
Abstract
Cancer cells rely on heightened protein quality control mechanisms, including the ubiquitin-proteosome system that is predominantly driven by ubiquitination comprising E1, E2, and E3 trienzyme cascades. Although E3s have been extensively studied, the implication of E2s in tumorigenesis is poorly defined. Here we reveal a critical E2 in the pathogenesis of hepatocellular carcinoma (HCC). Among all of E2s, UBE2O shows the strongest association with HCC survival prognosis, and its expression is increased in HCC tumors. Accordingly, UBE2O deficiency inhibits HCC growth and metastasis both in vitro and in vivo, while its overexpression has opposite effects. Depending on both E2 and E3 enzymatic activities, UBE2O can interact with and mediate the ubiquitination and degradation of HADHA, a mitochondrial β-oxidation enzyme, thereby modulating lipid metabolic reprogramming. HADHA is reduced in HCC tumors and inversely correlated with UBE2O levels. Importantly, HADHA acts as a tumor suppressor and primarily mediates UBE2O’s function on HCC. Moreover, liver-specific deletion of Ube2o in mice are resistant to DEN-induced hepatocarcinogenesis, along with HADHA upregulation and reduced hepatic lipid accumulation. These data reveal UBE2O as a novel oncogenic driver for metabolic reprogramming and HCC development, highlighting the potential of targeting UBE2O/HADHA axis for HCC therapy.
Similar content being viewed by others


Introduction
Protein quality control (PQC) is crucial for eukaryotic cells to monitor protein biogenesis and maintain cellular homeostasis [1]. It has been proposed that cancer cells are more dependent on PQC mechanisms than normal cells due to aberrant genomic alternations and environmental stresses [2], and thus PQC components have emerged as therapeutic targets for human cancers [3]. However, the PQC mechanisms underlying tumor initiation and progression remain incompletely defined.
The ubiquitin-proteasome system (UPS) is the major PQC mechanism responsible for degradation of proteins and plays an essential role in maintaining protein homeostasis [1]. The UPS is predominantly driven by ubiquitination, which is controlled by multilayered, reversible enzymatic reactions catalyzed by ubiquitin-activating enzymes (E1s), ubiquitin-conjugating enzymes (E2s), and ubiquitin-ligating enzymes (E3s). Given that substrate specificity is driven by E3s, it have been extensively studied compared with E1s and E2s [4]. Relative to E3s, the role of E2s in cancer is largely undetermined. Functionally, E2s generally interact with a number of E3s to transfer the activated ubiquitin to the protein substrate, and they are often considered as ‘ubiquitin carriers’ with a merely auxiliary role. However, accumulating evidence suggest that E2s can not only dictate the length and topology of ubiquitin chains and the processivity of the chain assembly reaction, but also determine substrate specificity in many cases [5, 6]. Notably, the human genome contains ~40 E2s and ~600 E3s, suggesting that an E2 might averagely have a large range of ubiquitination substrates and consequences compared to an E3. Under these circumstances, it is of great interest to explore the role and extended implication of E2s in tumorigenesis.
Ubiquitin-conjugating enzyme E2 O (UBE2O) is one of the largest E2s and has multiple functional domains in addition to the UBC domain, implying that it may execute diverse functions by targeting a broad spectrum of molecules. Indeed, UBE2O can interact with and regulate various proteins by mediating multiple types of ubiquitination in either E3-dependent or E3-independent (i.e., an E2/E3 hybrid enzyme) manner, and has been implicated in many human diseases, such as anemia, cancer and neurodegenerative disorder [7,8,9,10,11,12]. Interestingly, human UBE2O cDNA was first cloned from liver tissue, in which UBE2O is preferentially expressed [13], indicating potential functional significance. However, the functions and mechanisms of UBE2O in liver diseases are not well defined.
HCC, the most common primary malignant liver tumor, is the sixth highest cause of cancer-related death [14, 15]. Intriguingly, early observations suggest that ubiquitination appears to be upregulated in HCC tumors compared to normal liver tissues [16,17,18], making HCC as an excellent model to delineate the link between the PQC and tumorigenesis. However, the involvement of E2s in HCC has not been investigated systematically and remains largely unknown. Given the diverse roles and outcomes of E2s in ubiquitination and UPS, it is of significance to characterize critical E2s underlying HCC pathogenesis, which would provide novel therapeutic strategies for this disease.
Here, we identify UBE2O as a crucial UPS enzyme for HCC development. Among all of 40 E2s, UBE2O exhibits the strongest association with HCC survival prognosis. UBE2O is consistently increased in HCC tumors and can significantly promote the growth and metastasis of HCC cells in vitro and in vivo. Mechanistically, UBE2O mediates K48-linked polyubiquitination of HADHA at lysine 129, leading to its proteasomal degradation, and therefore promotes lipid metabolism reprogramming. Opposite to UBE2O, HADHA is post-transcriptionally reduced in HCC and capable of suppressing HCC growth and metastasis. Furthermore, liver-specific deletion of Ube2o in mice profoundly impairs HCC initiation and growth by regulating lipid reprogramming. These findings reveal the function and mechanism of UBE2O-HADHA axis in hepatocarcinogenesis and provide potential therapeutic targets for HCC.
Results
UBE2O is upregulated in HCC and associated with poor prognosis
To identify potential E2s associated with HCC, we initially analyzed the correlation between E2s expression levels and patient survival by using the Human Protein Atlas database [19]. 16 of 40 E2s were significantly correlated with HCC patient survival, and more intriguingly, all of them were found to be unfavorable prognostic predictors (Table S1). Among them, UBE2O showed the strongest association with survival in HCC patients (Fig. 1a). Analysis of the GEPIA data [20] also found that UBE2O mRNA expression was negatively correlated with overall survival (Fig. 1b) and diseases-free survival (DFS) (Fig. 1c). Therefore, we chose UBE2O for further investigation.

a Correlation of UBE2O mRNA levels and HCC overall survival. Data from Human Protein Atlas. Correlation of UBE2O mRNA levels with HCC overall survival (b) and disease-free survival (c). Data from TCGA. d UBE2O mRNA levels in normal human liver tissues (NT, n = 50) and HCC tumors (T, n = 371). Data from TCGA. UBE2O mRNA levels in GSE36376 (NT, n = 193; T, n = 240) (e) and GSE54236 (NT, n = 77; T, n = 78) (f), two publicly available expression datasets of HCC. g UBE2O mRNA levels in HCC patients with (n = 95) or without (n = 40) microvascular invasion (MVI). Data from GSE20017. h UBE2O mRNA levels in HCC tumors and paired adjacent peritumoral tissues (n = 49), as measured by qPCR analysis. UBE2O protein levels in HCC tumors and paired adjacent peritumoral tissues, as measured by western blotting (i) (n = 9) and immunohistochemistry staining (j) (n = 8). Scale bars, 100 μm. Data are shown as mean ± SEM. *P < 0.05, **P < 0.01, ***P < 0.001, two-tailed Student t test (d–j). Log-rank P value is shown for Kaplan–Meier plots (a–c).
Next, the expression of UBE2O transcripts in HCC was analyzed by using publicly available human HCC datasets. Analysis of the TCGA revealed an upregulation of UBE2O transcripts in HCC tumors compared to control liver tissues (Fig. 1d). This increase was further validated by two independent GEO datasets (GSE36376 and GSE54236) [21, 22] (Fig. 1e, f). Moreover, UBE2O mRNA levels were significantly increased in HCC patients with microvascular invasion (MVI) [23] (Fig. 1g), which is an important risk factor related to post-surgery recurrence, suggesting a potential involvement of UBE2O in HCC metastasis.
Furthermore, we measured the expression of UBE2O in primary HCC samples by qPCR, western blotting, and immunohistochemistry (IHC). UBE2O mRNA expression was higher in HCC tumors than paired adjacent peritumoral tissues (Fig. 1h). More importantly, UBE2O protein expression was dramatically elevated in HCC tumors (Fig. 1i, j). Taken together, these results strongly suggest a potential role of UBE2O in HCC.
UBE2O promotes proliferation of HCC cells in vitro and in vivo
We determined the potential effects of UBE2O on HCC cell proliferation. Knockdown of UBE2O by small interfering RNAs (siRNAs) significantly inhibited the growth of HCC cells (Supplementary Fig. S1a and Fig. 2a, b). The inhibitory effect of UBE2O knockdown on HCC cell growth was recapitulated by using short hairpin RNA (shRNA) (Supplementary Fig. S1a and Fig. 2c, d). Because shUBE2O#2 exerted a stronger inhibitory effect on UBE2O expression than shUBE2O#1, it was chosen to establish stable hepatic cell lines with low UBE2O expression by using the lentivirus system, which were used for subsequent functional studies. As expected, shRNA-mediated UBE2O inhibition led to obvious decreases in EdU incorporation (Supplementary Fig. S1b) and colony forming ability (Supplementary Fig. S1c), supporting a role of UBE2O in HCC cell proliferation.

a Western blotting analysis to detect the knockdown efficiency of siRNA for UBE2O in Huh7, SK-Hep1 and SNU449. b MTS assays of HCC cells (Huh7, SK-Hep1, and SNU449) transfected with negative control (NC) or siRNA for UBE2O (siUBE2O). c The protein levels of UBE2O in HCC cells transfected with negative control (shNC) or UBE2O shRNAs (shUBE2O#1 and shUBE2O#2). d MTS assays of HCC cells (Huh7, SK-Hep1, and SNU449) transfected with shNC or UBE2O shRNAs. HCC cells with stable UBE2O knockdown (e, f and i) or overexpression (g, h and j) were injected subcutaneously into nude mice, and tumor volume was monitored over time. Tumor growth rate (e), tumor weight and tumor image (f) of SK-Hep1 tumor xenografts stably expressing either shNC or shUBE2O#2 (n = 8 mice/group). Tumor growth rate (g), tumor weight and tumor image (h) of Huh7 tumor xenografts stably expressing either Ctrl or UBE2O (n = 6 mice/group). Representative IHC staining of KI67 of lungs from mice bearing SK-Hep1 (i) or Huh7 (j) tumor xenografts. Scale bars, 100 μm. Data are shown as mean ± SEM. *P < 0.05, **P < 0.01, ***P < 0.001, two-tailed ANOVA (e, g), two-tailed Student t test (b, d, f and h).
Furthermore, we investigated the impact of UBE2O overexpression in cell growth by using a pcDNA3.1 vector containing the full-length UBE2O cDNA. A combination of MTS, EdU staining and colony formation assays consistently revealed that UBE2O overexpression significantly enhanced the proliferation of HCC cells (Supplementary Fig. S1d–g). These data collectively suggest that UBE2O can promote the proliferation of HCC cells in vitro.
We then confirmed the tumorigenic role of UBE2O in vivo by the mouse-xenograft system. We subcutaneously injected HCC cells with stable UBE2O knockdown or overexpression into nude mice respectively, and tumor growth was monitored regularly. Knockdown of UBE2O in SK-Hep1 cells remarkably decreased tumor weight and volume (Fig. 2e, f), while overexpression of UBE2O in Huh7 cells dramatically increased tumor growth rate and tumor size (Fig. 2g, h), demonstrating that UBE2O can promote tumor growth in vivo. In line with these observations, the percentage of KI67-positive cells was lower in UBE2O-deficient tumor xenografts (Fig. 2i), but higher in UBE2O-overexpressing tumor xenografts (Fig. 2j), than their respective controls. Taken together, these data suggest that UBE2O promotes HCC growth in vitro and in vivo.
UBE2O enhances migration and invasion of HCC cells in vitro and in vivo
Next, we investigated the effect of UBE2O in HCC metastasis. Knockdown of UBE2O by shRNA significantly inhibited the migration of HCC cells (Fig. 3a, b), as evidenced by wound-healing assays. Transwell analysis revealed that UBE2O knockdown reduced the number of migrated cells (Fig. 3c). These effects were recapitulated in siRNA-mediated UBE2O knockdown cells (Supplementary Fig. S2a, b), further supporting a role of UBE2O in cell migration. Invasion assay showed that UBE2O knockdown noticeably reduced the invasive capacity of SK-Hep1 and SNU449 cells by 74% and 57%, respectively (Fig. 3d), indicating a role of UBE2O in cell invasion. Conversely, overexpression of UBE2O significantly promoted HCC cell migration and invasion (Supplementary Fig. S2c–f).

Wound healing assays of SK-Hep1 (a) and SNU449 (b) cells transfected with shNC or UBE2O shRNA (shUBE2O#2). Transwell (c) and invasion (d) assays of HCC cells transfected with shNC or UBE2O shRNA (shUBE2O#2). e Western blotting of EMT markers in shNC or shUBE2O#2-treated Huh7 and SNU449 cells. E-CAD, E-CADHERIN. f Western blotting of MMPs in shNC or shUBE2O#2-treated Huh7 and SNU449 cells. Representative SLUG staining of lungs from mice bearing SK-Hep1 (g) or Huh7 (h) tumor xenografts. Scale bars, 100 μm. i, j SK-Hep1 cells stably expressing either shNC or shUBE2O#2 were injected into left ventricle of nude mice (n = 6 mice/group). Metastasis was assessed three weeks after the injection. i Number of total metastases in the lung. j Representative pictures of lungs with xenografts (left panel), and H&E staining of the pulmonary metastatic foci (right panel). Arrows indicate metastases. Scale bars, 100 μm. Data are shown as mean ± SEM. *P < 0.05, **P < 0.01, two-tailed Student t test (a–d), and wilcoxon rank sum test (i).
Epithelial-mesenchymal transition (EMT), a crucial step of tumor metastasis, is hallmarked by upregulation of the mesenchymal markers (e.g., SLUG) and downregulation of the epithelial markers (e.g., E-CADHERIN) [24]. In line with reduced metastasis phenotype, UBE2O knockdown markedly decreased SLUG expression, but increased E-CADHERIN expression (Fig. 3e and Supplementary Fig. S2g, h). Similarly, the levels of MMP9 and MMP14, two markers of tumor invasion, were attenuated by UBE2O knockdown (Fig. 3f). Altogether, these results demonstrate that UBE2O can facilitate migration and invasion of HCC cells in vitro.
In addition, IHC analyses showed that expression of metastatic markers (SLUG) was decreased in UBE2O-deficient tumor xenografts (Fig. 3g), but increased in UBE2O-overexpressing tumor xenografts (Fig. 3h), indicating a role of UBE2O in metastasis in vivo. To further verify this notion, we established the lung metastatic model by implanting SK-Hep1 cells with stable UBE2O knockdown into left ventricle of nude mice. It showed that the metastases in the lungs of mice injected with UBE2O-deficient cells were less frequent and much smaller than those of control mice (Fig. 3i, j). Collectively, these data suggest that UBE2O promotes HCC metastasis in vitro and in vivo.
UBE2O interacts with HADHA
Next, we investigated the molecular mechanism underlying UBE2O’s function in HCC. Given that UBE2O can act as an E2/E3 hybrid enzyme, we first determined the correlation between UBE2O function and its E2/E3 enzymatic activity. It has been shown that Cys1040 and Cys617 are essential for the E2 and E3 activity of human UBE2O [25, 26]. Therefore, we generated two UBE2O mutants, namely C1040S (inactive E2) and C617S (inactive E3) (Supplementary Fig. S3a), and found that both mutations nearly abolished the impact of UBE2O on HCC cell proliferation (Fig. 4a) and migration (Fig. 4b), suggesting that UBE2O functions as an E2/E3 hybrid enzyme in HCC development.

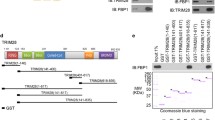
MTS (a) and transwell assays (b) of HCC cells transfected with pcDNA3.1 empty vector (Ctrl), UBE2O WT (UBE2OWT) or mutant vectors (UBE2OC617S, UBE2OC1040S). c Veen diagram of 494 potential UBE2O-associated proteins in HEK293T, HepG2 and Huh7 cells. HEK293T, HepG2 and Huh7 cells were transfected with Flag-tagged UBE2O vectors, and the anti-Flag immunoprecipitates were detected by mass-spectrometric peptide sequencing. The number of potential UBE2O-associated proteins are shown, and three proteins that were consistently identified in these three cell lines are indicated. Co-immunoprecipitation assays of the association of UBE2O with HADHA in HEK293T and Huh7 cells with either exogenous (d) or endogenous (e) expression of UBE2O and HADHA. Western blotting (f) and qPCR assays (g) of HADHA in shUBE2O#2-treated Huh7, SK-Hep1, and SNU449 cells. Western blotting (h) and qPCR assays (i) of HADHA in Huh7, SK-Hep1 and SNU449 cells transfected with pcDNA 3.1 empty vectors (Ctrl) or UBE2O-overexpressing vectors (UBE2O). Western blotting assays of UBE2O and HADHA in SK-Hep1 (j) and Huh7 (k) tumor xenografts. Data are shown as mean ± SEM. *P < 0.05, **P < 0.01, ***P < 0.001, two-tailed Student t test.
To characterize potential UBE2O substrates, we expressed Flag-tagged UBE2O in HEK293T, HepG2 and Huh7 cells respectively and analyzed the anti-Flag immunoprecipitates using mass spectrometry (MS). The combination of MS data from these three cell lines was designed to identify the potential common substrates independent of cellular origin. This integrative analysis revealed three protein candidates, namely HADHA, NACA and SLC25A3 (Fig. 4c, Supplementary Tables S2–S4). Interestingly, our analysis failed to identify AMPKα2, a known substrate of UBE2O [9]. To verify this observation, we performed the immunoprecipitation assay and did not find a clear interaction between UBE2O and AMPKα2 in HEK293T cells (Supplementary Fig. S3b). Moreover, the protein level of AMPKα2 was not affected obviously by UBE2O knockdown or overexpression in HCC cells (Supplementary Fig. S3c, d) and xenograft tumors (Supplementary Fig. S3e, f), further arguing against an effect of UBE2O on AMPKα2 expression in HCC.
Among three aforementioned potential substrates of UBE2O, HADHA, a mitochondrial functional enzyme, has been implicated in several cancers [27, 28]. Specifically, HADHA expression was negatively correlated with the de-differentiation of HCC [29], indicating a potential involvement in hepatocarcinogenesis. In contrast, the implication of NACA and SLC25A3 in tumorigenesis remains unknown. Therefore, we chose HADHA for further investigation and clarified the interaction between HADHA and UBE2O by co-immunoprecipitation assay. Notably, UBE2O co-immunoprecipitated with HADHA ectopically expressing Flag-tagged UBE2O and Myc-tagged HADHA (Fig. 4d). Importantly, this association was further verified with endogenous proteins in Huh7 cells (Fig. 4e), suggesting that HADHA can directly interact with UBE2O.
We then determined the consequence of this interaction on HADHA expression. The protein level of HADHA was consistently induced by UBE2O knockdown in all HCC cell lines that we examined (Fig. 4f), while its mRNA level remained unchanged (Fig. 4g). Conversely, overexpression of UBE2O decreased HADHA at the protein level (Fig. 4h), but not at the mRNA level (Fig. 4i). Intriguingly, this reduction was not observed by overexpression of E2 or E3 inactive mutant (Supplementary Fig. S3g). In addition, HADHA was decreased in UBE2O-overexpressing tumor xenografts, but increased in UBE2O-deficient tumor xenografts (Fig. 4j, k), suggesting an UBE2O-mediated HADHA inhibition in vivo.
Collectively, these results indicate that UBE2O can physically interact with HADHA and subsequently suppress its expression at the post-transcriptional level.
UBE2O targets HADHA for ubiquitination and degradation
Given that the function of UBE2O depends on both E2 and E3 enzymatic activities, and that UBE2O post-transcriptionally inhibits HADHA expression, we therefore speculated that HADHA might be a ubiquitination substrate of UBE2O. Indeed, the ubiquitination assay showed that UBE2O promoted ubiquitination of HADHA in the presence of HA-tagged wild-type ubiquitin (HA-Ub-WT) (Fig. 5a). Lysine 48 (K48)-linked polyubiquitin chains usually serve as major proteolytic signals for degradation by the proteasome. In line with this, K48-mutant ubiquitin (HA-Ub-K48R), in which K48 of ubiquitin has been mutated to arginine (R), dramatically diminished HADHA ubiquitination, even in the presence of ectopic UBE2O expression (Fig. 5a). Moreover, HA-Ub-K48 was functional equivalent to WT-Ub for supporting UBE2O-mediated ubiquitination of HADHA (Fig. 5b). Thus, HADHA may undergo K48-linked polyubiquitination in cells. Additionally, proteasomal inhibition by MG-132 strikingly blocked UBE2O-mediated HADHA degradation (Fig. 5c), and cycloheximide (CHX) treatment remarkably altered UBE2O-mediated HADHA protein turnover rate (Fig. 5d), further supporting that HADHA could be degraded via a ubiquitin-proteasome pathway.

a UBE2O and HADHA overexpressing plasmids were co-transfected with HA-tagged ubiquitin WT (Ub-HA-WT) or Ub-HA-K48R (K48 mutated to arginine) plasmids into Huh7 cells. 48 h later, cells were treated with MG-132 (10 μM) and lysed. HADHA was immunoprecipitated and then immunoblotted for ubiquitin. The cell lysates were immunoblotted for the indicated antibodies. b UBE2O and Myc-tagged HADHA overexpressing plasmid were co-transfected with Ub-HA-WT or Ub-HA-K48 (K48 only, other lysines mutated to arginines) plasmids into HEK293T cells. Then cells were treated, lysed, immunoprecipitated, and immunoblotted as (a). c Myc-HADHA plasmids were transfected alone or together with HA-Ub-WT, Flag-UBE2O into Huh7 cells. 24 h later, cells were treated with MG132 for 6 h. Cell lysates were subjected to immunoblotting against indicated proteins. d Immunoblotting in lysates from Huh7 cells expressing UBE2O shUBE2O#2 treated with cycloheximide (CHX) for the indicated times. e Predicted HADHA ubiquitination sites (●, all lysine residues; ▲, putative ubiquitination sits; ◊, K129). f Immunoblotting in lysates from HEK293T cells transfected with the indicated plasmids. g UBE2O, Ub-HA-WT, and HADHA WT or mutants, were co-transfected into Huh7 cells respectively, followed by MG-132 treatment. Immunoprecipitation (left panel) and immunoblotting assays (right panel) were performed.
To further elucidate the molecular mechanism underlying UBE2O-mediated HADHA degradation, potential ubiquitination sites of HADHA were predicted. Based on the prediction, we generated fifteen site-specific mutants of HADHA, in which a single K residue of HADHA was replaced with R residue (Fig. 5e). Compared to the HADHA WT, three mutants (K129R, K303R, and K309R) markedly increased HADHA protein levels (Fig. 5f). Then, HADHA WT and mutants were co-expressed with UBE2O and Ub in Huh7 cells respectively, and HADHA ubiquitination was assessed. It found that UBE2O overexpression did not initiate the ubiquitination of the K129R mutant of HADHA (Fig. 5g). These results collectively suggest that UBE2O mediates the ubiquitination of the K129 residue of HADHA and results in its degradation.
HADHA expression is reduced in human HCC and negatively correlated with UBE2O expression
We then evaluated the expression of HADHA, as well as its correlation with UBE2O levels, in human HCC tumors. HADHA mRNA levels were comparable in HCC tumors and control liver tissues (Supplementary Fig. S4a, b), and did not significantly correlate with HCC prognosis (Supplementary Fig. S4c, d) by analyzing several available human HCC datasets. However, western blotting and IHC staining analyses revealed a reduction of HADHA protein in HCC tumors (Fig. 6a, b), consistent with the post-transcriptional regulation of UBE2O-mediated ubiquitination on HADHA expression. Furthermore, by analyzing a recent proteomic profiling [30], we found that UBE2O was increased, while HADHA was decreased in HCC tumors compared with normal liver tissues (Fig. 6c, d), and their levels were inversely correlated (Fig. 6e). These results further support that HADHA is regulated post-translationally by UBE2O, and suggest a clinical relevance of UBE2O-HADHA axis in human HCC pathogenesis.

HADHA protein levels in HCC tumors and paired adjacent peritumoral tissues, as measured by western blotting (a) and immunohistochemistry staining (b). Scale bars, 100 μm. The protein levels of UBE2O (c) and HADHA (d), and their correlation (e), in a publicly available HCC proteomic dataset determined by mass spectroscopy (n = 159). Tumor growth rate (f), tumor weight and tumor image (g) of SK-Hep1 tumor xenografts stably expressing either Ctrl or HADHA (n = 6 mice/group). h Representative IHC staining of HADHA and KI67 in SK-Hep1 tumor xenografts. Scale bars, 100 μm. i, j SK-Hep1 cells stably expressing either Ctrl or HADHA were injected into left ventricle of nude mice (n = 6 mice/group). Metastasis was assessed three weeks after the injection. i Number of total metastases in the lung. j Representative pictures of lungs with xenografts (left panel), and H&E staining of the pulmonary metastatic foci (right panel). Arrows indicate metastases. Scale bars, 100 μm. MTS (k) and transwell (l) assays of the indicated HCC cells transfected with pcDNA3.1 empty vectors (Ctrl), UBE2O overexpressing vectors (UBE2O), or UBE2O plus HADHA overexpressing vectors (HADHA). Data are shown as mean ± SEM. *P < 0.05, **P < 0.01, ***P < 0.001, two-tailed Student t test (c, d, g, k and l), two-tailed ANOVA (f), and wilcoxon rank sum test (i).
HADHA suppresses HCC growth and metastasis and predominantly mediates the function of UBE2O on HCC
The implication of HADHA in human cancer, including HCC, is poorly understood. Therefore, we investigated the role of HADHA in HCC development. HADHA knockdown by siRNAs significantly promoted the growth and migration of HCC cells (Supplementary Fig. S5a–c). Reversely, HADHA overexpression led to noticeable decreases in HCC cell growth and migration (Supplementary Fig. S5d–g), confirming a suppressive role of HADHA in HCC in vitro.
Next, the in vivo function of HADHA in HCC growth and metastasis was verified by mouse xenograft models. SK-Hep1 cells stably overexpressing HADHA were injected subcutaneously into nude mice, and tumor growth was monitored. Notably, SK-Hep1 tumor growth was dramatically inhibited by overexpression of HADHA (Fig. 6f, g). Moreover, HADHA-overexpressing tumors had reduced KI67 expression, along with increased HADHA expression (Fig. 6h and Supplementary Fig. S5h). Furthermore, intracardiac metastasis model showed that mice receiving HADHA-overexpressing SK-Hep1 cells exhibited less and smaller lung metastases than control mice (Fig. 6i, j). Collectively, these data suggest that HADHA inhibits HCC growth and metastasis in vivo and in vitro.
Given that UBE2O and HADHA have opposite effects on HCC growth and metastasis, and that UBE2O interacts with and degrades HADHA, we reasoned that HADHA may mediate the impact of UBE2O on HCC. To test it, ectopic expression of HADHA was introduced into UBE2O-overexpressing HCC cells. Strikingly, HADHA overexpression markedly abolished the promotional effects of UBE2O on the proliferation and migration of different HCC cells (Fig. 6k, l and Supplementary Fig. S6a, b). At the molecular level, HADHA overexpression significantly counteracted the effect of UBE2O overexpression on the expression of EMT markers (Supplementary Fig. S6c), highlighting the functional importance of UBE2O-HADHA regulatory axis in HCC progression. These data strongly suggest that HADHA plays an essential role in mediating the oncogenic phenotype of UBE2O in HCC growth and metastasis.
UBE2O promotes lipid metabolism reprogramming of HCC cells through regulating HADHA
HADHA plays an essential role in fatty acid metabolism, whose dysregulation is a well-established cancer hallmark. Therefore, we determined the potential function of UBE2O on lipid metabolism. QPCR analysis showed that crucial lipogenic genes, such as FASN, SCD and SREBF1, were significantly downregulated, while critical regulators of fatty acid oxidation (FAO), including ACOX1, CPT1A and CPT2, were upregulated in UBE2O-deficient Huh7 cells (Fig. 7a). In contrast, overexpression of UBE2O resulted in an opposite expression pattern of these genes (Fig. 7a). Correspondingly, UBE2O-deficient HCC cells exhibited reduced lipid accumulation, as measured by the lipophilic dye Bodipy 493/503 staining (Fig. 7b). Lipid accumulation can increase energy production, which supports cancer cell proliferation and metastasis. In line with this, cellular oxygen consumption rate (OCR) analysis revealed that silencing of UBE2O inhibited ATP generation, basal and maximal respiration prominently (Fig. 7c, d).

a Heat map of mRNA levels of hepatic genes involved in lipid metabolism. Red and blue depict higher and lower gene expression respectively. Color intensity indicates magnitude of expression differences. b Bodipy staining in Huh7 cells transfected with shNC or shUBE2O#2. c Oxygen consumption rate (OCR) of Huh7 cells transfected with shNC or shUBE2O#2. Oligomycin, an inhibitor of ATP synthase (complex V). FCCP, carbonyl cyanide-4 (trifluoromethoxy) phenylhydrazone. Ret/AA, a mixture of rotenone (a complex I inhibitor) and antimycin A (a complex III inhibitor). d ATP generation, basal and maximal respiration of Huh7 cells transfected with shNC or shUBE2O#2. QPCR analysis of UBE2O and HADHA (e), and lipogenic genes (f) in Huh7 cells transfected with indicated plasmids. Bodipy staining (g) and triglyceride content (h) of Huh7 cells transfected with indicated plasmids. i Oxygen consumption rate (OCR) of Huh7 cells transfected with indicated plasmids. j ATP generation, basal and maximal respiration of Huh7 cells transfected with indicated plasmids. Data are shown as mean ± SEM. *P < 0.05, ***P < 0.001, two-tailed Student t test.
We further explored whether UBE2O-mediated lipid reprogramming is mediated by HADHA. HADHA overexpression remarkably counteracted the promotive effects of UBE2O on lipogenic gene expression (Fig. 7e, f), and reversed UBE2O-induced lipid accumulation (Fig. 7g), triglyceride content (Fig. 7h), and mitochondrial respiration including ATP generation, basal and maximal respiration (Fig. 7i, j). Taken together, these results suggest a modulatory role UBE2O-HADHA axis in lipid reprogramming and HCC progression.
It has recently shown that HADHA can repress glucagon-stimulated hepatic gluconeogenesis and is implicated in the treatment of diabetes [31]. Given the close association between lipid metabolism and gluconeogenesis [32], we tested the potential effect of UBE2O axis on gluconeogenesis in HCC. Expressions of gluconeogenic genes FBP1 and G6PC (Supplementary Fig. S7a–d), as well as cellular glucose production (Supplementary Fig. S7e, f), remained unchanged in both UBE2O-deficient and -overexpressing HCC cells. These results suggest a minor effect of UBE2O/HADHA axis on gluconeogenesis in HCC cells, albeit its outcome in vivo awaits further investigation.
Ube2o knockout mice are resistant to DEN-induced hepatocarcinogenesis
Finally, we verified the above findings in primary liver hepatocarcinogenesis. We generated Ube2o knockout mice (Ube2ofl/fl) (Supplementary Fig. S8a, b), which were then crossed with Alb-cre transgenic mice to obtain hepatocyte-specific Ube2o knockout mice (Ube2oAlb−/−) (Supplementary Fig. S8c, d). As expected, the expression of UBE2O was completely abolished in livers of Ube2oAlb−/− mice (Supplementary Fig. S8e), indicating an efficient knockout.
To define the direct role of UBE2O in hepatocarcinogenesis, we next performed our previously established diethylnitrosamine (DEN)-induced HCC model [33], in which Ube2oAlb−/− mice and their littermate controls (Ube2ofl/fl) were injected the mutagen DEN, followed by 8 times of 1,4-Bis-[2-(3,5-dichloropyridyloxy)] benzene (TCPOBOP) intraperitoneally injections every two weeks (Fig. 8a). Notably, Ube2oAlb−/− mice had fewer and smaller tumors on the liver surface, and less liver weight than Ube2ofl/fl mice (Fig. 8b–e), suggesting that Ube2o deficiency are resistant to DEN-induced hepatocarcinogenesis.

a Ube2ofl/fl (WT) and Ube2oAlb−/− (KO) mice were injected with DEN at 6–8 weeks old, then induced chronic liver injury with biweekly TCPOBOP injections for 16 weeks. At 6 months after injection, the mice were sacrificed. b Representative livers of DEN-treated male mice. c Number of visible tumors on liver surface (n = 7 WT and 7 KO mice). d The tumor number (>3 mm) were calculated. e Liver weight and liver-to-body weight ratios (n = 7 WT and 7 KO mice). QPCR (f) and western blotting (g) analysis of HADHA in livers from Ube2ofl/fl and Ube2oAlb−/− mice. h QPCR analysis of hepatic genes involved in lipid metabolism in livers from Ube2ofl/fl and Ube2oAlb−/− mice. i IHC staining of UBE2O, HADHA and FASN in livers from Ube2ofl/fl and Ube2oAlb−/− mice, Scale bars, 100 μm. j H&E and oil red O staining in livers from Ube2ofl/fl and Ube2oAlb−/− mice. Scale bars, 100 μm. Data are shown as mean ± SEM. *P < 0.05, **P < 0.01, two-tailed Student t test (e, f and h), wilcoxon rank sum test (c and d).
We then validated the correlation between the phenotype, HADHA expression, and lipid metabolism reprogramming in the DEN-induced HCC model. The protein levels of HADHA were markedly increased in livers of Ube2oAlb−/− mice, and its mRNA levels were comparable in both genotypes (Fig. 8f, g), consistent with the notion that UBE2O ubiquitinates and degrades HADHA. In line with increased HADHA expression, the lipogenic markers Fasn, Scd1, and Srebf1 were decreased in Ube2oAlb−/− mice, whereas the expression of Cpt1a, a critical regulator promoting FAO, was increased (Fig. 8h, i), indicative of reduced lipogenesis and elevated lipid oxidation respectively. Accordingly, Ube2oAlb−/− mice had less lipid accumulation than their WT littermates (Fig. 8j), as evidenced by both H&E and oil red staining, further supporting the UBE2O/HADHA/lipid reprogramming axis in HCC.
Taken together, these results demonstrate that hepatocyte-specific deletion of Ube2o is sufficient to protect mice from DEN-induced hepatocarcinogenesis, and also suggest the existence of UBE2O/HADHA/lipid reprogramming regulatory axis in the development of murine liver cancer.
Discussion
In this study, we show that UBE2O acts as an oncogene in HCC both in vitro and in vivo. UBE2O, an E2/E3 hybrid enzyme, is upregulated in HCC tumors and positively correlated with poor survival of HCC patients. We demonstrate that UBE2O deficiency inhibits, while its overexpression promotes HCC growth and metastasis. Mechanistically, HADHA is directly targeted by UBE2O for ubiquitination and degradation, which predominantly mediates the effects of UBE2O on HCC, and HADHA reduction results in lipid metabolic reprogramming. More intriguingly, liver-specific deletion of Ube2o in mice is sufficient to prevent lipid metabolic reprogramming and DEN-induced hepatocarcinogenesis. Our findings collectively reveal a critical role of UBE2O-HADHA regulatory axis in liver tumorigenesis and thus provide novel opportunities for treating HCC.
Cancer cells may rely on heightened PQC mechanisms, including the UPS, to support rapid growth [1, 2]. Specifically, elevated ubiquitin immunoreactivity has been observed in HCC tumors, suggesting that an increased capacity of UPS might contribute to hepatocarcinogenesis [16,17,18]. Indeed, numerous UPS components, particularly E3s, have been shown to play diverse roles in hepatocarcinogenesis [34,35,36,37,38]. However, the pathogenic functions of E2s that were traditionally treated as “ubiquitin carriers” on HCC remain largely elusive. In this study, all of 16 E2s that significantly associate with HCC patient survival function as unfavorable prognostic predictors. These observations not only support a potential implication of increased UPS in HCC proteome remodeling, but also suggest E2s as promising druggable targets for treating this malignancy. Of note, UBE2O exhibits the strongest association with poor survival rate of HCC patients, and can significantly enhance HCC initiation and progression both in vitro and in vivo. Interestingly, although UBE2O is indispensable for remodeling the vast proteome during erythropoiesis [8], we found that artificial overexpression or knockdown of UBE2O in HCC cells was not sufficient to result in obvious changes in global ubiquitination and proteome (data not shown). Given the broad association of numerous E2s with HCC, it is possible that multiple E2s might cooperate to remodel the proteome during hepatocarcinogenesis, which awaits future investigation.
UBE2O has been shown to play an important role in several types of cancer through ubiquitinating a variety of substrates. Many of these substrates are ubiquitinated by UBE2O without E3s, consistent with its property as an E2/E3 hybrid enzyme. For instance, UBE2O can directly interact with and ubiquitinate MLL and AMPKα2 in an E3-independent manner, whereby promote the development of leukemia, breast and prostate cancer [9, 12, 39], Alternatively, UBE2O mediates the ubiquitination of WASH and NLRP1B by acting as an E2 [40, 41], thereby regulating endosomal protein trafficking and inflammasome activation, respectively. Here we provided several lines of evidence to delineate the role and underlying mechanism of UBE2O in HCC. First, UBE2O was consistently increased in HCC tumors, and this increase can predict poor survival of HCC patients, establishing a strong link between UBE2O and HCC. Second, UBE2O overexpression or knockdown significantly affected the growth and metastasis of HCC in vitro and in vivo, demonstrating its oncogenic function on hepatocarcinogenesis. Third, a combination of biochemical and functional analyses illustrated that UBE2O can directly interact with and ubiquitinate HADHA at the K129 residue without E3s, leading to proteasomal degradation of HADHA. Fourth, HADHA protein levels were markedly reduced in HCC tumors and exhibited an inverse correlation with UBE2O protein levels, supporting the existence of this regulatory axis in human pathogenesis. Fifth, HADHA can significantly suppress HCC progression, and its overexpression was able to abolish the consequences mediated by UBE2O overexpression. Sixth, we generated liver-specific Ube2o knockout mice and found that its deletion can significantly prevent DEN-induced hepatocarcinogenesis. These results validate for the first time, to our knowledge, the significance of UBE2O in primary HCC progression. During the preparation of this manuscript, Shi et al. showed that UBE2O enhanced HCC proliferation and invasion by ubiquitinating AMPKα2 [42], recapitulating the regulatory axis in other systems reported previously [9, 12, 43]. However, our study failed to observe the interaction between UBE2O and AMPKα2, as well as the negative correlation of their expressions, in HCC cells, suggesting a possible specificity of UBE2O/AMPKα2 axis in certain conditions. Consistent with this notion, Vila et al. recently showed a unique interaction of UBE2O and AMPKα2 in skeletal muscle, but not in adipose tissue and liver [43]. Although we cannot exclude the possibility that the unknown target genes of UBE2O contribute to metabolic reprogramming and HCC development, our combined biochemical, cellular, molecular analyses on HCC cells in vitro and mouse models in vivo strongly suggest HADHA as a major downstream target to mediate the function of UBE2O on hepatocarcinogenesis. Nevertheless, it is of interest for future study to characterize additional substrates of UBE2O and to expand the oncogenic function of UBE2O in HCC by utilizing other animal models, such as the in vivo extravasation assay [44]. Taken together, these findings not only elucidate a role of UBE2O in liver diseases, but also exemplify a novel E2-associated PQC mechanism controlling tumorigenesis.
HADHA encodes the alpha subunit of the mitochondrial trifunctional protein (MTP) and catalyzes the middle two steps of long-chain fatty acids oxidation in mitochondria [45]. Accordingly, HADHA plays an important role in energy metabolism and metabolic homeostasis, and participants in the pathogenesis of human disease, especially metabolic disorder [31, 46, 47]. Recently, the role of HADHA in tumorigenesis is emerging. For example, HADHA is likely to be an oncogene in lymphoma and lung cancer [48, 49], while functions as a tumor suppressor in clear cell renal cell carcinoma [28], albeit the underlying mechanisms are far from elucidated. Specifically, the pathological role of HADHA in liver cancer remains controversial. On the one hand, HADHA was found to be downregulated in HCC [50], and this decrease was positively correlated with the de-differentiation of HCC [29]. More importantly, mice with heterozygous deletion of LCHAD, the mouse synonym for the human HADHA gene, had a much higher risk for autonomous HCC development than WT controls [27], strongly suggesting HADHA as a suppressor for hepatocarcinogenesis. In this study, we provided substantial evidence to demonstrate the suppressive role of HADHA in HCC in vitro and in vivo, consistent with this notion. On the other hand, a recent study showed that HADHA can be directly targeted by microRNA-612 and thus promote HCC metastasis [51]. In this respect, it is of importance for future studies to clarify the precise role and underlying mechanism of HADHA in HCC by using other appropriate models, such as organoids, patient-derived xenograft models, and chemical-induced mouse HCC models.
Notably, UBE2O overexpression resulted in a moderate increase in the proliferation of Huh7 cells in vitro, but it dramatically increased Huh7 xenograft weight and volume in vivo. A similar great discrepancy between the in vitro and in vivo effects was recapitulated in UBE2O-deficient HCC cells. Given that the emerging significance of tumor microenvironment (TME) in tumorigenesis, it is conceivable that TME might contribute to UBE2O-driven HCC growth in vivo. Metabolic reprogramming for adaptation to the TME has been recognized as a hallmark of cancer [52]. Deregulation of lipid metabolism, partially due to alternations in fatty acid synthesis and oxidation, is a crucial determinant of metabolic reprogramming in cancer cells. HADHA, the newly identified substrate of UBE2O, is an important regulator of fatty acid oxidation and energy metabolism in various pathological processes, including HCC. In line with this, our findings in this study suggest that UBE2O can promote lipid accumulation, primarily attributable to HADHA degradation, to meet tumor growth and metastasis. Moreover, HADHA can be acetylated by HDAC3 in macrophages [53], which leads to restricted fatty acid oxidation and in turn modulates inflammation, a hallmark of cancer. Recently, HADHA was reported to inhibit glucagon-stimulated hepatic gluconeogenesis by regulating production of the ketone body β-hydroxybutyrate (BHB) via β-oxidation [31], which suggests HADHA may play a dual role in regulating glucose and lipid energy metabolism. In these circumstances, it is possible that the role of HADHA in various cell types within the TME, as well as their crosstalk, may contribute to the potent in vivo effect of UBE2O. In addition to HADHA, we and others have shown that UBE2O can interact with and target numerous substrates, which could also augment UBE2O’s effects in vivo. Noteworthy, UBE2O mRNAs have been found in exosomes [54], which are emerging as novel mediators of intercellular communication and play diverse roles in diverse pathological processes [32, 55], making it an attractive potential mechanism amplifying the in vivo function of UBE2O. Importantly, the role of UBE2O and its downstream mechanism have been further confirmed in the DEN-induced mouse HCC model.
In summary, this study exemplifies the E2/E3 hybrid enzyme UBE2O as a crucial PQC factor and demonstrates the significance of the new UBE2O-HADHA regulatory axis in HCC growth and metastasis, providing novel therapeutic strategies for HCC.
Materials and methods
Human liver samples
Clinical HCC tumors and normal human liver tissues were harvested from West China Hospital, Sichuan University with informed consent. The study was approved by the Ethics Committee of Sichuan University (WCH/SCU) (2022, no. 1040) and conducted in accordance with the Declaration of Helsinki.
Generation of Ube2o Alb−/− mice
To generate Ube2ofl/fl mice, two loxP fragments, were inserted into the introns downstream of the ATG-containing-exon and the removal of the flanked exon(s) will lead to protein reading frame-shift. After scanning the gene structure and the size of exons, exon 4–18 can be conditionally removed and the deletion of exon 4–18 will result in null protein. Hepatocyte-specific Ube2o knockout (Ube2oAlb−/−) mice were generated by crossing Alb-Cre mice with Ube2ofl/fl mice.
DEN-induced hepatocarcinogenesis
The diethylnitrosamine (DEN)-induced HCC mouse model was conducted as described previously [33, 56]. Briefly, Ube2ofl/fl and Ube2oAlb−/−male mice were randomized and allocated for treatment to avoid cage or injection order effect, and then intraperitoneally injected with DEN (100 mg/kg body weight) at 6–8-week-old. One week later, 1,4-bis[2-(3,5-dichloropyridyloxy)] benzene (TCPOBOP) (3 mg/kg body weight) was injected intraperitoneally for once every two weeks for 8 times. Mice were euthanized 6 months after the DEN injection. The authors who did the experiments were blinded to group allocation during data collection and/or analysis.
Xenograft models
See Supplementary Information.
Cell culture
See Supplementary Information.
Histology, immunohistochemistry and bodipy staining
See Supplementary Information.
Mass spectrometry
Immunoprecipitations were separated by SDS/PAGE. Gel sections were harvested, detained, dehydrated, followed by reductive alkylation, trypsin digestion and desalination. Samples were then loaded on Q Exactive Plus System (ThermoFisher, USA), and data were processed with proteome Discover 2.0 software and searched in associated human protein database.
Immunoprecipitation and in vivo ubiquitination assay
See Supplementary Information.
Seahorse XF mito stress test
The cellular OCR (Oxygen Consumption Rate) was determined using the Seahorse XFp Extracellular Flux Analyzer (Seahorse Bioscience). Experiments were performed according to the manufacturer’s protocols. OCR was examined using Seahorse XF Cell Mito Stress Test Kit. Briefly, 2.0 × 104 cells per well were seeded into a Seahorse XFp cell culture microplate, followed by transfection with indicated plasmids. After baseline measurements, for OCR, oligomycin, the reversible inhibitor of oxidative phosphorylation FCCP (p-trifluoromethoxy carbonyl cyanide phenylhydrazone), and the mitochondrial complex I inhibitor rotenone plus the mitochondrial complex III inhibitor antimycin A (Rotenone/Antimycin) were sequentially injected. Data were assessed by Seahorse XFp Wave software. OCR is shown in pmol/min and normalized to cell counts.
Plasmids construction, transfection and lentivirus transduction
See Supplementary Information.
Cell proliferation, EdU staining and colony formation assay
See Supplementary Information.
Cell migration and invasion assays
Wound-healing assay, transwell and invasion assay were carried out as described previously [24].
RNA extraction and quantitative real-time PCR
See Supplementary Information.
Western blotting
See Supplementary Information.
Statistical analysis
All data represents at least 3 independent experiments and are shown as mean ± SEM. Statistical analysis was performed with GraphPad Prism 8.0. If not mentioned otherwise in the figure legends, statistical significance (*P < 0.05; **P < 0.01; ***P < 0.001) was determined by unpaired, two-tailed Student’s t tests, two-way ANOVA tests or wilcoxon rank sum test where appropriate.
References
Pohl C, Dikic I. Cellular quality control by the ubiquitin-proteasome system and autophagy. Science. 2019;366:818–22.
Deshaies RJ. Proteotoxic crisis, the ubiquitin-proteasome system, and cancer therapy. BMC Biol. 2014;12:94.
Costa-Mattioli M, Walter P. The integrated stress response: from mechanism to disease. Science. 2020;368:eaat5314.
Zheng N, Shabek N. Ubiquitin ligases: structure, function, and regulation. Annu Rev Biochem. 2017;86:129–57.
Stewart MD, Ritterhoff T, Klevit RE, Brzovic PS. E2 enzymes: more than just middle men. Cell Res. 2016;26:423–40.
Osborne HC, Irving E, Forment JV, Schmidt CK. E2 enzymes in genome stability: pulling the strings behind the scenes. Trends Cell Biol. 2021;31:628–43.
Yanagitani K, Juszkiewicz S, Hegde RS. UBE2O is a quality control factor for orphans of multiprotein complexes. Science. 2017;357:472–5.
Nguyen AT, Prado MA, Schmidt PJ, Sendamarai AK, Wilson-Grady JT, Min M, et al. UBE2O remodels the proteome during terminal erythroid differentiation. Science. 2017;357:eaan0218.
Vila IK, Yao Y, Kim G, Xia W, Kim H, Kim SJ, et al. A UBE2O-AMPKalpha2 axis that promotes tumor initiation and progression offers opportunities for therapy. Cancer Cell. 2017;31:208–24.
Faust TB, Li Y, Bacon CW, Jang GM, Weiss A, Jayaraman B, et al. The HIV-1 Tat protein recruits a ubiquitin ligase to reorganize the 7SK snRNP for transcriptional activation. eLife. 2018;7:e31879.
Huang Y, Yang X, Lu Y, Zhao Y, Meng R, Zhang S, et al. UBE2O targets Mxi1 for ubiquitination and degradation to promote lung cancer progression and radioresistance. Cell Death Differ. 2021;28:671–84.
Liu X, Ma F, Liu C, Zhu K, Li W, Xu Y, et al. UBE2O promotes the proliferation, EMT and stemness properties of breast cancer cells through the UBE2O/AMPKalpha2/mTORC1-MYC positive feedback loop. Cell Death Dis. 2020;11:10.
Yokota T, Nagai H, Harada H, Mine N, Terada Y, Fujiwara H, et al. Identification, tissue expression, and chromosomal position of a novel gene encoding human ubiquitin-conjugating enzyme E2-230k. Gene. 2001;267:95–100.
McGlynn KA, Petrick JL, El-Serag HB. Epidemiology of hepatocellular carcinoma. Hepatology. 2021;73:4–13.
Siegel RL, Miller KD, Fuchs HE, Jemal A. Cancer statistics, 2022. CA Cancer J Clin. 2022;72:7–33.
Osada T, Sakamoto M, Nishibori H, Iwaya K, Matsuno Y, Muto T, et al. Increased ubiquitin immunoreactivity in hepatocellular carcinomas and precancerous lesions of the liver. J Hepatol. 1997;26:1266–73.
Lectez B, Migotti R, Lee SY, Ramirez J, Beraza N, Mansfield B, et al. Ubiquitin profiling in liver using a transgenic mouse with biotinylated ubiquitin. J Proteome Res. 2014;13:3016–26.
Shirahashi H, Sakaida I, Terai S, Hironaka K, Kusano N, Okita K. Ubiquitin is a possible new predictive marker for the recurrence of human hepatocellular carcinoma. Liver. 2002;22:413–8.
Uhlen M, Zhang C, Lee S, Sjostedt E, Fagerberg L, Bidkhori G, et al. A pathology atlas of the human cancer transcriptome. Science. 2017;357:aan2507.
Tang Z, Li C, Kang B, Gao G, Li C, Zhang Z. GEPIA: a web server for cancer and normal gene expression profiling and interactive analyses. Nucleic Acids Res. 2017;45:W98–W102.
Villa E, Critelli R, Lei B, Marzocchi G, Camma C, Giannelli G, et al. Neoangiogenesis-related genes are hallmarks of fast-growing hepatocellular carcinomas and worst survival. Results from a prospective study. Gut. 2016;65:861–9.
Lim HY, Sohn I, Deng S, Lee J, Jung SH, Mao M, et al. Prediction of disease-free survival in hepatocellular carcinoma by gene expression profiling. Ann Surg Oncol. 2013;20:3747–53.
Minguez B, Hoshida Y, Villanueva A, Toffanin S, Cabellos L, Thung S, et al. Gene-expression signature of vascular invasion in hepatocellular carcinoma. J Hepatol. 2011;55:1325–31.
Ma M, Xu H, Liu G, Wu J, Li C, Wang X, et al. Metabolism-induced tumor activator 1 (MITA1), an energy stress-inducible long noncoding RNA, promotes hepatocellular carcinoma metastasis. Hepatology. 2019;70:215–30.
Mashtalir N, Daou S, Barbour H, Sen NN, Gagnon J, Hammond-Martel I, et al. Autodeubiquitination protects the tumor suppressor BAP1 from cytoplasmic sequestration mediated by the atypical ubiquitin ligase UBE2O. Mol Cell. 2014;54:392–406.
Chen S, Yang J, Zhang Y, Duan C, Liu Q, Huang Z, et al. Ubiquitin-conjugating enzyme UBE2O regulates cellular clock function by promoting the degradation of the transcription factor BMAL1. J Biol Chem. 2018;293:11296–309.
Khare T, Khare S, Angdisen JJ, Zhang Q, Stuckel A, Mooney BP, et al. Defects in long-chain 3-hydroxy acyl-CoA dehydrogenase lead to hepatocellular carcinoma: a novel etiology of hepatocellular carcinoma. Int J Cancer. 2020;147:1461–73.
Liu S, Liu X, Wu F, Zhang X, Zhang H, Gao D, et al. HADHA overexpression disrupts lipid metabolism and inhibits tumor growth in clear cell renal cell carcinoma. Exp Cell Res. 2019;384:111558.
Tanaka M, Masaki Y, Tanaka K, Miyazaki M, Kato M, Sugimoto R, et al. Reduction of fatty acid oxidation and responses to hypoxia correlate with the progression of de-differentiation in HCC. Mol Med Rep. 2013;7:365–70.
Gao Q, Zhu H, Dong L, Shi W, Chen R, Song Z, et al. Integrated proteogenomic characterization of HBV-related hepatocellular carcinoma. Cell. 2019;179:561–77.
Pan A, Sun XM, Huang FQ, Liu JF, Cai YY, Wu X, et al. The mitochondrial beta-oxidation enzyme HADHA restrains hepatic glucagon response by promoting beta-hydroxybutyrate production. Nat Commun. 2022;13:386.
Liu J, Zhang Y, Tian Y, Huang W, Tong N, Fu X. Integrative biology of extracellular vesicles in diabetes mellitus and diabetic complications. Theranostics. 2022;12:1342–72.
Meng Z, Ma X, Du J, Wang X, He M, Gu Y, et al. CAMK2gamma antagonizes mTORC1 activation during hepatocarcinogenesis. Oncogene. 2017;36:2446–56.
Liu X, Liu J, Xiao W, Zeng Q, Bo H, Zhu Y, et al. SIRT1 regulates N(6) -methyladenosine RNA modification in hepatocarcinogenesis by inducing RANBP2-dependent FTO SUMOylation. Hepatology. 2020;72:2029–50.
Wang J, Wang H, Peters M, Ding N, Ribback S, Utpatel K, et al. Loss of Fbxw7 synergizes with activated Akt signaling to promote c-Myc dependent cholangiocarcinogenesis. J Hepatol. 2019;71:742–52.
Liu Y, Tao S, Liao L, Li Y, Li H, Li Z, et al. TRIM25 promotes the cell survival and growth of hepatocellular carcinoma through targeting Keap1-Nrf2 pathway. Nat Commun. 2020;11:348.
Yu H, Li M, He R, Fang P, Wang Q, Yi Y, et al. Major vault protein promotes hepatocellular carcinoma through targeting interferon regulatory factor 2 and decreasing p53 activity. Hepatology. 2020;72:518–34.
Muto Y, Moroishi T, Ichihara K, Nishiyama M, Shimizu H, Eguchi H, et al. Disruption of FBXL5-mediated cellular iron homeostasis promotes liver carcinogenesis. J Exp Med. 2019;216:950–65.
Liang K, Volk AG, Haug JS, Marshall SA, Woodfin AR, Bartom ET, et al. Therapeutic targeting of MLL degradation pathways in MLL-rearranged leukemia. Cell. 2017;168:59–72.
Hao YH, Doyle JM, Ramanathan S, Gomez TS, Jia D, Xu M, et al. Regulation of WASH-dependent actin polymerization and protein trafficking by ubiquitination. Cell. 2013;152:1051–64.
Xu H, Shi J, Gao H, Liu Y, Yang Z, Shao F, et al. The N-end rule ubiquitin ligase UBR2 mediates NLRP1B inflammasome activation by anthrax lethal toxin. EMBO J. 2019;38:e101996.
Shi Z, Liu R, Lu Q, Zeng Z, Liu Y, Zhao J, et al. UBE2O promotes hepatocellular carcinoma cell proliferation and invasion by regulating the AMPKalpha2/mTOR pathway. Int J Med Sci. 2021;18:3749–58.
Vila IK, Park MK, Setijono SR, Yao Y, Kim H, Badin PM, et al. A muscle-specific UBE2O/AMPKalpha2 axis promotes insulin resistance and metabolic syndrome in obesity. JCI Insight. 2019;4:e128269.
Kim Y, Williams KC, Gavin CT, Jardine E, Chambers AF, Leong HS. Quantification of cancer cell extravasation in vivo. Nat Protoc. 2016;11:937–48.
Xia C, Fu Z, Battaile KP, Kim JP. Crystal structure of human mitochondrial trifunctional protein, a fatty acid beta-oxidation metabolon. Proc Natl Acad Sci USA. 2019;116:6069–74.
Ljubkovic M, Gressette M, Bulat C, Cavar M, Bakovic D, Fabijanic D, et al. Disturbed fatty acid oxidation, endoplasmic reticulum stress, and apoptosis in left ventricle of patients with type 2 diabetes. Diabetes. 2019;68:1924–33.
Miklas JW, Clark E, Levy S, Detraux D, Leonard A, Beussman K, et al. TFPa/HADHA is required for fatty acid beta-oxidation and cardiolipin re-modeling in human cardiomyocytes. Nat Commun. 2019;10:4671.
Yamamoto K, Abe S, Honda A, Hashimoto J, Aizawa Y, Ishibashi S, et al. Fatty acid beta oxidation enzyme HADHA is a novel potential therapeutic target in malignant lymphoma. Lab Invest. 2020;100:353–62.
Amoedo ND, Sarlak S, Obre E, Esteves P, Begueret H, Kieffer Y, et al. Targeting the mitochondrial trifunctional protein restrains tumor growth in oxidative lung carcinomas. J Clin Invest. 2021;131:e133081.
Yokoyama Y, Kuramitsu Y, Takashima M, Iizuka N, Toda T, Terai S, et al. Proteomic profiling of proteins decreased in hepatocellular carcinoma from patients infected with hepatitis C virus. Proteomics. 2004;4:2111–6.
Liu Y, Lu LL, Wen D, Liu DL, Dong LL, Gao DM, et al. MiR-612 regulates invadopodia of hepatocellular carcinoma by HADHA-mediated lipid reprogramming. J Hematol Oncol. 2020;13:12.
Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–74.
Chi Z, Chen S, Xu T, Zhen W, Yu W, Jiang D, et al. Histone deacetylase 3 couples mitochondria to drive IL-1beta-dependent inflammation by configuring fatty acid oxidation. Mol Cell. 2020;80:43–58.
Mi B, Chen L, Xiong Y, Yan C, Xue H, Panayi AC, et al. Saliva exosomes-derived UBE2O mRNA promotes angiogenesis in cutaneous wounds by targeting SMAD6. J Nanobiotechnology. 2020;18:68.
Xu H, Du X, Xu J, Zhang Y, Tian Y, Liu G, et al. Pancreatic beta cell microRNA-26a alleviates type 2 diabetes by improving peripheral insulin sensitivity and preserving beta cell function. PLoS Biol. 2020;18:e3000603.
Tian Y, Zhang M, Fan M, Xu H, Wu S, Zou S, et al. A miRNA-mediated attenuation of hepatocarcinogenesis in both hepatocytes and Kupffer cells. Mol Ther Nucleic Acids. 2022;30:1–12.
Acknowledgements
We thank Dr. Hongbo Hu for kindly providing WT and mutant ubiquitin-overexpression plasmids.
Funding
This work was supported by the National Natural Science Foundation of China (82103128, 92157205, 81970561, and 81802836), the National Key Research and Development Program of China (2018YFC2000305), the Ministry of Science and Technology of China (2018ZX09201018-005), China National Postdoctoral Program for Innovative Talents (BX2021201), China Postdoctoral Science Foundation (2021M692302), the 1.3.5 Project for Disciplines Excellence, West China Hospital, Sichuan University (ZYJC18049), Sichuan Science & Technology Program (2019JDTD0013), and the Post-Doctor Research Project, West China Hospital, Sichuan University (2020HXBH121).
Author information
Authors and Affiliations
Contributions
XF and YT conceived the project. MM and CZ designed the experiments and analyzed the data. RC, DT, XS, SZ, XW, HX and GL performed the experiments, in part, and analyzed the data. LD helped perform the analysis with constructive discussions. XF, YT and XG supervised the study. XF, YT and MM wrote and revised the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Ma, M., Zhang, C., Cao, R. et al. UBE2O promotes lipid metabolic reprogramming and liver cancer progression by mediating HADHA ubiquitination. Oncogene 41, 5199–5213 (2022). https://doi.org/10.1038/s41388-022-02509-1
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41388-022-02509-1
- Springer Nature Limited
This article is cited by
-
UBE2O reduces the effectiveness of interferon-α via degradation of IFIT3 in hepatocellular carcinoma
Cell Death & Disease (2023)