
Abstract
SUMOylation is an important protein post-translational modification (PTM) process, in which the small ubiquitin-like modifier (SUMO) protein covalently binds to the target protein and regulates stability, subcellular localization, and protein–protein interaction of the target protein. Protein SUMOylation exerts crucial regulatory function in the liver, and its abnormalities are associated with various liver-related disease processes. This review focuses on the biological functions of protein SUMOylation in liver-related diseases in recent years, summarizes the molecular mechanisms of SUMOylation in the replication of hepatitis viruses and the occurrence of hepatocellular carcinoma, and discusses the significance of SUMOylation in liver-related disorders, which is essential for understanding liver biological processes and formulating therapeutic strategies.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Small ubiquitin-like modifiers (SUMO) are conserved members of the ubiquitin-related protein family. Known SUMO proteins include SUMO1, SUMO2, SUMO3, SUMO4, and SUMO5. SUMOylation is the attachment of SUMO proteins to substrates, which involves several key enzymes. The SUMO E1 activating enzyme is the initiating enzyme for SUMOylation, which activates SUMO proteins and generates a thioester. The SUMO E2 conjugating enzyme is an intermediary enzyme that transfers the activated SUMO protein from E1 to substrates. The SUMO E3 ligases are crucial enzymes in the SUMOylation process. E3 ligases determine and assist in the covalent attachment of SUMOs to substrates. Each E3 ligase typically interacts with specific target proteins, providing selectivity in the modification process. The SUMO-specific proteases (SENPs) are enzymes responsible for deSUMOylation, which removes SUMOs from target proteins. They restore the target proteins to an un-modified state, maintaining the dynamic nature of SUMOylation. Six family members have been characterized, including SENP1-3 and SENP5-7. In general, SUMOylation is a dynamic and reversible process that involves activation of SUMO precursor proteins, covalent attachment to target proteins, and subsequent deSUMOylation.
Protein SUMOylation occurs mainly in the nucleus and is involved in regulating nucleoplasmic shuttling, subcellular localization, RNA/protein interactions, and cell cycle progression of target proteins. Under physiological conditions, cell cycle-related proteins and RNA-binding proteins are dynamically regulated by SUMOylation to maintain the balance of physiological functions. However, alterations in the expression activity of SUMOylation-related enzymes and abnormal levels of target protein SUMOylation can cause disorders and contribute to the development of disease.
The liver, as a vital metabolic organ in the body, is involved in multiple physiological processes such as lipid metabolism, glucose metabolism, and amino acid metabolism. Recently, increasing evidence suggests that SUMOylation is participating in multiple biological procedures, such as hepatic cell proliferation, apoptosis, and stress response. However, abnormal SUMOylation is strongly related to the progression of liver-related diseases. For instance, SUMOylation is closely associated with tumor suppressor genes, transcription factor activity, and protein stability in hepatocellular carcinoma (HCC). Research on hepatitis viruses has also revealed the role of SUMOylation in regulating viral replication and host immune responses. Additionally, metabolic diseases are also correlated with SUMOylation, which regulates the expression of lipid metabolism-related genes.
To conclude, an in-depth study of the biological functions and molecular mechanisms of protein SUMOylation may provide new insights and targets for the treatment and prevention of liver diseases. This article provides a comprehensive overview of the latest research on the biological functions of protein SUMOylation. Based on our current understanding, we discuss and prospect the clinical application potential of target SUMOylation for HCC therapy.
The expression levels of SUMOylation-related enzymes in liver diseases
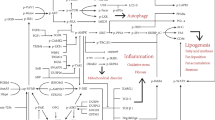
Many studies suggested that an imbalance between SUMOylation and deSUMOylation leads to various pathological changes in liver disease. Table 1 summarizes the profile of SUMO pathway components in human liver diseases. The expression of SUMO E1 activating enzymes (SAE1 and SAE2) [1, 2], SUMO E2 ligase (Ubc9) [3,4,5], or SUMO E3 ligase (e.g., Cbx4) [6] appears to be enhanced in a variety of liver diseases. Studies have shown enhanced expression of some SENPs in HCC [7,8,9,10,11,12]. Notably, the levels of SUMOylation and deSUMOylation enzymes are elevated in some liver diseases, which may indicate a need for accelerated SUMO cycling (Fig. 1). Thus, it is suggested that changes in the expression levels of SUMOylation-related enzymes can predict the occurrence of liver disease.

Relationship of SUMOylation-related enzymes with liver diseases. SUMOylation promotes the replication of hepatitis virus. The SUMOylated HBV core is essential for establishing viral persistence. SUMOylated S-HDAg is closely related to HDV replication. SUMOylated NS5A positively affects HCV replication. Abnormal SUMO pathway promotes the development of fatty liver. SUMOylation of Prox1 is a nutritionally sensitive determinant of hepatic fasting metabolism. SUMOylated RIPK1 regulates the ubiquitination and activation of RIPK1, leading to the progression of MASH. SUMOylation promotes the development and progression of HCC. SUMOylation of Mettl3 regulates HCC invasion, metastasis, and progression. SUMOylation of PKM2 promotes HCC progression by activating EMT and STAT3 signaling pathways. SUMOylation of HSP27 promotes HCC cell proliferation and invasion
The link between SUMOylation and liver disease
The liver is the largest digestive gland and the central station for the metabolism of substances and energy in the body. It is estimated that the liver is the largest detoxification organ, performing more than 500 chemical reactions. Depending on the pathogenesis, liver diseases can be categorized into viral and non-viral liver diseases. Protein SUMOylation promotes HCC and hepatitis virus replication; involves in alcoholic liver disease (ALD), metabolic dysfunction-associated steatotic liver disease (MASLD), and liver fibrosis; and exerts an influential regulatory role in the pathogenic mechanisms of biliary cirrhosis and alcoholic liver disease.
SUMOylation holds a pivotal role in non-viral liver diseases
With the successive introduction of hepatitis B immunization and hepatitis C curative drugs in China, significant achievements have been made in the prevention and control of hepatitis B and C. The overall incidence rates of these diseases have shown a downward trend. However, non-viral liver diseases such as fatty liver and drug-induced liver damage are showing a more noticeable high-incidence trend, which requires attention and concern. Non-viral liver diseases primarily include fatty liver, ALD, MASLD, drug-induced liver diseases, autoimmune liver diseases, and other liver disorders.
SUMOyaltion in ALD
ALD is one of the common liver diseases in our country and is caused by prolonged and heavy drinking. Alcohol causes ALD through a variety of pathways. Cytochrome P450 2E1 (CYP2E1) is the major enzyme involved in alcohol metabolism and plays an important role in ALD. It was found that ethanol-induced SUMOylation of CYP2E1 at the K410 site contributes to CYP2E1 stabilization and promotes its activity, accelerating the process of ALD [3]. Methionine adenosyltransferase alpha1(MATα1) plays a crucial role in maintaining the mitochondrial function of hepatocytes by negatively regulating CYP2E1 expression in ALD. However, MATα1 is highly SUMOylated by SUMO2 in ALD, and SUMOylation of MATα1 promotes its degradation, leading to mitochondrial dysfunction [13]. Preventing MATα1 SUMOylation may represent a potential treatment strategy for ALD.
SUMOyaltion in MASLD
MASLD, which was previously named nonalcoholic fatty liver disease (NAFLD), is becoming the most common cause of liver diseases. MASLD has a complex disease profile that can be categorized into stages of simple fatty liver, metabolic-associated steatohepatitis (MASH), and cirrhosis. Hepatic SENP2 governs metabolic homeostasis by regulating liver-adipose tissue crosstalk, linking the SUMO pathway to metabolic regulation. Mechanistically, SENP2 was dramatically increased in fatty liver. SENP2 was conjugated to PPARα and deSUMOylated it, thereby promoting ubiquitination and degradation of PPARα, which in turn inhibited FGF21 expression and fatty acid oxidation [10]. Excess nutrient accumulation impedes normal liver function and is associated with MASLD in obesity. SUMOylation of Prox1 was a nutritionally sensitive detector of hepatic fasting metabolism. Prox1 was modified by SUMO2 at lysine 556 in free-feeding mice and mouse livers, and this modification was abolished during fasting [14]. This suggests that SUMOylation of Prox1 modulates different gene subgroups involved in the hepatic cholesterol detoxification system in response to fasting. Sterol regulatory element-binding protein-1c (SREBP-1c) is a crucial transcription factor for cholesterol and lipid homeostasis. The SUMO ligase PIASy promoted SUMOylation of SREBP1c at Lys98, thereby inhibiting the hepatic lipogenic program in response to fasting-induced signals. This led to a disruption of the balance between lipogenesis and lipolysis, resulting in lipid accumulation and triggering the development of MASLD [15]. Moreover, SUMOylation of hepatic E4 promoter-binding protein 4 (E4BP4) is essential for the downregulation of Fsp27 and lipid droplets by cAMP signaling in hepatocytes. However, HFD feeding induces deSUMOylation of E4BP4, which promotes lipid droplet formation and liver steatosis in mice [16].
MASH is the progressive form of MASLD. RIPK1 regulates the process of MASH, whereas SENP1 acts as an endogenous inhibitor of RIPK1 to suppress its expression. Mechanistically, SENP1 interacts with RIPK1 and deSUMOylates RIPK1 in the TNF-R1 signaling complex (TNF-RSC). Loss of SENP1 leads to elevated levels of RIPK1 SUMOylation, which re-orchestrates TNF-RSC and regulates the ubiquitination patterns and activity of RIPK1 [7].
SUMOylation mediates hepatitis virus replication
Viral liver disease is an infectious disease caused by a variety of different hepatitis viruses, with a predominance of liver damage. Viruses rely on persistence to spread and establish long-term infection by deploying potent immune evasion mechanisms within host cells [17]. Oncogenic viruses can induce cancer characteristics directly in infected cells through anti-apoptotic and proliferative programs. The underlying mechanisms include three main processes [18]. Firstly, viruses infect the host and encode a series of viral proteins that can dominantly disrupt signaling mechanisms responsible for cell growth and survival. Second, the host cell recognizes the viral genome or replication intermediates and triggers a DNA damage response, which is a protective mechanism against viral invasion. However, a prolonged DNA damage response can lead to genetic instability in host cells, increasing mutation rates and accelerating host chromosome alterations. Another important process is that during viral infection, the infected organs accumulate a large number of inflammatory cells and cytokines, leading to an inflammatory environment that causes irreversible damage. The prolonged inflammatory damage can contribute to cancer development.
According to the etiological diagnosis, there are at least five forms of hepatitis viruses (A, B, C, D, and E, respectively). SUMOylation is involved in regulating the interaction between the hepatitis virus and the host. During the infection of host cells, the virus-host interaction can alter the SUMOylation of intracellular proteins, including interfering with the activation of SUMO and E1 enzymes, binding to E2 enzyme Ubc9, promoting the occurrence of deSUMOylation, and altering the quantity or stability of SUMOylated proteins in host cells [19]. Viruses can also use SUMOylation-related enzymes to modify their proteins to evade the host’s immune system, ensuring efficient replication and viral persistence in the host [20]. Taken together, virus-host interaction alters the protein SUMOylation.
Hepatitis B virus (HBV) manipulates the SUMO system in host cells
HBV, a partial double-stranded circular DNA virus, is closely linked to cirrhosis and HCC. During HBV infection of host cells, HBV utilized SUMOylation systems to modify their proteins to be able to replicate persistently in the host. For example, the HBV core protein is a substrate modified by SUMO2. SUMOylation of HBV core proteins mediates its recruitment to promyelocytic leukemia-nuclear bodies (PML-NBs) within host cells. SUMOylated HBV core induces HBV capsid decomposition, which is a prerequisite for HBV core to enter the nucleus. The binding of SUMOylated HBV core to PML-NBs is essential for efficient conversion of rcDNA to cccDNA and establishment of viral persistence [21].
Correspondingly, the SUMOylation of host proteins is altered to enhance the antiviral response when the host is exposed to viral invasion. IQGAP2 is a scaffold protein that interacts with key molecules in multiple signaling pathways and is a critical substrate for SENP3-mediated deSUMOylation. In HBV-infected cells, downregulation of SENP3 promotes SUMOylation of IQGAP2, thereby suppressing the expression of HBV viral proteins by regulating AKT phosphorylation. Hence, the process of deSUMOylation enzyme SENP3-mediated deSUMOylation of IQGAP2 protein enhances AKT phosphorylation as a defense mechanism of liver cells against the virus [22].
HBx is a viral product that acts as a transcriptional cofactor during viral replication and can promote cell transformation by altering key cellular pathways involved in cell growth, DNA repair, apoptosis, and cell cycle progression. The overexpression of centrosomal protein P4.1-associated protein (CPAP) is associated with SUMO1 modification. SUMO1-modified CPAP is essential for IKK-mediated NF-κB activation and enhancement of the HBx-induced NF-κB signaling pathway [23]. The host transcription factor Speckled 110 kDa (Sp110) can be modified by SUMO1. However, in the presence of HBV infection, Sp110 undergoes deSUMOylation modification and dissociates from the promyelocytic leukemia nuclear body (PML-NB) complex. This reduction in apoptosis and increase in the survival capacity of liver cells significantly affect genes related to the type I interferon pathway, favoring HBV replication within host cells [24]. Mechanistically, HBx may promote the formation of the Sp110-SENP1-HBx complex, which catalyzes the removal of SUMO1 from Sp110 and relocates HBx to the Sp110 gene promoter. This reprograms host gene expression and triggers viral proliferation [24]. This research emphasizes the importance of protein SUMOylation and deSUMOylation in mediating the HBV lifecycle. Furthermore, HBx expression in mouse and human cell lines can promote cell growth and alter the SUMOylation modification status of E-cadherin, a membrane protein crucial in epithelial-mesenchymal transition (EMT). In contrast to Sp110 SUMOylation, HBx expression promotes the binding of SUMO1 and 2/3 to E-cadherin, resulting in E-cadherin degradation, EMT transition, loss of intercellular communication, and hepatocyte overgrowth [25].
SUMOylation regulates hepatitis C virus (HCV) replication and metabolic homeostasis in host cells
HCV is a single-stranded RNA virus that encodes a multiprotein precursor, subsequently cleaved by the host cassette protease into biologically active proteins (including the core proteins E1, E2, and P7) and non-structural proteins (including NS1, NS3, NS4A, NS4B, NS5A, and NS5B). The expression of SUMO1 in host cells was increased during HCV infection. Silencing SUMO1 can inhibit the expression of adipose differentiation-associated protein (ADRP) and the formation of lipid droplets, thereby reducing HCV core protein and lipid droplet deposition, and thus inhibiting HCV replication [26]. Moreover, nonstructural 5A (NS5A), an essential component of the HCV replication complex, was SUMOylated at K348 residue. SUMOylation increased protein stability of the NS5A, thereby positively affecting the interaction of NS5A with NS5B protein and replication of HCV replication [20]. In contrast, the protein inhibitor of activated STAT2 (PIAS2), a SUMO E3 ligase, interacts and enhances the SUMOylation level of the HCV core, subsequently reducing its protein levels via the proteasome pathway in a ubiquitin-independent manner, thereby inhibiting HCV replication [19]. Moreover, HCV infection led to metabolic abnormality in the host. Orphan nuclear receptor small isoform partners (SHP) regulate hepatic lipogenesis and metabolic homeostasis by suppressing SUMOylation of liver X receptor at the SREBP-1c promoter and SREBP-1c expression. However, the expression levels of SHP are reduced in the livers of HCV patients and mice with persistent HCV infection, which results in abnormal lipogenesis [27].
Hepatitis D virus (HDV) utilizes SUMO pathway to modify own protein
Hepatitis delta antigen (HDAg) is a nucleoprotein closely related to HDV replication. There are two specific forms of HDAg, namely S-HDAg and L-HDAg. S-HDAg is one of the target proteins for SUMO1 modification. The binding of SUMO1 with S-HDAg selectively enhanced the synthesis of HDV genomic RNA and mRNA but did not enhance the synthesis of antigenic RNA [28].
Taken together, the level of SUMOylation-related enzyme or substrate protein SUMOylation can be altered during viral infection, thereby affecting viral replication and host antiviral properties.
SUMOylation involved the signaling pathway in HCC
HCC is the commonly malignant tumor in China with an advanced mortality rate. Chronic hepatitis infection, ALD, MASLD, carcinogenic toxins, and some environmental factors contribute to the development of HCC. Recent findings suggest that the protein SUMOylation plays an essential regulatory role in the carcinogenesis and progression of HCC (Table 2, Fig. 2). And SUMOylation levels are higher in HCC samples compared to normal liver tissues [29].

SUMOylation-mediated biological processes in HCC. A The effect of SUMOylation on the viral replication. Viruses can manipulate the whole process of SUMOylation by interacting with the SUMO pathway. B SUMOylation is implicated in tumorigenesis. Serine starvation increased the level of NRF2 SUMOylation. SUMOylated NRF2 promoted de novo serine synthesis via ROS-PHGDH signaling, which led to sustained HCC tumorigenesis. C SUMOylation is involved in the regulation of cell proliferation. PEPCK1 is SUMOylated at K124 residue, which resulted in the decrease of protein stability and degradation through the ubiquitin–proteasome pathway, ultimately leading to the downregulation of gluconeogenesis and the elevation of glycolysis, and accelerating the proliferation of HCC cells. D SUMOylation regulates cell migration, invasion, and metastasis. p65 SUMOylation is required for MANF recruitment and its interaction with p65, which suppresses NF-κB/Snail signal pathway and thereby inhibits EMT and HCC progression
SUMOylation-involved oncogenic signal pathway
Many tumor suppressors conjugate with SUMO in cancer, and the SUMO machinery plays a crucial regulatory role in many mechanisms of carcinogenesis. It has been shown that SUMOylation is involved in tumorigenesis and cell proliferation of HCC. For instance, nuclear factor erythroid-2 related factor 2 (NRF2) is SUMOylated at K110 by SUMO1. Serine starvation increased NRF2 SUMOylation levels, leading to the persistence of HCC tumorigenesis. Mechanically, SUMOylated NRF2 promoted de novo synthesis of serine through reactive oxygen species (ROS)-phosphoglycerate dehydrogenase (PHGDH) signaling in HCC [30]. SENP1 is upregulated in human HCC cells and tissues. SENP1 increased the stability and transcriptional activity of hypoxia-inducible factor (HIF)-1α under hypoxic conditions via deSUMOylation in HCC. These augment the stemness of cancer, increase liver cancer stem cell (CSC) subpopulations, and promote hepatocarcinogenesis [9]. SUMOylation of liver kinase B1 (LKB1) is higher in fast-growing HCC tumors. Mechanistically, LKB1 was SUMOylated at K178 residue, which facilitates LKB1 to recognize and activate the cell energy sensor AMP-activated protein kinase through the SUMO-interacting motif. And SUMOylation promotes the interaction of LKB1 with STRADα and nuclear export, which fuels the growth of hepatoma cells [31]. RNF146, a RING-type E3 ubiquitin ligase, plays an essential role in Wnt/β-catenin signaling pathway. RNF146 was modified by SUMO3 at K19, K61, K174, and K175 residues, which promotes its combination with Axin and accelerates Axin degradation, thereby enhancing β-catenin signaling and contributing to HCC progression [32]. Phosphoenolpyruvate carboxykinase PEPCK1 is an important rate-limiting enzyme in the gluconeogenic pathway. However, PEPCK1 is expressed at a low level in both clinical HCC samples and HCC cells. Mechanistically, PEPCK1 is SUMOylated at K124 residue, which results in the degradation of PEPCK1 protein via the ubiquitin–proteasome pathway, ultimately leading to the downregulation of gluconeogenesis and the elevation of glycolysis, and accelerating the proliferation of HCC cells. Therefore, the inhibition of PEPCK1 SUMOylation provides a new direction for the treatment of HCC [33].
SUMOylation is also involved in regulating cell invasion and migration of HCC. For instance, SUMOylation of methyltransferase-like 3 (Mettl3) was increased after mitogen stimulation, which correlated with the upregulation of Ubc9 and was positively associated with the high metastatic potential of HCC. SUMOylated Mettl3 regulated HCC invasion, metastasis, and progression via controlling Snail mRNA homeostasis in an m6A methyltransferase activity-dependent manner [34]. XPO5 was SUMOylated by SUMO2 at K125. SUMOylated XPO5 plays an oncogenic role in the proliferation, migration, and invasion of HCC cells by manipulating the nuclear-cytoplasmic translocation of miR-3184, thereby increasing PLCB1 expression [35]. Guanosine triphosphate binding protein 4 (GTPBP4) induced pyruvate kinase M2 (PKM2) SUMOylation in HCC. Then SUMO1-modified PKM2 translocated from the cytoplasm to the nucleus and promoted HCC progression through activating EMT and STAT3 signaling pathway [36]. Polycomb chromobox 4 (Cbx4), a SUMO E3 ligase, potentiated hypoxia-induced vascular endothelial growth factor (VEGF) expression and angiogenesis in HCC cells by elevating the level of SUMOylation and transcriptional activity of HIF-1α, which ultimately facilitates the cell migration in HCC [6].
SUMOylation also modulates anti-tumor immunity in HCC. Interleukin-33 (IL-33) plays multiple roles in cancer and immunity. IL-33 was SUMOylated by SUMO1 at K54, which promotes its nuclear translocation. Moreover, RanBP2-promoted SUMOylation of IL-33 disrupted ubiquitin-mediated degradation of IRF1, which promotes the abundance of PD-L1 and the secretion of the chemokine IL-8, thereby preventing the activation of cytotoxic T cells and promoting M2 polarization of macrophages, respectively [37]. This result suggests that SUMOylation of IL-33 in the nucleus of HCC cells impairs anti-tumor immunity.
Moreover, SUMOylation plays an important role in the switch from HBV to HCC. p65, the most important subunit of NF-κB, interacted with SUMO2/3 and enhances the stabilization of p65 in the cytoplasm, affecting the development of HCC and participating in the transition from chronic hepatitis B to HCC [38]. Interestingly, SUMO1-related p65 SUMOylation promotes the nuclear translocation of p65 and enhances the transcriptional activity of NF-κB by affecting p65 phosphorylation, which promotes HCC progression [39]. It can be observed that SUMO1-related or SUMO2/3-related p65 SUMOylation has different mechanisms to regulate HCC progression. HBx mediates HER2 protein expression via LASP1, which binds to HER2 in HBx-positive HCC cells. Mechanistically, LASP1 interacted with RANBP2 and RANGAP1 to increase SUMOylation of LASP1 and HER2 protein levels in HBx-positive HCC cells, promoted sensitization of insulin signaling pathway, and enhanced HCC cell growth and migration [40].
SUMOylation involved the signaling pathway of tumor suppression
SUMOylation does not necessarily exert pro-cancer functions; SUMOylation can also enhance the anti-tumor effects of target proteins. Mesencephalic astrocyte-derived neurotrophic factor (MANF) is an endoplasmic reticulum stress inducible secretion protein. p65 SUMOylation is required for MANF recruitment and its interaction with p65, which suppresses NF-κB/Snail signal pathway and thereby inhibits EMT and HCC progression [41]. The lncRNA p53 stabilizing and activating RNA (PSTAR) binds to heterogeneous nuclear ribonucleoprotein K (hnRNP K) and enhances its SUMOylation, which strengthens the interaction between hnRNP K and p53, ultimately leading to the accumulation and transcriptional activation of p53, and consequently inhibiting the proliferation and tumorigenicity of HCC cells [42].
Alpha-fetoprotein (AFP) is the most widely used biomarker for the diagnosis of HCC. However, a significant proportion of patients with HCC have normal serum levels of AFP. The underlying mechanism is that heat shock protein gp96 inhibited the interaction of RanBP2 with NR5A2, reduced RanBP2-mediated NR5A2 SUMOylation, and subsequent ubiquitination [43]. This result found that the detection of gp96 levels may increase the sensitivity of HCC diagnosis by AFP test.
Molecular mechanism of SUMOylation-mediated drug resistance in HCC
Due to the insidious character of HCC, the majority of HCC are diagnosed at an intermediate to advanced stage. Sorafenib was the first tyrosine kinase inhibitor approved by the FDA for the treatment of advanced HCC. Studies have shown that sorafenib can inhibit caspase-1 expression by inhibiting the TLR4/stat3/SUMO1 signaling cascade, which reduces the SUMOylation of p65 and inhibits the nuclear translocation of p65. This study provides new insights into the mechanisms of sorafenib treatment in HCC [44]. However, the low survival rate and poor prognosis of HCC are closely related to the development of drug resistance in HCC. The drug resistance in HCC was observed to be associated with increased SUMOylation, likely due to the increased expression of SAE2 (Uba2) and SENP1 [45]. FOXK2 is a key transcription factor that directly promotes the transcription of nucleotide synthetic genes and cancer cell resistance to chemotherapy. In addition, PIAS4 is a SUMO E3 ligase for FOXK2 SUMOylation and SUMOylates FOXK2 at K527 and K633. FOXK2 SUMOylation promotes nucleotide de novo synthesis in nucleus and causes resistance to 5-fluorouracil [46]. This showed that aberrant activation of the PIAS4-FOXK2 signaling axis is closely related to hepatocarcinogenesis, patient prognosis, and chemotherapy resistance. Moreover, SENP1 is a direct target of miR-122 and reversed drug sensitivity in miR-122 overexpressing HCC cells. Mechanistically, the miR-122/SENP1 axis can regulate β-catenin stability through deSUMOylation, thereby promoting stemness and chemoresistance in HCC [47].
Hypoxia is an essential feature of HCC and is associated with chemotherapy resistance. Saikosaponin-d (Ssd) significantly suppressed the malignant phenotype in HCC cells and enhanced the chemosensitivity of HCC by altering the expression of SENP5 and SUMOylation of GLI under hypoxia [48]. Dexamethasone (Dex) inhibited stemness maintenance and enhanced the chemosensitivity of HCC stem cells to HSVtk/GCV by inducing deSUMOylation of HIF-1α and Oct4 [49]. These results suggested that an appropriate combination of Ssd or Dex and chemotherapeutic agents is expected to improve the survival and prognosis of HCC patients.
Altogether, SUMOylation has multiple regulatory roles in the development and progression of HCC and can be developed as a novel marker. Moreover, a high expression of SUMOylation-related genes is closely associated with common cancer-related pathways, anti-cancer drug sensitivity, and vascular invasion [50]. Thus, further research could help to reveal the specific SUMO modification mechanisms and provide new insights and targets for the treatment of HCC. The current understanding of the role of SUMOylation in HCC is still limited, and more in-depth studies are needed to resolve its detailed molecular mechanisms.
Therapeutic potential for targeting protein SUMOylation
The impact of various PTMs in tumorigenesis, metastasis, and anti-cancer strategies is increasingly recognized. The altered states of many PTMs are corrected with cancer signature and behavior. This is an emerging area in cancer research that may lead to new mechanisms and therapeutic strategies to facilitate early detection and treatment of cancer. Dysregulation of the SUMO-related enzymes disrupts the equilibrium of protein SUMOylation and deSUMOylation, leading to carcinogenesis and drug resistance in liver diseases in an environmentally dependent manner.
SUMOs and SUMO-related enzymes have promotional roles in different stages of liver diseases, which makes SUMO pathway as a promising target. Therefore, the development of active compounds that block the SUMO pathway is very meaningful. Over the past two decades, the SUMO pathway has emerged as an attractive target for disease therapy. In the early days, several SUMO-activating enzyme E1 inhibitors have been identified, including ginkgolic acid, kerriamycin B, tannic acid, and davidiin, as well as SUMO-conjugating enzyme E2 inhibitors 2-D08, GSK145A, and spectomycin B1, but these molecules lack specificity and are inefficient (half-inhibitory concentrations in the µM range) [51].
Notably, the synthesized ML-792 inhibitor can form a complex with SUMO, selectively block SAE activity, and act as a competitive inhibitor of SUMO E1, which is a highly efficient and highly selective SUMO E1 inhibitor. ML-792 inhibits cancer cell proliferation in vitro and is particularly sensitive to tumor cells with higher MYC upregulation [52]. Another derivative of the ML-792 inhibitor is TAK-981, which made a breakthrough in targeting SUMO modification. TAK-981 has a similar half-inhibitory concentration as ML-792 and has higher potency and long-lasting potency [53]. TAK-981 has been proven to promote the innate immune response, as well as type I IFN-dependent activation of innate immune cells, including macrophages, NK cells, dendritic cells, and T cells [54]. Moreover, it was shown that TAK-981 and ML-792 reduced the global protein SUMOylation levels in HCC cells at the nanomolar level and successfully attenuated the tumor burden [29]. The discovery of drugs targeting SUMOylation is expected to improve the efficacy of HCC treatment.
There are no drugs related to SUMOylation, but the inhibitor TAK-981 is undergoing phase 1/2 clinical studies in multiple solid and hematological tumors, including combination and single-agent methods (NCT Numbers: NCT05976334, NCT04776018, NCT04381650, NCT04074330, NCT04065555, and NCT03648372) (Table 3). Preclinical data suggest potential synergistic mechanisms between TAK-981 and other immunomodulators.
Conclusions and perspectives
A growing number of findings strongly support a central role for protein SUMOylation in disease. Protein SUMOylation also appears to enhance or inhibit antiviral/antitumor effects, depending on substrate specificity and modification site. However, it is important to note that targeting SUMOylation induces the activation of certain latent viruses, and there is also the potential to interfere with DNA damage repair processes, increasing the risk of tumorigenesis. In addition, SUMO proteins rarely promote cancer development directly, but rather by binding to their target proteins and enhancing their oncogenic effects. Therefore, tumor microenvironment models in different contexts need to be refined to comprehensively illustrate the biological importance of SUMOylation-mediated pathways in cancer, as well as to explore their therapeutic potential. Moreover, the SUMO pathways should be analyzed individually in patients with different types of hematologic and solid tumor cancers.
In conclusion, further investigation of the multiple roles of protein SUMOylation in liver disease could make targeting SUMOylation as a therapeutic strategy for treating patients with immune dysregulation or cancer (Fig. 3).

Potential clinical value of SUMOylation for diagnosis, treatment, and prognosis of diseases. A Monitoring the level of protein SUMOylation in clinical samples (such as serum and tissues) is helpful for doctors to diagnose and treat patients better. B rotein SUMOylation level is related to patient survival. C The SUMOylation-related inhibitors in the treatment of HCC
Availability of data and materials
Not applicable.
References
Chen Y, Peng W, Tao Q et al (2023) Increased small ubiquitin-like modifier-activating enzyme SAE1 promotes hepatocellular carcinoma by enhancing mTOR SUMOylation. Lab Invest 103(1):100011
Ong JR, Bamodu OA, Khang NV et al (2021) SUMO-activating enzyme subunit 1 (SAE1) is a promising diagnostic cancer metabolism biomarker of hepatocellular carcinoma. Cells 10(1):178
Tomasi ML, Ramani K, Ryoo M et al (2018) SUMOylation regulates cytochrome P450 2E1 expression and activity in alcoholic liver disease. Faseb j 32(6):3278–3288
Yang H, Gao S, Chen J et al (2020) UBE2I promotes metastasis and correlates with poor prognosis in hepatocellular carcinoma. Cancer Cell Int 20:234
Tu J, Chen Y, Cai L et al (2015) Functional proteomics study reveals SUMOylation of TFII-I is involved in liver cancer cell proliferation. J Proteome Res 14(6):2385–2397
Li J, Xu Y, Long XD et al (2014) Cbx4 governs HIF-1α to potentiate angiogenesis of hepatocellular carcinoma by its SUMO E3 ligase activity. Cancer Cell 25(1):118–131
Yan L, Zhang T, Wang K et al (2022) SENP1 prevents steatohepatitis by suppressing RIPK1-driven apoptosis and inflammation. Nat Commun 13(1):7153
Tao Y, Li R, Shen C et al (2020) SENP1 is a crucial promotor for hepatocellular carcinoma through deSUMOylation of UBE2T. Aging (Albany NY) 12(2):1563–1576
Cui CP, Wong CC, Kai AK et al (2017) SENP1 promotes hypoxia-induced cancer stemness by HIF-1α deSUMOylation and SENP1/HIF-1α positive feedback loop. Gut 66(12):2149–2159
Liu Y, Dou X, Zhou WY et al (2021) Hepatic small ubiquitin-related modifier (SUMO)-specific protease 2 controls systemic metabolism through SUMOylation-dependent regulation of liver-adipose tissue crosstalk. Hepatology 74(4):1864–1883
Jin ZL, Pei H, Xu YH et al (2016) The SUMO-specific protease SENP5 controls DNA damage response and promotes tumorigenesis in hepatocellular carcinoma. Eur Rev Med Pharmacol Sci 20(17):3566–3573
Qian J, Luo Y, Gu X et al (2013) Inhibition of SENP6-induced radiosensitization of human hepatocellular carcinoma cells by blocking radiation-induced NF-κB activation. Cancer Biother Radiopharm 28(3):196–200
Floris A, Chandla S, Lim Y et al (2023) Sumoylation of methionine adenosyltransferase alpha 1 promotes mitochondrial dysfunction in alcohol-associated liver disease. Hepatology. Epub ahead of print
Alfaro AJ, Dittner C, Becker J et al (2023) Fasting-sensitive SUMO-switch on Prox1 controls hepatic cholesterol metabolism. EMBO Rep 24(10):e55981
Lee GY, Jang H, Lee JH et al (2014) PIASy-mediated sumoylation of SREBP1c regulates hepatic lipid metabolism upon fasting signaling. Mol Cell Biol 34(6):926–938
Wang S, Yang M, Li P et al (2023) High-fat diet-Iinduced deSUMOylation of E4BP4 promotes lipid droplet biogenesis and liver steatosis in mice. Diabetes 72(3):348–361
Fan Y, Li X, Zhang L et al (2022) SUMOylation in viral replication and antiviral defense. Adv Sci (Weinh) 9(7):e2104126
Krump NA, You J (2018) Molecular mechanisms of viral oncogenesis in humans. Nat Rev Microbiol 16(11):684–698
Guo J, Chen D, Gao X et al (2017) Protein inhibitor of activated STAT2 restricts HCV replication by modulating viral proteins degradation. Viruses 9(10):285
Lee HS, Lim YS, Park EM et al (2014) SUMOylation of nonstructural 5A protein regulates hepatitis C virus replication. J Viral Hepat 21(10):e108–e117
Hofmann S, Plank V, Groitl P et al (2023) SUMO modification of hepatitis B virus core mediates nuclear entry, promyelocytic leukemia nuclear body association, and efficient formation of covalently closed circular DNA. Microbiol Spectr 11(3):e0044623
Xi R, Kadur Lakshminarasimha Murthy P, Tung KL et al (2019) SENP3-mediated host defense response contains HBV replication and restores protein synthesis. PLoS One 14(1):e0209179
Yen CJ, Yang ST, Chen RY et al (2019) Hepatitis B virus X protein (HBx) enhances centrosomal P4.1-associated protein (CPAP) expression to promote hepatocarcinogenesis. J Biomed Sci 26(1):44
Sengupta I, Das D, Singh SP et al (2017) Host transcription factor Speckled 110 kDa (Sp110), a nuclear body protein, is hijacked by hepatitis B virus protein X for viral persistence. J Biol Chem 292(50):20379–20393
Ha HL, Kwon T, Bak IS et al (2016) IGF-II induced by hepatitis B virus X protein regulates EMT via SUMO mediated loss of E-cadherin in mice. Oncotarget 7(35):56944–56957
Akil A, Wedeh G, Zahid Mustafa M et al (2016) SUMO1 depletion prevents lipid droplet accumulation and HCV replication. Arch Virol 161(1):141–148
Chen J, Zhou Y, Zhuang Y et al (2019) The metabolic regulator small heterodimer partner contributes to the glucose and lipid homeostasis abnormalities induced by hepatitis C virus infection. Metabolism 100:153954
Tseng CH, Cheng TS, Shu CY et al (2010) Modification of small hepatitis delta virus antigen by SUMO protein. J Virol 84(2):918–927
Wang Z, Pan B, Su L et al (2023) SUMOylation inhibitors activate anti-tumor immunity by reshaping the immune microenvironment in a preclinical model of hepatocellular carcinoma. Cell Oncol (Dordr). Epub ahead of print
Guo H, Xu J, Zheng Q et al (2019) NRF2 SUMOylation promotes de novo serine synthesis and maintains HCC tumorigenesis. Cancer Lett 466:39–48
Zubiete-Franco I, García-Rodríguez JL, Lopitz-Otsoa F et al (2019) SUMOylation regulates LKB1 localization and its oncogenic activity in liver cancer. EBioMedicine 40:406–421
Li W, Han Q, Zhu Y et al (2023) SUMOylation of RNF146 results in Axin degradation and activation of Wnt/β-catenin signaling to promote the progression of hepatocellular carcinoma. Oncogene 42(21):1728–1740
Bian XL, Chen HZ, Yang PB et al (2017) Nur77 suppresses hepatocellular carcinoma via switching glucose metabolism toward gluconeogenesis through attenuating phosphoenolpyruvate carboxykinase sumoylation. Nat Commun 8:14420
Xu H, Wang H, Zhao W et al (2020) SUMO1 modification of methyltransferase-like 3 promotes tumor progression via regulating Snail mRNA homeostasis in hepatocellular carcinoma. Theranostics 10(13):5671–5686
Lin D, Fu Z, Yang G et al (2020) Exportin-5 SUMOylation promotes hepatocellular carcinoma progression. Exp Cell Res 395(2):112219
Zhou Q, Yin Y, Yu M et al (2022) GTPBP4 promotes hepatocellular carcinoma progression and metastasis via the PKM2 dependent glucose metabolism. Redox Biol 56:102458
Wang Z, Pan B, Qiu J et al (2023) SUMOylated IL-33 in the nucleus stabilizes the transcription factor IRF1 in hepatocellular carcinoma cells to promote immune escape. Sci Signal 16(776):eabq3362
Liu J, Sha M, Wang Q et al (2015) Small ubiquitin-related modifier 2/3 interacts with p65 and stabilizes it in the cytoplasm in HBV-associated hepatocellular carcinoma. BMC Cancer 15:675
Jiang C, Zhang C, Dai M et al (2024) Interplay between SUMO1-related SUMOylation and phosphorylation of p65 promotes hepatocellular carcinoma progression. Biochim Biophys Acta Mol Cell Res 1871(1):119595
You H, Yuan D, Li Q et al (2023) Hepatitis B virus X protein increases LASP1 SUMOylation to stabilize HER2 and facilitate hepatocarcinogenesis. Int J Biol Macromol 226:996–1009
Liu J, Wu Z, Han D et al (2020) Mesencephalic astrocyte-derived neurotrophic factor inhibits liver cancer through small ubiquitin-related modifier (SUMO)ylation-related suppression of NF-κB/Snail signaling pathway and epithelial-mesenchymal transition. Hepatology 71(4):1262–1278
Qin G, Tu X, Li H et al (2020) Long noncoding RNA p53-stabilizing and activating RNA promotes p53 signaling by inhibiting heterogeneous nuclear ribonucleoprotein K deSUMOylation and suppresses hepatocellular carcinoma. Hepatology 71(1):112–129
Qian L, Liang Z, Wang Z et al (2023) Cellular gp96 upregulates AFP expression by blockade of NR5A2 SUMOylation and ubiquitination in HCC. J Mol Cell Biol 15(5):mjad027
Li J, Zhou Y, Liu Y et al (2018) Sorafenib inhibits caspase-1 expression through suppressing TLR4/stat3/SUMO1 pathway in hepatocellular carcinoma. Cancer Biol Ther 19(11):1057–1064
Qin Y, Bao H, Pan Y et al (2014) SUMOylation alterations are associated with multidrug resistance in hepatocellular carcinoma. Mol Med Rep 9(3):877–881
Li Y, Chen J, Wang B et al (2023) FOXK2 affects cancer cell response to chemotherapy by promoting nucleotide de novo synthesis. Drug Resist Updat 67:100926
Dai J, Hao Y, Chen X et al (2023) miR-122/SENP1 axis confers stemness and chemoresistance to liver cancer through Wnt/β-catenin signaling. Oncol Lett 26(3):390
Zhang CY, Jiang ZM, Ma XF et al (2019) Saikosaponin-d inhibits the hepatoma cells and enhances chemosensitivity through SENP5-dependent inhibition of Gli1 SUMOylation under hypoxia. Front Pharmacol 10:1039
Jiang Z, Zhang C, Liu X et al (2020) Dexamethasone inhibits stemness maintenance and enhances chemosensitivity of hepatocellular carcinoma stem cells by inducing deSUMOylation of HIF-1α and Oct4. Int J Oncol 57(3):780–790
Wang J, Cong P, Jin Z et al (2023) A novel prognostic signature for hepatocellular carcinoma based on SUMOylation-related genes. Sci Rep 13(1):11233
Yang Y, Xia Z, Wang X et al (2018) Small-molecule inhibitors targeting protein SUMOylation as novel anticancer compounds. Mol Pharmacol 94(2):885–894
He X, Riceberg J, Soucy T et al (2017) Probing the roles of SUMOylation in cancer cell biology by using a selective SAE inhibitor. Nat Chem Biol 13(11):1164–1171
Langston SP, Grossman S, England D et al (2021) Discovery of TAK-981, a first-in-class inhibitor of SUMO-activating enzyme for the treatment of cancer. J Med Chem 64(5):2501–2520
Kumar S, Schoonderwoerd MJA, Kroonen JS et al (2022) Targeting pancreatic cancer by TAK-981: a SUMOylation inhibitor that activates the immune system and blocks cancer cell cycle progression in a preclinical model. Gut 71(11):2266–2283
Guo WH, Yuan LH, Xiao ZH et al (2011) Overexpression of SUMO-1 in hepatocellular carcinoma: a latent target for diagnosis and therapy of hepatoma. J Cancer Res Clin Oncol 137(3):533–541
Chen J, Chen C, Lin Y et al (2021) Downregulation of SUMO2 inhibits hepatocellular carcinoma cell proliferation, migration and invasion. FEBS Open Bio 11(6):1771–1784
Zhang Z, Wen H, Peng B et al (2021) CDKN2A deregulation in fatty liver disease and its accelerative role in the process of lipogenesis. Faseb j 35(4):e21230
Liu Q, Zhou B, Liao R et al (2020) PIAS4, upregulated in hepatocellular carcinoma, promotes tumorigenicity and metastasis. J Cell Biochem 121(5–6):3372–3381
Shen Y, Li Y, Ma X et al (2018) Connexin 43 SUMOylation improves gap junction functions between liver cancer stem cells and enhances their sensitivity to HSVtk/GCV. Int J Oncol 52(3):872–880
Ge H, Du J, Xu J et al (2017) SUMOylation of HSP27 by small ubiquitin-like modifier 2/3 promotes proliferation and invasion of hepatocellular carcinoma cells. Cancer Biol Ther 18(8):552–559
Mei L, Yuan L, Shi W et al (2017) SUMOylation of large tumor suppressor 1 at Lys751 attenuates its kinase activity and tumor-suppressor functions. Cancer Lett 386:1–11
Funding
This work was financially supported by grants from the National Natural Science Foundation of China (82360389).
Author information
Authors and Affiliations
Contributions
YYF wrote the manuscript and created schematic representations. YFX reviewed and edited the manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Competing interests
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Yang, Y., Yu, F. Abnormal protein SUMOylation in liver disease: novel target for therapy. J Mol Med 102, 719–731 (2024). https://doi.org/10.1007/s00109-024-02440-w
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00109-024-02440-w