (2020) Fine-mapping of a major quantitative trait locus Qdff3-1 controlling flowering time in watermelon. Mol Breed 40:3
Guo S, Xu Y, Zhang H, Gong G, Huang S, Yi H, Wu M, Zheng Y, Fei Z (2010) Latest advances in watermelon genomics (In Proceedings of the 4th IS on Cucurbits Ed.: Xiaowu Sun). Acta Hort 871:599–606
Guo S, Zhang J, Sun H, Salse J, Lucas WJ, Zhang H, Zheng Y, Mao L, Ren Y, Wang Z, Min J, Guo X, Murat F, Ham B, Zhang Z, Gao S, Huang M, Xu Y, Zhong S, Bombarely A, Mueller LA, Zhao H, He H, Zhang Y, Zhang Z, Huang S, Tan T, Pang E, Lin K, Hu Q, Kuang H, Ni P, Wang B, Liu J, Kou Q, Hou W, Zou X, Jiang J, Gong G, Klee K, Schoof H, Huang Y, Hu X, Dong S, Liang D, Wang J, Wu K, Xia Y, Zhao X, Zheng Z, Xing M, Liang X, Huang B, Lv T, Wang J, Yin Y, Yi H, Li R, Wu M, Levi A, Zhang X, Giovannoni JJ, Wang J, Li Y, Fei Z, Xu Y (2013) The draft genome of watermelon (Citrullus lanatus) and resequencing of 20 diverse accessions. Nat Genet 45(1):51–58
Guo S, Zhao S, Sun H, Wang X, Wu S, Lin T, Ren Y, Gao L, Deng Y, Zhang J, Lu X, Zhang H, Shang J, Gong G, Wen C, He N, Tian S, Li M, Liu J, Wang Y, Zhu Y, Jarret R, Levi A, Zhang X, Huang S, Fei Z, Liu W, Xu Y (2019) Resequencing of 414 cultivated and wild watermelon accessions identifies selection for fruit quality traits. Nat Genet 51:1616-1623
Hashizume T, Shimamoto I, Hirai M (2003) Construction of a linkage map and QTL analysis of horticultural traits for watermelon [Citrullus lanatus (Thunb.) Matsum & Nakai] using RAPD, RFLP and ISSR markers. Theor Appl Genet 106:779–785
Hawkins LK, Hawkins TL, Rhodes BB, Jarret RL (2001) Linkage Mapping in a Watermelon Population Segregating for Fusarium Wilt Resistance. J Amer Soc Hort Sci 126(3):344–350
Hobbs HA, Reddy DVR, Rajeshwari R, Reddy AS (1987) Use of direct antigen coating and protein A coating ELISA procedures for detection of three peanut viruses. Plant Dis 71:747–749
Holkar SK, Kumar R, Yogita M, Katiyar A, Jain RK, Mandal B (2017) Diagnostic assays for two closely related tospovirus species, watermelon bud necrosis virus and groundnut bud necrosis virus and identification of new natural hosts. J Plant Biochem Biotechnol 26:43–51
Jahn M, Paran I, Hoffmann K, Radwanski ER, Livingstone KD, Grube RC, Aftergoot E, Lapidot M, Moyer J (2000) Genetic mapping of the Tsw locus for resistance to the Tospovirus tomato spotted wilt virus in Capsicum spp. and Its relationship to the Sw-5 gene for resistance to the same pathogen in tomato. Mol Plant-Microbe Interact 13(6):673–682
Jain RK, Bag S, Umamaheswaran K, Mandal B (2007) Natural infection by tospoviruses of cucurbitaceous and fabaceous vegetable crops in India. J Phytopathol 155:22–25
Jain RK, Mandal B, Pappu HR, Holkar, SK (2015) ICTV Taxonomic Proposal 2014. 005aV. A. v2. Tospovirus_sp. Create 1 New Species in the Genus Tospovirus, Family Bunyaviridae. https://www.ictvonline.org/proposals-14/2014
Jain RK, Pappu HR, Pappu SS, Krishnareddy M, Vani A (1998) Watermelon bud necrosis tospovirusis a distinct virus species belonging to serogroup IV. Arch Virol 143:1637–1644
Joehanes R, Nelson JC (2008) QGene 4.0 An extensible Java QTL-analysis platform. Bioinformatics 24:2788–2789
Keb-Llanes M, Gonzalez G, Chi B, Infante D (2002) A rapid and simple method for small-scale DNA extraction in Agavaceae and other tropical plants. Plant Mol Biol Rep 20:299–303
Khera P, Pandey MK, Wang H, Feng S, Qiao L, Culbreath AK, Kale S, Wang J, Holbrook CC, Zhuang W, Varshney RK, Guo B (2016) Mapping quantitative trait loci of resistance to tomato spotted wilt virus and leaf spots in a recombinant inbred line population of peanut (Arachis hypogaea L.) from SunOleic 97R and NC94022. PLoS ONE 11(7):e0158452. https://doi.org/10.1371/journal.pone.0158452
Kim KH, Hwang JH, Han DY, Park M, Kim S, Choi D, Kim Y, Lee GP, Kim ST, Park YH (2015) Major quantitative trait loci and putative candidate genes for powdery mildew resistance and fruit-related traits revealed by an intraspecific genetic map for watermelon (Citrullus lanatus var. lanatus). PLoS ONE 10(12):e0145665
Kosambi DD (1944) The estimation of map distances from recombination values. Ann Eugenics 12:172–175
Kumar NKK, Venkatesh N, Kalleshwaraswamy CM, Ranganath HR (2006) Seasonal incidence of thrips and bud necrosis virus on watermelon. Pest Man Hort Ecosyst 12(2):85–92
Kumar R, Mandal B, Geetanjali AS, Jain RK, Jaiwal PK (2010) Genome organisation and sequence comparison suggest intraspecies incongruence in M RNA of Watermelon bud necrosis virus. Arch Virol 155:1361–1365
Kunkalikar SR, Sudarsana P, Arun BM, Rajagopalan P, Chen TC, Yeh SD, Naidu RA, Zehr UB, Ravi KS (2011) Importance and genetic diversity of vegetable-infecting tospoviruses in India. Phytopathology 101:367–376
Lambel S, Lanini B, Vivoda E, Fauve J, Wechter WP, Harris-Shultz KR, Massey L, Levi A (2014) A major QTL associated with Fusarium oxysporum race1 resistance identified in genetic populations derived from closely related watermelon lines using selective genotyping and genotyping-by-sequencing for SNP discovery. Theor Appl Genet 127:2105–2115
Lee HJ, Cho HJ, Lee KA, Lee MS, Shin YS, Harn CH, Yang SG, Nahm SH (2007) Development of sequence-based dna markers for evaluation of phylogenetic relationships in Korean watermelon varieties. J Crop Sci Biotech 10(2):98–105
Levi A, Thies JA, Wechter WP, Harrison HW, Simmons AM, Reddy UK, Nimmakayala P, Fei Z (2013) High frequency oligonucleotides:targeting active gene (HFO-TAG) markers revealed wide genetic diversity among Citrullus spp. accessions useful for enhancing disease or pest resistance in watermelon cultivars. Genet Resour Crop Evol 60:427–440
Levi A, Thomas CE, Keinath AP, Wehner TC (2001a) Genetic diversity among watermelon (Citrullus lanatus and Citrullus colocynthis) accessions. Genet Res Crop Evol 48:559–566
Levi A, Thomas CE, Trebitsh T, Salman A, King J, Karaliaus J, Newman M, Reddy OUK, Xu Y, Zhang X (2006) An extended linkage map for watermelon based on SRAP, AFLP, SSR, ISSR and RAPD markers. J American Soc Hort Sci 131(3):393–402
Levi A, Thomas CE, Zhang X, Joobeur T, Davis A (2002) A genetic linkage map for watermelon derived from a testcross population: (Citrullus lanatus var. citroides × C. lanatus var. lanatus) × Citrullus colocynthis. Theor Appl Genet 105(4):555–563
Levi A, Thomas CE, Zhang X, Joobeur T, Dean RA, Wehner TC, Carle BR (2001b) A genetic linkage map for watermelon based on Randomly Amplified polymorphic DNA (RAPD) markers. J Amer Soc Hort Sci 126:730–737
Levi A, Wechter P, Massey L, Carter L, Hopkins D (2011) An extended genetic linkage map for watermelon based on a testcross and a BC2F2 population. American J Plant Sci 2:93–110
Liu S, Gao P, Wang X, Davis AR, Baloch AM, Luan F (2015) Mapping of quantitative trait loci for lycopene content and fruit traits in Citrullus lanatus. Euphytica 202(3):411–426
MAFW (2017) Horticultural Statistics at a Glance 2017 (https://agricoop.nic.in) Horticulture Statistics Division, Department of Agriculture, Cooperation and Farmers Welfare, Ministry of Agriculture and Farmers Welfare, Government of India
Mandal B, Jain RK, Chaudhary V, Varma A (2003) First report of natural infection of Luffa acutangula by Watermelon bud necrosis virus in India. Plant Dis 87:598
Mandal B, Jain RK, Krishnareddy M, Krishna Kumar NK, Ravi KS, Pappu HR (2012) Emerging problems of Tospoviruses (Bunyaviridae) and their management in the Indian subcontinent. Plant Dis 96(4):468–479
Maule AJ, Caranta C, Boulton MI (2007) Sources of natural resistance to plant viruses: status and prospects. Mol Plant Pathol 8(2):223–231
Nagesh GC, Rao ES, Pitchaimuthu M, Varalakshmi B, Lakshmana Reddy DC, Samuel DK, Rekha A, Krishna Reddy M (2018) Genetic analysis of resistance to watermelon bud necrosis orthotospovirus in watermelon [Citrullus lanatus (Thunb.) Matsum & Nakai]. Plant Breed 137:814–822. https://doi.org/10.1111/pbr.12639
Nimmakayala P, Abburi VL, Bhandary A, Abburi L, Vajja VG, Reddy R, Malkaram S, Venkatramana P, Wijeratne A, Tomason YR, Levi A, Wehner TC, Reddy UK (2014) Use of Vera Code 384-plex assays for watermelon diversity analysis and integrated genetic map of watermelon with single nucleotide polymorphisms and simple sequence repeats. Mol Breed 34:537–548
Park SW, Kim KT, Kang SC, Yang HB (2016) Rapid and practical molecular marker development for rind traits in watermelon. Hort Env Biotech 57:385–391
Prothro J, Sandlin K, Gill R, Bachlava E, White V, Knapp SP, McGregor C (2012) Mapping of the egusi seed trait locus (eg) and quantitative trait loci associated with seed oil percentage in watermelon. J American Soc Hort Sci 137(5):311–315
Rajasekharam T (2010) Biological and molecular characterization and management of Watermelon bud necrosis virus. Ph. D Thesis, UAS, Dharawad
Rebijith KB, Asokan R, Hande HR, Kumar NK (2016) The first report of miRNAs from a thysanopteran insect, Thrips palmi Karny using high-throughput sequencing. PLoS ONE 11:e0163635
Rebijith KB, Asokan R, Krishna Kumar NK, Krishna V, Ramamurthy VV (2012) Development of species-specific markers and molecular differences in mtDNA of Thrips palmi Karny and Scirtothrips dorsalis Hood (Thripidae: Thysanoptera), vectors of tospoviruses Bunyaviridae) in India. Entomol News 122:201–213
Reddy U, Aryal N, Islam-Faridi N, Tomason Y, Levi A, Nimmakayala P (2013) Cytomolecular characterization of rDNA distribution in various Citrullus species using fluorescent in situ hybridization. Genet Resour Crop Evol 60(7):2091–2100. https://doi.org/10.1007/s10722-013-9976-1
Reddy UK, Nimmakayala P, Levi A, Abburi VL, Saminathan T, Tomason YR, Vajja G, Reddy R, Abburi L, Wehner TC, Ronin Y, Karol A (2014) High-resolution genetic map for understanding the effect of genome-wide recombination rate on nucleotide diversity in watermelon. G3 4(11):2219–2230
Ren R, Ray R, Li P, Xu J, Zhang M, Liu G, Yao X, Kilian A, Yang X (2015a) Construction of a high-density DArTseq SNP-based genetic map and identification of genomic regions with segregation distortion in a genetic population derived from a cross between feral and cultivated-type watermelon. Mol Genet Genom 290:1457–1470
Ren Y, Jiao D, Gong G, Zhang H, Guo S, Zhang J, Xu Y (2015b) Genetic analysis and chromosome mapping of resistance to Fusarium oxysporum f. sp. niveum (FON) race 1 and race 2 in watermelon (Citrullus lanatus L.). Mol Breeding 35:183–191
Ren Y, McGregor C, Zhang Y, Gong G, Zhang H, Guo S, Sun H, Cai W, Zhang J, Xu Y (2014) An integrated genetic map based on four mapping populations and quantitative trait loci associated with economically important traits in watermelon (Citrullus lanatus). BMC Plant Biol 14:1471–2229
Ren Y, Zhao H, Kou Q, Jiang J, Guo S, Zhang H, Hou W, Zou X, Sun H, Gong G, Levi A, Xu Y (2012) A High Resolution Genetic Map Anchoring Scaffolds of the Sequenced Watermelon Genome. PLoS ONE 7(1):1–10
Riley DG, Pappu HR (2000) Evaluation of tactics for management of thrips-vectored Tomato spotted wilt virus in tomato. Plant Dis 84(8):847–852
Sandlin K, Prothro J, Heesacker A, Khalilian N, Okashah R, Xiang W, Bachlava E, Caldwell DG, Taylor CA, Seymour DK, White V, Chan E, Tolla G, White C, Safran D, Graham E, Knapp S, McGregor C (2012) Comparative mapping in watermelon [Citrullus lanatus (Thunb.) Matsum. & Nakai]. Theor Appl Genet 125:1603–1618
Shang J, Li N, Li N, Xu Y, Ma S, Wang J (2016) Construction of a high-density genetic map for watermelon (Citrullus lanatus L.) based on large-scale SNP discovery by specific length amplified fragment sequencing (SLAF-seq). Sci Hort 203:38–46
Singh SJ, Krishnareddy M (1996) Watermelon bud necrosis: new Tospovirus disease. Acta Hort 431:68–77
Stevens MR, Lamb EM, Rhoads DD (1995) Mapping the Sw-5 locus for tomato spotted wilt virus resistance in tomatoes using RAPD and RFLP analyses. Theor Appl Genet 90:451–456
Sugiyama M, Kawazu Y, Fukino N, Yoshioka Y, Shimomura K, Sakata Y, Okuda M (2015) Mapping of quantitative trait loci for Melon yellow spot virus resistance in cucumber (Cucumis sativus L.). Euphytica 205:615–625
Sugiyama M, Okuda M, Sakata Y (2009) Evaluation of resistance to melon yellow spot virus in a cucumber germplasm collection. Plant Breeding 128:696–700
Sun X, Mumm RH (2016) Method to represent the distribution of QTL additive and dominance effects associated with quantitative traits in computer simulation. BMC Bioinform 17:73. https://doi.org/10.1186/s12859-016-0906-z
Wang C, Qiao A, Fang X, Sun L, Peng P, Davis AR, Liu S, Luan F (2019) Fine mapping of lycopene content and flesh color related gene and development of molecular marker-assisted selection for flesh color in watermelon (Citrullus lanatus). Front Plant Sci 10:1240. https://doi.org/10.3389/fpls.2019.01240
Wang J, Li H, Zhang L, Meng L (2016) Users manual of QTL IciMapping. The Quantitative Genetics Group, Institute of Crop Science, Chinese Academy of Agricultural Sciences (CAAS), Beijing 100081, China, and Genetic Resources Program, International Maize and Wheat Improvement Center (CIMMYT), Apdo. Postal 6-641, 06600 Mexico, D.F., Mexico, p 274
Wu S, Wang X, Reddy U, Sun H, Bao K, Gao L, Mao L, Patel T, Ortiz C, Abburi V, Nimmakayala P, Branham S, Wechter P, Massey L, Ling KS, Kousik C, Hammar S, Tadmor Y, Portnoy V, Fei Z (2019) Genome of ‘Charleston Gray’, the principal American watermelon cultivar, and genetic characterization of 1365 accessions in the U. S. National Plant Germplasm System watermelon collection. Plant Biotech J 17(12):2246–2258
Zhang H, Wang H, Guo S, Ren Y, Gong G, Weng Y, Xu Y (2012) Identification and validation of a core set of microsatellite markers for genetic diversity analysis in watermelon (Citrullus lanatus Thunb. Matsum Nakai). Euphytica 186(2):329–342
Zhang R, Xu Y, Yi K, Zhang H, Liu L, Gong G, Levi A (2004) A genetic linkage map for watermelon derived from recombinant inbred lines. J American Soc Hort Sci 129:237–324
Zhang Z, Zhang Y, Sun L, Qiu G, Sun Y, Zhu Z, Luan F, Wang X (2018) Construction of a genetic map for Citrullus lanatus based on CAPS markers and mapping of three qualitative traits. Sci Hort 233:532–538
Acknowledgements
The authors would like to acknowledge Ministry of Social Justice and Empowerment, Government of India, for providing the Ph.D fellowship for first author and Department of Biotechnology, Government of India, for funding the program.
Author information
Authors and Affiliations
Contributions
Conceived and designed the experiments: ESR; NGC, TRN, SKM and MR performed the experiments with assistance from MKR for phenotyping; LRDC for genotyping; AR for viral diagnostic assays; ESR and NGC wrote the paper. All the authors have discussed the results and commented on the manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Nagesh, G.C., Thontadarya, R.N., Swamy, K.M. et al. Mapping quantitative trait loci for resistance to watermelon bud necrosis orthotospovirus in watermelon [Citrullus lanatus (Thunb.) Matsum & Nakai]. Euphytica 216, 104 (2020). https://doi.org/10.1007/s10681-020-02632-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s10681-020-02632-8