
Abstract
Developments of many renal diseases are substantially influenced by epigenetic modifications of numerous genes, mainly mediated by DNA methylations, histone modifications, and microRNA interference; however, not all gene modifications causally affect the disease onset or progression. Klotho is a critical gene whose repressions in various pathological conditions reportedly involve epigenetic regulatory mechanisms. Klotho is almost unexceptionally repressed early after acute or chronic renal injuries and its levels inversely correlated with the disease progression and severity. Moreover, the strategies of Klotho derepression via epigenetic modulations beneficially change the pathological courses both in vitro and in vivo. Hence, Klotho is not only considered a biomarker of the renal disease but also a potential or even an ideal target of therapeutic epigenetic intervention. Here, we summarize and discuss studies that investigate the Klotho repression and intervention in renal diseases from an epigenetic point of view. These information might shed new sights into the effective therapeutic strategies to prevent and treat various renal disorders.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Klotho (α-Klotho) is a kidney-enriched membrane protein accidently discovered from mice of gene-targeting in 1997 [1]. Mice with Klotho gene knockout display systemic human age-sensitive traits, whereas overexpression or exogenous supplementation of Klotho extends mouse lifespan [1, 2], suggesting that Klotho protein functions primarily as an aging suppressor in mammals. Later, two other paralogs β-Klotho and γ-Klotho were identified based on their homologies to α-Klotho, but less studied [3, 4]. This review will discuss the evidence of epigenetic modifications of α-Klotho, briefly Klotho, in kidney diseases.
Klotho is predominantly expressed in kidney distal tubule, parathyroid gland, and brain choroid plexus [1, 5]. Klotho exists in two general forms, a type 1 transmembrane protein and a secreted form derived from the same gene by alternative mRNA splicing [6]. The membrane-bound Klotho is known to serve as an obligatory co-receptor for fibroblast growth factor 23 (FGF23), through which it plays a critical role in the maintenance of mineral ion and vitamin D homeostasis [7, 8]. The extracellular domain of membrane Klotho consists of two repeated Klotho domains Kl1 and Kl2 that can be cleaved by metalloproteinases and released into blood, urine, and cerebrospinal fluid [9,10,11], whereby regulating the functions of renal and extrarenal organs. The cleaved KL fragments together with secreted Klotho are jointly called soluble Klotho, which are structurally homologous to family 1 glycosidase and possess purported intrinsic glycosidase activities [12]. Klotho beneficially regulates various cellular processes including aging, renal ion transport, oxidative stress, fibrosis, inflammation, autophagy, and apoptosis by targeting various signaling molecules, cell membrane receptors and ion channels via physical interactions or its enzymatic activities [13, 14]. Hence, Klotho is considered a key gene controlling aging and renal homeostasis and an ideal target of intervention for a number of kidney diseases and the extrarenal complications.
One of the critical features of Klotho expression is that its level drastically declined early in response to almost all renal damaging stimuli under various acute or chronic pathological conditions [15, 16]. Accumulating evidence indicates that acute Klotho repressions can be regulated by epigenetic or non-epigenetic mechanisms [17], but its sustained suppressions in chronic renal disorders mainly involve epigenetic modifications [18]. Epigenetic modifications generally refer to DNA methylation, protein/histone acetylation, and miRNA interference that influence many physiological and pathological processes such as embryo development, aging, carcinogenesis, and various chronic or degenerative disorders [19,20,21]. Epigenetic involvements in renal diseases have been evidenced by genome-wide association studies (GWASs), gene-linkage studies, and epigenome analysis [22,23,24]. Because epigenetic modifications occur in a tissue, cell, or gene-specific manner and epigenetic drugs can reversibly regulate the modifications [25], it is appealing that identification of the key genes of modifications and understanding the underlying regulatory mechanisms might lead to clinical benefits.
Klotho expression regulations by epigenetic or non-epigenetic mechanisms have attracted tremendous research attentions, and its epigenetic regulations also involve other pathological conditions, such as aging, carcinogenesis, and chronic disorders of other organs. This review will only focus on its epigenetic regulations in kidney diseases.
DNA methylation modification of Klotho in kidney diseases
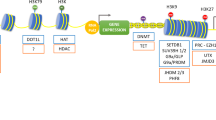
DNA methylation is a process that adds methyl (–CH3) groups onto the C5 position of cytosine to form 5-methylcytosine on cytosine-phosphate-guanine (CpG) dinucleotide [26]. The reaction is catalyzed by maintenance DNA methyltransferase DNMT1 that copies DNA methylation pattern from a parental DNA strand onto newly synthesized daughter strand during DNA replication, and by de novo DNA methyltransferase DNMT3a and DNMT3b that are responsible for the establishment of a new methylation pattern [27]. Conversely, DNA demethylation is processed by Ten-Eleven translocation family protein TET1, TET2, and TET3 in sequential 5-hydroxymethylcytosine (5-hmC), 5-formylcytosine (5-fC), and 5-carboxylcytosine (5-caC) steps and eventually finished via a base excision repair procedure [28,29,30]. The methylated cysteine located in CpG island of gene promoters or enhancers can recruit methyl-CpG binding domain (MBD) proteins, histone deacetylases (HDACs), and/or polycomb group (PcG) proteins that form a transcriptional repressor complex and actively silence the downstream gene transcription [31].
Klotho promoter lacks ordinary TATA box but contains evolutionally conserved CpG islands, providing a structural basis for DNA methylation modification [32,33,34,35]. Klotho promoter hypermethylation and the associated Klotho repression have been found in several renal diseases of animal models [36,37,38] and in renal patients [39, 40].
Acute kidney injury
AKI (acute kidney injury) may reflect a transient deficiency of renal Klotho, whose reduction might be an important pathological feature of AKI development and progression to CKD [17, 41, 42]. Intriguingly, the Klotho levels are not alike in all forms of AKI. Urinary Klotho levels were significantly lower in patients with prerenal AKI than that of intrinsic AKI [43], suggesting that the level of Klotho varies under different acute pathological conditions.
It is likely that Klotho suppression is regulated in part by DNA methylation modification during the progression of AKI to CKD. Some demethylating agents such as hydrogen-rich saline is protective against AKI via correcting aberrant DNA methylation of Klotho gene, thus retaining Klotho expression [44]. We found that LPS-induced AKI was effectively alleviated by rhein, a small compound isolated from medicinal plants, via Klotho restoration and subsequent inhibition of TLR4 and the downstream NF-κB signaling [45], which was likely due to Klotho promoter demethylation since rhein possesses DNA demethylating capacity in kidney [38, 46]. It was reported that in LPS triggered AKI, NF-κB interacted with PRMT6 (protein arginine methyltransferase 6) that suppressed Klotho expression, which could be restored by AMI-1, an inhibitor of protein arginine N-methyltransferase and demethylating agent 5-Aza-2′-deoxycytidine (decitabine) [47], suggesting that both protein and DNA methylation aberrations are involved in the process. Notably, use of DNA demethylating agents such as decitabine to treat AKI has produced inconsistent results. Decitabine prevented cisplatin-induce nephrotoxicity in rats [48], but another study reported that decitabine enhanced cisplatin-induced apoptosis of proximal tubular cells and mice with renal proximal tubule-specific DNMT1 knockout had more serious AKI after cisplatin treatment [49]. The reason for the discrepancy is currently unclear.
CKD and the extrarenal complications
Chronic kidney disease (CKD) is a systemic disorder of chronic and progressive renal damage in which Klotho loss is considered a biomarker of its progression and severity [50]. Epigenetic Klotho repression due to aberrant DNA methylation is likely a causal event that promotes and expedites the disease course [51]. Renal tissues of CKD animals and blood cells from CKD patients showed marked increases of Klotho promoter methylation that correlated with the Klotho loss. In an animal model of uremic toxin-induced CKD, accumulation of uremic toxins can induce abnormal DNMT expression and Klotho promoter hypermethylation, leading to subsequent Klotho suppression [52]. In particular, indoxyl sulfate (IS) and p-cresyl sulfate (PCS), two protein-bound uremic toxins, caused CKD by increasing the expression of DNMT 1, DNMT3a, and DNMT3b, Klotho promoter hypermethylation, and Klotho repression in the mouse kidney [53]. IS also increased Klotho promoter methylation in vascular smooth muscle cells, exacerbating vascular calcification in the CKD mice [54]. Similarly, urinary cadmium exposure enhanced Klotho promoter methylation to adversely influence liver and kidney functions [40, 55]. These results indicate that DNMT aberration-mediated promoter hypermethylation limits Klotho expression, thus exacerbating CKD pathologies. In addition, persistent inflammation and homocysteinemia in CKD is associated with DNA hypermethylation [56,57,58], but whether these processes involve Klotho promoter hypermethylation needs further investigation.
Emerging evidence indicates that endogenous Klotho restorations via demethylating agents are beneficial for CKD animals. Downregulation of Klotho in uremic toxin-induced CKD could be reverted by demethylating agent decitabine [52,53,54]. Rosiglitazone, an antagonist of peroxisome proliferator-activated receptor γ (PPARγ), increased Klotho expression and protected against high phosphate-induced vascular calcification in CKD mice, which might involve reversal of MeCP2 (methyl-CpG binding protein 2) abnormal expression [59]. Several extrarenal CKD complications such as soft-tissue and cardiovascular calcifications, hypertension, cardiomyopathy, disturbed mineral metabolism, and bone injuries could be partially ameliorated via Klotho recovery by demethylating drugs [38, 54, 60, 61]. In studies of CKD mice with mineral and bone disorder (CKD-MBD) incurred by adenine feeding, we observed that aberrant DNMT1/DNMT3a elevations, Klotho promoter hypermethylation, and Klotho suppression were reversed by rhein, a chemical compound isolated from medicinal herb, resulting in improved renal and bone pathologies [38] (Fig. 1), supporting that some components from medicinal plants or other natural sources possess strong epigenetic modulating capacities.

A diagram illustrating DNMT1/3a aberrations, Klotho promoter hypermethylation, and subsequent Klotho repression as well as rhein intervention in adenine-incurred CKD/MBD mice
Renal fibrosis
Renal fibrosis is a common pathohistological feature of CKD independent of the underlying etiologies. Its development is characterized by activation of myofibroblast trans-differentiation (MTD) and excessive extracellular matrix (ECM) deposition [62], the processes that are profoundly affected by aberrant DNA methylation modifications [63].
As demonstrated by several studies, Klotho deficiency potentiates renal fibrosis in animal models [64]. Consistently, Klotho reportedly suppressed basic fibroblast growth factor-2 signaling and inhibited TGFβ (transforming growth factor-beta)/Smad and Wnt/β-catenin signaling to relieve EMT (epithelial–mesenchymal transition) and myofibroblast activation, and subsequently improved renal fibrotic lesions [65,66,67,68]. Klotho inhibitions of oxidative stress and excessive inflammation also contribute to the anti-fibrotic activities [69, 70]. Administrations of decitabine or medicinal plant–derived products or “epigenetic diets” including rhein, genistein, or curcumin attenuated renal fibrosis by demethylating Klotho promoter. In particular, rhein and genistein were capable of inhibiting aberrant DNMT1/3a elevations, resulting in Klotho restoration and reduced renal fibrosis in UUO (unilateral ureteral obstruction) mice [36, 37, 46, 71]. In mice with renal fibrosis after IRI, hydrogen-rich saline retained Klotho expression and reduced renal fibrotic damage likely via demethylating Klotho promoter [44]. These studies suggest that TGFβ-induced DNMT1/3a aberrations are responsible for the Klotho promoter hypermethylation and Klotho suppression in renal fibrotic mice [37].
TET-mediated hydroxymethylation also plays an important role to induce DNA demethylation on Klotho promoter. Accordingly, High-fidelity CRISPR/Cas9-based gene-specific hydroxymethylation by TET3 catalytic domain was shown to rescue Klotho repression and attenuate renal fibrosis [72]. Hydrogen sulfide also attenuates renal fibrosis induced by hypoxia microenvironment by inducing TET-dependent DNA demethylation on Klotho promoter [73]. Those results suggest that TET family proteins can be potential therapeutic targets for reversing Klotho fibrotic suppression.
Diabetic nephropathy
Diabetic nephropathy (DN) is a main cause of CKD [74] and Klotho repressions are observed in DN animals [75]. On the other hand, exogenous Klotho supplementations protected against DN by attenuating glomerular inflammation, fibrosis, oxidative stress, albuminuric activity, and abnormal lipid metabolisms [76,77,78,79]. Moreover, epigallocatechin-3-gallate, a green tea extract, reduced DNMT3a binding to Klotho promoter and hypomethylated the promoter, leading to improved DN pathologies [80]. Similarly, administration of baicalin, a flavone glycoside, alleviated the renal injury of DN partially through modulating Klotho promoter methylation [81].
Kidney transplantation
Kidney transplantation is an optimal choice for treatment of patients with the end-stage renal disease (ESRD). Post-transplant ischemic AKI secondary to ischemia reperfusion injury (IRI) is a major problem that affects graft and patient survivals [82]. As seen in AKI, Klotho is also suppressed in patients with kidney transplantation [83]. Patients who experienced delayed graft function were found to have significant lower serum Klotho levels compared with the control group. IRI induces excessive inflammatory responses and oxidative stress [84] that might contribute to the Klotho suppression through aberrant DNA methylation [41, 82]. Clinical studies demonstrated that kidney transplant recipients showed Klotho promoter hypomethylation and Klotho recovery after paricalcitol treatment, which beneficially affected the clinical course [82, 85], suggesting that kidney transplant recipients might benefit from endogenous Klotho restoration via DNA methylation intervention.
It seems that aberrant DNMT elevations are the major causes of most renal diseases reported since DNMT inhibitions by various strategies of DNA demethylation effectively reduced the renal pathologies. It should be also emphasized that DNMTs are not the only factors dictating the methylation status of Klotho promoter. The abnormal expressions of other DNA methylation regulatory proteins such as DNA insulator protein, methylated DNA binding protein, and transcriptional repressors likely play significant roles in the epigenetic Klotho repression, which are important but less explored areas of research.
Histone acetylation modulation of Klotho expression in kidney diseases
Post-translational protein acetylation is another epigenetic regulatory mode that modulates chromatin structure and transcriptional status to affect gene expression. Typically, histone acetylation is regulated by two groups of enzymes of opposite functions, namely histone acetyl-transferases (HAT) and histone deacetylases (HDAC), that consist of at least 4 major classes of 18 members, namely class I (HDAC1, 2, 3, 8), class II (IIa 4, 5, 7, 9, and IIb 6,10), class III (SIRT1–7), and class IV (HDAC11) [86]. Histone acetylation reduces chromatin condensation, allowing access of transcription factors and facilitating gene transcription. On the contrary, HDACs remove the acetyl groups from lysine on the core histone tails and restore the positive charge of lysine, resulting in chromatin compaction and gene transcription inhibition [87]. Comparing to the extensive investigations of histone/protein acetylations and the therapeutic application of HDAC inhibitors in cancer research [88], the protein acetylation modifications in renal diseases are less explored; however, some interesting data are emerging.
Acute kidney injury
In the setting of AKI induced by folic acid, HDAC1 and HDAC2 were aberrantly elevated, which physically interacted with NF-κB, leading to transcriptional downregulation of Klotho [17], whereas HDAC inhibitor trichostatin A and valproate inhibited the downregulation and improved AKI [17, 56]. A number of studies have proposed that persistent inflammation is increasingly recognized as an important determinant factor for AKI progression to CKD [89]. Thus, restoration of Klotho through HDAC inhibitors is considered as potential therapeutic intervention to mitigate the progression of AKI-to-CKD transition [90].
CKD and the extrarenal complications
The specific roles of HDACs in CKD progression are only incompletely understood. It has been reported that Klotho preservation via HDAC inhibition could attenuate CKD and the associated bone injury [91, 92]. In CKD/MBD model of adenine-fed mice, a non-selective HDAC inhibitor trichostatin A (TSA) increased acetylation of PPARγ and PPARγ-dependently alleviated Klotho loss and kidney injury [92]. Moreover, renal HDAC3 was preferentially upregulated in the mouse kidney and a HDAC3-selective inhibitor RGFP966 extent-similarly alleviated Klotho loss and renal injuries as TSA, suggesting that increased HDAC3 is a major driving force for the pathological effects [92].
CKD is often associated with cardiovascular complications [93]. Activation of SIRT1 by SRT1720, a specific SIRT1 activator, could attenuate Klotho deficiency-induced arterial stiffness and hypertension [94]. Similarly, resveratrol, a known SIRT1 activator, can upregulate Klotho or even reduce vascular calcification in ESRD animals likely through correcting aberrant SIRT1 activities [95, 96].
Renal fibrosis
Aberrant HDAC activities are observed in almost all animal models of renal fibrosis [97, 98]. Various pan- and class-selective inhibitors of histone deacetylase (HDAC) exhibit impressive anti-renal fibrosis properties [99,100,101,102,103,104,105,106,107]. In the fibrotic kidneys of various animal models, Klotho is markedly depressed due to, at least in a significant part, the aberrant HDAC activities. HDAC inhibitions by known HDAC inhibitor TSA and genistein from soy products with epigenetic modulating capacity displayed anti-renal fibrosis activities by recovering histone acetylation-associated Klotho loss [36, 92]. MC1568, a selective class IIa HDAC inhibitor, increased renal expression of Klotho in UUO mice [103]. Similarly, selective inhibitions of HDAC8 by either a HDAC8-selective inhibitor PCI34051or siRNA interference were reportedly effective in inhibiting multiple profibrotic signaling with concomitant Klotho recovery and renal fibrosis mitigation [108]. Conversely, aldosterone-induced renal fibrosis was accompanied by HDAC1-incurred H3K9 deacetylation and Klotho transcriptional repression [109].
These studies demonstrate that fibrotic Klotho repressions are associated with aberrant expressions or activities of multiple HDAC isoforms; however, the key HDAC isoforms that are causally involved in Klotho repression during renal fibrogenesis might need confirmation by gene-specific deletion investigations. We have recently showed that genomic HDAC3 gene knockout exacerbated renal fibrosis in UUO mice. HDAC3 was aberrantly and preferentially elevated in UUO kidney, likely induced by TGFβ. The upregulated HDAC3 formed a transcriptional repressor complex with NcoR and NF-κB that inhibited Klotho transcription. Conversely, HDAC3-selective inhibitor RGFP966 derepressed Klotho and Klotho dependently mitigated the renal fibrotic damage, suggesting that HDAC3 aberration and the associated Klotho suppuration play essential roles in renal fibrotic pathologies [110] (Fig. 2).

A schematic of sequential regulations of TGFβ-incurred HDAC3 aberration, Klotho transcription inhibition, and renal fibrogenesis, as well as the epigenetic intervention by HDAC3-selective inhibitor
Past research studies have accumulated ample evidence indicating that aberrant protein acetylation alterations due to increased HADC expression or activity contribute significantly to the development of kidney diseases. It is likely that different HDAC isoforms and mediators function at different disease stages of different pathological processes. Identifications of the causal HDAC isoforms and the key mediators causally involved in the processes are the key to fully understand the precise underlying mechanisms.
miRNA modifications of Klotho expression in kidney diseases
Micro-RNA (miRNA) interference represents an additional layer of epigenetic gene expression regulation. MiRNAs are endogenous short non-coding RNAs of 22–25 base pairs that regulate gene expression through post-transcriptional repression of target mRNAs [111, 112]. Generally, miRNA binds to the 3′-untranslational region (UTR) of a target mRNA through base-pairing mechanism to suppress target gene expression by either inhibiting protein translation or mRNA degradation [112]. Of note, some novel class of RNA regulators for gene expression including long non-coding RNAs (lncRNAs) and circular RNAs (circRNAs) have been recently discovered through functional genomics studies [113, 114], adding the complexity of this epigenetic regulatory scheme.
The interplay of miRNAs and Klotho has been extensively investigated in cancer studies [115]. It turned out that Klotho is a target of a diversity of miRNAs as demonstrated by renal cell [116,117,118] and animal studies [37, 119,120,121,122,123,124,125,126,127] (see Table 1), suggesting that miRNAs might play essential roles in the expression of Klotho in renal diseases.
AKI
The information regarding miRNA modulation of Klotho in AKI is increasing. Upregulation of miR-29a and miR-34a in mesenchymal stromal cells might act synergistically with Klotho to prevent IRI-induced AKI.[119]. miR-130a promoted PI3K/AKT pathway but inhibited Wnt and NF-κB pathways through upregulation of Klotho to protect against LPS-induced glomerular cell injury [120]. EVs isolated from normal urine transferred their microRNA cargo containing miR-30s/miR-151 to kidney, reduced the endogenous Klotho loss, and exhibited renal protection in a murine model of acute injury generated by glycerol injection [121]. These studies demonstrate that miRNA inhibition of Klotho expression plays critical roles in AKI development and could be targeted for AKI intervention.
CKD and its extrarenal complications
The information regarding altered miRNA regulation of CKD is limited. The evidence for the important role of miRNAs in the physiological regulation of parathyroid function and its dysregulation in the secondary hyperparathyroidism in CKD are recently reported [122, 128]. The studies showed that many miRNAs were aberrantly expressed in experimental uremic hyperparathyroidism, and inhibition of let-7 family increased parathyroid hormone (PTH) secretion in normal and uremic rats. Conversely, inhibition of the upregulated miRNA-148 family blocked the increase of serum PTH in uremic rats, suggesting that miRNA dysregulation represents a crucial step in the pathogenesis of secondary hyperparathyroidism in CKD.
Renal fibrosis
MiRNA aberrations potentially affect renal fibrosis [129]. MiRNA-34a was shown to promote renal fibrosis by directly downregulating Klotho in tubular epithelial cells [123]. On the other hand, miR-152 and miR-30a inhibited by TGFβ can indirectly regulate Klotho expression via targeting DNMT1 and DNMT3a, respectively, leading to Klotho promoter hypomethylation and Klotho recovery in UUO mice, which might serve as a regulatory loop mediating TGFβ’s profibrotic activities [37, 130].
Diabetic nephropathy
Diabetic nephropathy (DN) is a main cause of CKD [74] and Klotho repressions are observed in diabetic nephropathy [75], in which altered miRNA expression might be a causal factor. In particular, miRNA-199a-5p functioned as a key mediator of Klotho expression in DN. In renal cell assays, high glucose reportedly increased miRNA-199a-5p expression accompanied by significant decrease of Klotho at both mRNA and protein levels. High glucose also activated NF-κB signaling and promoted fibrotic and inflammatory reactions, which could be restrained by inhibition of miR-199a-5p or exogenous addition of Klotho [124]. Likewise, miRNA-199a-5p from HK-2 cell-derived extracellular vesicles (EVs) induced macrophage M1 polarization by targeting the Klotho/TLR4 signaling pathway and further accelerated DN development [125]. In agreement with these, a selective endothelin-A receptor antagonist atrasentan increased Klotho expression by lowering miRNA-199b-5p and prevented renal tubular injury of DN, and overexpression of miR-199b-5p disrupted the influences of atrasentan on Klotho expression and apoptosis of renal tubular cells both in vivo and in vitro [126], suggesting that miRNA-199a-5p might represent a potential target for DN therapeutic intervention.
Nephritis
Lupus nephritis (LN) is considered a life-threatening complication of systemic lupus erythematosus [131] and characterized by exaggerated inflammation and fibrosis progression [132]. As a crucial pathway involved, NF-κB activation is closely related to the initiation and progression of LN through the transcriptional regulation of pro-inflammatory cytokines [133, 134]. Several studies suggested that miRNAs adversely regulated Klotho expression, NF-κB signaling activation, and inflammatory cytokine expression during LN progression [120, 127]. MiR-199a reportedly activated NF-κB signaling and promoted TNF-α and IL-1β expressions by targeting Klotho directly, explaining in part the lupus nephritis pathogenesis [127]. Therefore, Klotho restoration through miRNA intervention is considered a promising treatment option for treatment of nephritis.
Although studies on miRNAs and kidney diseases are continuously expanding, it is noteworthy that analysis of human and mouse Klotho coding regions by miDBD online software (http://mirdb.org) predicts 130 and 72 putative miRNA binding sites, respectively. MiRNA lacks specificity. A single miRNA can regulate many target genes and a single mRNA can be targeted by multiple miRNAs [135]. Therefore, when translating the results from cell and animal assays to a particular clinical setting, the miRNA specificity and the off-target effects on other target gene expressions have to be carefully evaluated.
Other epigenetic modifications of Klotho expression in kidney diseases
Other less-studied epigenetic modifications also affect Klotho expressions in kidney diseases, although the evidence is sparse. A recent study reported that Klotho mRNA was hypermethylated in IS-induced calcified arteries of CKD mice that was mediated by Mettl14 (methyltransferase-like 14) overexpression [136]. Blocking the histone3/lysine 79 methyltransferase DOT1L (disruptor of telomeric silencing-1 like) alleviated renal fibrosis through recovering Klotho loss [137]. H3K27me3 level was increased in kidneys of aged WT and Klotho mutant mice likely due to downregulation of the H3K27 (histone H3 Lys 27)-specific demethylase JMJD3 (the Jumonji domain containing-3). Inhibition of PRC2 (polycomb repressive complex C2; histone trimethyltransferase) decreased the H3K27me3 levels, leading to increased expression of Klotho in cultured primary renal tubule cells [138]. Notably, lncRNA MALAT1 could mediate high glucose-induced glomerular endothelial injury by epigenetically inhibiting Klotho via methyltransferase G9a [139]. It is expected that future investigations of new epigenetic Klotho regulations will bring more details of the molecular mechanisms of Klotho expressions that potentially lead clinical benefits.
Summary and perspectives
Current evidence supports that restoration of endogenous Klotho by epigenetic intervention represents a new direction of therapeutic approaches for kidney disease treatment. Up to now, at least four pan- or class HDAC inhibitors, namely vorinostat, romidepsin, belinostat, and panobinostat, and one demethylating agent decitabine are approved by the US Food and Drug Administration for treating cutaneous and peripheral T-cell lymphomas [140], and myelodysplastic syndromes (MDS) and acute myeloid leukemia (AML), respectively, making the applications easier in treatment of kidney diseases. However, long-term uses of synthetic epigenetic drugs inhibiting all HDAC and DNMT activities are potentially cytotoxic and might cause intolerable side effects in certain patients [141]. Moreover, epigenetic modifications are often mechanistically connected. For example, DNMT and HDAC expressions are regulated by miRNAs, which in turn are subjected to DNA methylation and protein acetylation regulations. Methylated CpGs on gene promoter are recognized and bound by methyl-binding proteins complexed with transcription co-repressors and HDACs to facilitate the gene transcriptional silencing [142]. Therefore, a combination of epigenetic drugs targeting multiple epigenetic alterations might lower the drug dosage and incur fewer side effects. In addition, recent studies have demonstrated that many components from natural food products or medicinal plants display previously unrecognized epigenetic modulating capacities with tolerable side effects. It is anticipated that future research identifying the key causal factors and dissecting the contribution of each epigenetic modification to Klotho expression will provide more effective prophylactic and therapeutic options for the managements of various kidney diseases.
References
Kuro-o M, Matsumura Y, Aizawa H, Kawaguchi H, Suga T, Utsugi T, Ohyama Y, Kurabayashi M, Kaname T, Kume E et al (1997) Mutation of the mouse klotho gene leads to a syndrome resembling ageing. Nature 390:45–51
Kurosu H, Yamamoto M, Clark JD, Pastor JV, Nandi A, Gurnani P, McGuinness OP, Chikuda H, Yamaguchi M, Kawaguchi H et al (2005) Suppression of aging in mice by the hormone Klotho. Science 309:1829–1833
Ito S, Fujimori T, Hayashizaki Y, Nabeshima Y (2002) Identification of a novel mouse membrane-bound family 1 glycosidase-like protein, which carries an atypical active site structure. Biochim Biophys Acta 1576:341–345
Ito S, Kinoshita S, Shiraishi N, Nakagawa S, Sekine S, Fujimori T, Nabeshima YI (2000) Molecular cloning and expression analyses of mouse betaklotho, which encodes a novel Klotho family protein. Mech Dev 98:115–119
Li SA, Watanabe M, Yamada H, Nagai A, Kinuta M, Takei K (2004) Immunohistochemical localization of Klotho protein in brain, kidney, and reproductive organs of mice. Cell Struct Funct 29:91–99
Xu Y, Sun Z (2015) Molecular basis of Klotho: from gene to function in aging. Endocr Rev 36:174–193
Lu X, Hu MC (2017) Klotho/FGF23 axis in chronic kidney disease and cardiovascular disease. Kidney Dis (Basel) 3:15–23
Erben RG (2016) Update on FGF23 and Klotho signaling. Mol Cell Endocrinol 432:56–65
Imura A, Iwano A, Tohyama O, Tsuji Y, Nozaki K, Hashimoto N, Fujimori T, Nabeshima Y (2004) Secreted Klotho protein in sera and CSF: implication for post-translational cleavage in release of Klotho protein from cell membrane. FEBS Lett 565:143–147
Akimoto T, Yoshizawa H, Watanabe Y, Numata A, Yamazaki T, Takeshima E, Iwazu K, Komada T, Otani N, Morishita Y et al (2012) Characteristics of urinary and serum soluble Klotho protein in patients with different degrees of chronic kidney disease. BMC Nephrol 13:155
Lim K, Groen A, Molostvov G, Lu T, Lilley KS, Snead D, James S, Wilkinson IB, Ting S, Hsiao LL et al (2015) alpha-Klotho expression in human tissues. J Clin Endocrinol Metab 100:E1308–E1318
Cha SK, Ortega B, Kurosu H, Rosenblatt KP, Kuro OM, Huang CL (2008) Removal of sialic acid involving Klotho causes cell-surface retention of TRPV5 channel via binding to galectin-1. Proc Natl Acad Sci U S A 105:9805–9810
Hu MC, Kuro-o M, Moe OW (2012) Secreted klotho and chronic kidney disease. Adv Exp Med Biol 728:126–157
Hu MC, Kuro-o M, Moe OW (2013) Klotho and chronic kidney disease. Contrib Nephrol 180:47–63
Neyra JA, Hu MC (2017) Potential application of klotho in human chronic kidney disease. Bone 100:41–49
Christov M, Neyra JA, Gupta S, Leaf DE (2019) Fibroblast growth factor 23 and Klotho in AKI. Semin Nephrol 39:57–75
Moreno JA, Izquierdo MC, Sanchez-Nino MD, Suarez-Alvarez B, Lopez-Larrea C, Jakubowski A, Blanco J, Ramirez R, Selgas R, Ruiz-Ortega M et al (2011) The inflammatory cytokines TWEAK and TNFalpha reduce renal klotho expression through NFkappaB. J Am Soc Nephrol 22:1315–1325
Azuma M, Koyama D, Kikuchi J, Yoshizawa H, Thasinas D, Shiizaki K, Kuro-o M, Furukawa Y, Kusano E (2012) Promoter methylation confers kidney-specific expression of the Klotho gene. FASEB J 26:4264–4274
Arrowsmith CH, Bountra C, Fish PV, Lee K, Schapira M (2012) Epigenetic protein families: a new frontier for drug discovery. Nat Rev Drug Discov 11:384–400
Shiels PG, McGuinness D, Eriksson M, Kooman JP, Stenvinkel P (2017) The role of epigenetics in renal ageing. Nat Rev Nephrol 13:471–482
Pal S, Tyler JK (2016) Epigenetics and aging. Sci Adv 2:e1600584. https://doi.org/10.1126/sciadv.1600584
Wuttke M, Kottgen A (2016) Insights into kidney diseases from genome-wide association studies. Nat Rev Nephrol 12:549–562
Smyth LJ, McKay GJ, Maxwell AP, McKnight AJ (2014) DNA hypermethylation and DNA hypomethylation is present at different loci in chronic kidney disease. Epigenetics 9:366–376
Wanner N, Bechtel-Walz W (2017) Epigenetics of kidney disease. Cell Tissue Res 369:75–92
Dawson MA, Kouzarides T (2012) Cancer epigenetics: from mechanism to therapy. Cell 150:12–27
Horvath S, Raj K (2018) DNA methylation-based biomarkers and the epigenetic clock theory of ageing. Nat Rev Genet 19:371–384
Moore LD, Le T, Fan G (2013) DNA methylation and its basic function. Neuropsychopharmacology 38:23–38
Hu L, Li Z, Cheng J, Rao Q, Gong W, Liu M, Shi YG, Zhu J, Wang P, Xu Y (2013) Crystal structure of TET2-DNA complex: insight into TET-mediated 5mC oxidation. Cell 155:1545–1555
Bochtler M, Kolano A, Xu GL (2017) DNA demethylation pathways: additional players and regulators. Bioessays 39:1–13
Melamed P, Yosefzon Y, David C, Tsukerman A, Pnueli L (2018) Tet enzymes, variants, and differential effects on function. Front Cell Dev Biol 6:22
Koch A, Joosten SC, Feng Z, de Ruijter TC, Draht MX, Melotte V, Smits KM, Veeck J, Herman JG, Van Neste L et al (2018) Analysis of DNA methylation in cancer: location revisited. Nat Rev Clin Oncol 15:459–466
Rubinek T, Shulman M, Israeli S, Bose S, Avraham A, Zundelevich A, Evron E, Gal-Yam EN, Kaufman B, Wolf I (2012) Epigenetic silencing of the tumor suppressor klotho in human breast cancer. Breast Cancer Res Treat 133:649–657
Xie B, Zhou J, Yuan L, Ren F, Liu DC, Li Q, Shu G (2013) Epigenetic silencing of Klotho expression correlates with poor prognosis of human hepatocellular carcinoma. Hum Pathol 44:795–801
Lee J, Jeong DJ, Kim J, Lee S, Park JH, Chang B, Jung SI, Yi L, Han Y, Yang Y et al (2010) The anti-aging gene KLOTHO is a novel target for epigenetic silencing in human cervical carcinoma. Mol Cancer 9:109
King GD, Rosene DL, Abraham CR (2012) Promoter methylation and age-related downregulation of Klotho in rhesus monkey. Age (Dordr) 34:1405–1419
Li Y, Chen F, Wei A, Bi F, Zhu X, Yin S, Lin W, Cao W (2019) Klotho recovery by genistein via promoter histone acetylation and DNA demethylation mitigates renal fibrosis in mice. J Mol Med (Berl) 97:541–552
Yin S, Zhang Q, Yang J, Lin W, Li Y, Chen F, Cao W (2017, 1864) TGFbeta-incurred epigenetic aberrations of miRNA and DNA methyltransferase suppress Klotho and potentiate renal fibrosis. Biochim Biophys Acta, Mol Cell Res:1207–1216. https://doi.org/10.1016/j.bbamcr.2017.03.002
Zhang Q, Liu L, Lin W, Yin S, Duan A, Liu Z, Cao W (2017) Rhein reverses Klotho repression via promoter demethylation and protects against kidney and bone injuries in mice with chronic kidney disease. Kidney Int 91:144–156
Hu MC, Shi M, Gillings N, Flores B, Takahashi M, Kuro OM, Moe OW (2017) Recombinant alpha-Klotho may be prophylactic and therapeutic for acute to chronic kidney disease progression and uremic cardiomyopathy. Kidney Int 91:1104–1114
Yu D, Zhang L, Yu G, Nong C, Lei M, Tang J, Chen Q, Cai J, Chen S, Wei Y et al (2019) Association of liver and kidney functions with Klotho gene methylation in a population environment exposed to cadmium in China. Int J Environ Health Res 30:38–48
Hu MC, Shi M, Zhang J, Quinones H, Kuro-o M, Moe OW (2010) Klotho deficiency is an early biomarker of renal ischemia-reperfusion injury and its replacement is protective. Kidney Int 78:1240–1251
Seo MY, Yang J, Lee JY, Kim K, Kim SC, Chang H, Won NH, Kim MG, Jo SK, Cho W et al (2015) Renal Klotho expression in patients with acute kidney injury is associated with the severity of the injury. Korean J Intern Med 30:489–495
Kim AJ, Ro H, Kim H, Chang JH, Lee HH, Chung W, Jung JY (2016) Klotho and S100A8/A9 as discriminative markers between pre-renal and intrinsic acute kidney injury. PLoS One 11:e0147255. https://doi.org/10.1371/journal.pone.0147255
Chen J, Zhang H, Hu J, Gu Y, Shen Z, Xu L, Jia X, Zhang X, Ding X (2017) Hydrogen-rich saline alleviates kidney fibrosis following AKI and retains Klotho expression. Front Pharmacol 8:499
Bi F, Chen F, Li Y, Wei A, Cao W (2018) Klotho preservation by Rhein promotes toll-like receptor 4 proteolysis and attenuates lipopolysaccharide-induced acute kidney injury. J Mol Med (Berl) 96:915–927
Zhang Q, Yin S, Liu L, Liu Z, Cao W (2016) Rhein reversal of DNA hypermethylation-associated Klotho suppression ameliorates renal fibrosis in mice. Sci Rep 6:34597
Tsai KD, Lee WX, Chen W, Chen BY, Chen KL, Hsiao TC, Wang SH, Lee YJ, Liang SY, Shieh JC et al (2018) Upregulation of PRMT6 by LPS suppresses Klotho expression through interaction with NF-kappaB in glomerular mesangial cells. J Cell Biochem 119:3404–3416
Tikoo K, Ali IY, Gupta J, Gupta C (2009) 5-Azacytidine prevents cisplatin induced nephrotoxicity and potentiates anticancer activity of cisplatin by involving inhibition of metallothionein, pAKT and DNMT1 expression in chemical induced cancer rats. Toxicol Lett 191:158–166
Guo C, Pei L, Xiao X, Wei Q, Chen JK, Ding HF, Huang S, Fan G, Shi H, Dong Z (2017) DNA methylation protects against cisplatin-induced kidney injury by regulating specific genes, including interferon regulatory factor 8. Kidney Int 92:1194–1205
Zou D, Wu W, He Y, Ma S, Gao J (2018) The role of klotho in chronic kidney disease. BMC Nephrol 19:285
Chen J, Zhang X, Zhang H, Lin J, Zhang C, Wu Q, Ding X (2013) Elevated Klotho promoter methylation is associated with severity of chronic kidney disease. PLoS One 8:e79856. https://doi.org/10.1371/journal.pone.0079856
Young GH, Wu VC (2012) KLOTHO methylation is linked to uremic toxins and chronic kidney disease. Kidney Int 81:611–612
Sun CY, Chang SC, Wu MS (2012) Suppression of Klotho expression by protein-bound uremic toxins is associated with increased DNA methyltransferase expression and DNA hypermethylation. Kidney Int 81:640–650
Chen J, Zhang X, Zhang H, Liu T, Zhang H, Teng J, Ji J, Ding X (2016) Indoxyl sulfate enhance the hypermethylation of Klotho and promote the process of vascular calcification in chronic kidney disease. Int J Biol Sci 12:1236–1246
Zhang C, Liang Y, Lei L, Zhu G, Chen X, Jin T, Wu Q (2013) Hypermethylations of RASAL1 and KLOTHO is associated with renal dysfunction in a Chinese population environmentally exposed to cadmium. Toxicol Appl Pharmacol 271:78–85
Ruiz-Andres O, Sanchez-Nino MD, Moreno JA, Ruiz-Ortega M, Ramos AM, Sanz AB, Ortiz A (2016) Downregulation of kidney protective factors by inflammation: role of transcription factors and epigenetic mechanisms. Am J Physiol Ren Physiol 311:F1329–F1340
Larkin BP, Glastras SJ, Chen H, Pollock CA, Saad S (2018) DNA methylation and the potential role of demethylating agents in prevention of progressive chronic kidney disease. FASEB J 32:5215–5226
Dwivedi RS, Herman JG, McCaffrey TA, Raj DS (2011) Beyond genetics: epigenetic code in chronic kidney disease. Kidney Int 79:23–32
Liu L, Liu Y, Zhang Y, Bi X, Nie L, Liu C, Xiong J, He T, Xu X, Yu Y et al (2018) High phosphate-induced downregulation of PPARgamma contributes to CKD-associated vascular calcification. J Mol Cell Cardiol 114:264–275
Jung D, Xu Y, Sun Z (2017) Induction of anti-aging gene klotho with a small chemical compound that demethylates CpG islands. Oncotarget 8:46745–46755
Chen K, Sun Z (2018) Activation of DNA demethylases attenuates aging-associated arterial stiffening and hypertension. Aging Cell 17:e12762. https://doi.org/10.1111/acel.12762
Nastase MV, Zeng-Brouwers J, Wygrecka M, Schaefer L (2018) Targeting renal fibrosis: mechanisms and drug delivery systems. Adv Drug Deliv Rev 129:295–307
Morgado-Pascual JL, Marchant V, Rodrigues-Diez R, Dolade N, Suarez-Alvarez B, Kerr B, Valdivielso JM, Ruiz-Ortega M, Rayego-Mateos S (2018) Epigenetic modification mechanisms involved in inflammation and fibrosis in renal pathology. Mediat Inflamm 2018:2931049
Lindberg K, Amin R, Moe OW, Hu M-C, Erben RG, Östman Wernerson A, Lanske B, Olauson H, Larsson TE (2014) The kidney is the principal organ mediating klotho effects. J Am Soc Nephrol 25:2169–2175
Doi S, Zou Y, Togao O, Pastor JV, John GB, Wang L, Shiizaki K, Gotschall R, Schiavi S, Yorioka N et al (2011) Klotho inhibits transforming growth factor-beta1 (TGF-beta1) signaling and suppresses renal fibrosis and cancer metastasis in mice. J Biol Chem 286:8655–8665
Zhou L, Li Y, Zhou D, Tan RJ, Liu Y (2013) Loss of Klotho contributes to kidney injury by derepression of Wnt/beta-catenin signaling. J Am Soc Nephrol 24:771–785
Satoh M, Nagasu H, Morita Y, Yamaguchi TP, Kanwar YS, Kashihara N (2012) Klotho protects against mouse renal fibrosis by inhibiting Wnt signaling. Am J Physiol Ren Physiol 303:F1641–F1651
Guan X, Nie L, He T, Yang K, Xiao T, Wang S, Huang Y, Zhang J, Wang J, Sharma K et al (2014) Klotho suppresses renal tubulo-interstitial fibrosis by controlling basic fibroblast growth factor-2 signalling. J Pathol 234:560–572
Yamamoto M, Clark JD, Pastor JV, Gurnani P, Nandi A, Kurosu H, Miyoshi M, Ogawa Y, Castrillon DH, Rosenblatt KP et al (2005) Regulation of oxidative stress by the anti-aging hormone klotho. J Biol Chem 280:38029–38034
Ortiz Arduan A (2012) Aging and inflammation: Klotho, diet and the kidney connection. An R Acad Nac Med (Madr) 129:231–242 discussion 242-234
Hu Y, Mou L, Yang F, Tu H, Lin W (2016) Curcumin attenuates cyclosporine A induced renal fibrosis by inhibiting hypermethylation of the klotho promoter. Mol Med Rep 14:3229–3236
Xu X, Tan X, Tampe B, Wilhelmi T, Hulshoff MS, Saito S, Moser T, Kalluri R, Hasenfuss G, Zeisberg EM et al (2018) High-fidelity CRISPR/Cas9- based gene-specific hydroxymethylation rescues gene expression and attenuates renal fibrosis. Nat Commun 9:3509
Gu Y, Chen J, Zhang H, Shen Z, Liu H, Lv S, Yu X, Zhang D, Ding X, Zhang X (2020) Hydrogen sulfide attenuates renal fibrosis by inducing TET-dependent DNA demethylation on Klotho promoter. FASEB J 34:11474–11487
Thomas MC, Brownlee M, Susztak K, Sharma K, Jandeleit-Dahm KA, Zoungas S, Rossing P, Groop PH, Cooper ME (2015) Diabetic kidney disease. Nat Rev Dis Primers 1:15018
Kim SS, Song SH, Kim IJ, Lee EY, Lee SM, Chung CH, Kwak IS, Lee EK, Kim YK (2016) Decreased plasma alpha-Klotho predict progression of nephropathy with type 2 diabetic patients. J Diabetes Complicat 30:887–892
Wu C, Ma X, Zhou Y, Liu Y, Shao Y, Wang Q (2019) Klotho restraining Egr1/TLR4/mTOR axis to reducing the expression of fibrosis and inflammatory cytokines in high glucose cultured rat mesangial cells. Exp Clin Endocrinol Diabetes 127:630–640
Wu C, Wang Q, Lv C, Qin N, Lei S, Yuan Q, Wang G (2014) The changes of serum sKlotho and NGAL levels and their correlation in type 2 diabetes mellitus patients with different stages of urinary albumin. Diabetes Res Clin Pract 106:343–350
Liu YN, Zhou J, Li T, Wu J, Xie SH, Liu HF, Liu Z, Park TS, Wang Y, Liu WJ (2017) Sulodexide protects renal tubular epithelial cells from oxidative stress-induced injury via upregulating Klotho expression at an early stage of diabetic kidney disease. J Diabetes Res 2017:4989847
Navarro-Gonzalez JF, Sanchez-Nino MD, Donate-Correa J, Martin-Nunez E, Ferri C, Perez-Delgado N, Gorriz JL, Martinez-Castelao A, Ortiz A, Mora-Fernandez C (2018) Effects of pentoxifylline on soluble Klotho concentrations and renal tubular cell expression in diabetic kidney disease. Diabetes Care 41:1817–1820
Yang XH, Zhang BL, Zhang XM, Tong JD, Gu YH, Guo LL, Jin HM (2020) EGCG attenuates renal damage via reversing Klotho hypermethylation in diabetic db/db mice and HK-2 cells. Oxidative Med Cell Longev 2020:6092715
Zhang XT, Wang G, Ye LF, Pu Y, Li RT, Liang J, Wang L, Lee KKH, Yang X (2020) Baicalin reversal of DNA hypermethylation-associated Klotho suppression ameliorates renal injury in type 1 diabetic mouse model. Cell Cycle 1–19. https://doi.org/10.1080/15384101.2020.1843815
Panah F, Ghorbanihaghjo A, Argani H, Asadi Zarmehri M, Nazari Soltan Ahmad S (2018) Ischemic acute kidney injury and klotho in renal transplantation. Clin Biochem 55:3–8
Castellano G, Intini A, Stasi A, Divella C, Gigante M, Pontrelli P, Franzin R, Accetturo M, Zito A, Fiorentino M et al (2016) Complement modulation of anti-aging factor Klotho in ischemia/reperfusion injury and delayed graft function. Am J Transplant 16:325–333
Soleymanian T, Ranjbar A, Alipour M, Ganji MR, Najafi I (2015) Impact of kidney transplantation on biomarkers of oxidative stress and inflammation. Iran J Kidney Dis 9:400–405
Donate-Correa J, Henriquez-Palop F, Martin-Nunez E, Perez-Delgado N, Muros-de-Fuentes M, Mora-Fernandez C, Navarro-Gonzalez JF (2016) Effect of paricalcitol on FGF-23 and Klotho in kidney transplant recipients. Transplantation 100:2432–2438
Van Beneden K, Mannaerts I, Pauwels M, Van den Branden C, van Grunsven LA (2013) HDAC inhibitors in experimental liver and kidney fibrosis. Fibrogenesis Tissue Repair 6:1
Chrun ES, Modolo F, Daniel FI (2017) Histone modifications: a review about the presence of this epigenetic phenomenon in carcinogenesis. Pathol Res Pract 213:1329–1339
McClure JJ, Li X, Chou CJ (2018) Advances and challenges of HDAC inhibitors in cancer therapeutics. Adv Cancer Res 138:183–211
Sato Y, Yanagita M (2018) Immune cells and inflammation in AKI to CKD progression. Am J Physiol Ren Physiol 315:F1501–F1512
Shi M, Flores B, Gillings N, Bian A, Cho HJ, Yan S, Liu Y, Levine B, Moe OW, Hu MC (2016) alphaKlotho mitigates progression of AKI to CKD through activation of autophagy. J Am Soc Nephrol 27:2331–2345
Lin W, Li Y, Chen F, Yin S, Liu Z, Cao W (2017) Klotho preservation via histone deacetylase inhibition attenuates chronic kidney disease-associated bone injury in mice. Sci Rep 7:46195
Lin W, Zhang Q, Liu L, Yin S, Liu Z, Cao W (2017) Klotho restoration via acetylation of peroxisome proliferation–activated receptor γ reduces the progression of chronic kidney disease. Kidney Int 92:669–679
Liu M, Li XC, Lu L, Cao Y, Sun RR, Chen S, Zhang PY (2014) Cardiovascular disease and its relationship with chronic kidney disease. Eur Rev Med Pharmacol Sci 18:2918–2926
Gao D, Zuo Z, Tian J, Ali Q, Lin Y, Lei H, Sun Z (2016) Activation of SIRT1 attenuates Klotho deficiency-induced arterial stiffness and hypertension by enhancing AMP-activated protein kinase activity. Hypertension 68:1191–1199
Zhang P, Li Y, Du Y, Li G, Wang L, Zhou F (2016) Resveratrol ameliorated vascular calcification by regulating Sirt-1 and Nrf2. Transplant Proc 48:3378–3386
Hsu SC, Huang SM, Chen A, Sun CY, Lin SH, Chen JS, Liu ST, Hsu YJ (2014) Resveratrol increases anti-aging Klotho gene expression via the activating transcription factor 3/c-Jun complex-mediated signaling pathway. Int J Biochem Cell Biol 53:361–371
Brilli LL, Swanhart LM, de Caestecker MP, Hukriede NA (2013) HDAC inhibitors in kidney development and disease. Pediatr Nephrol 28:1909–1921
Fontecha-Barriuso M, Martin-Sanchez D, Ruiz-Andres O, Poveda J, Sanchez-Nino MD, Valino-Rivas L, Ruiz-Ortega M, Ortiz A, Sanz AB (2018) Targeting epigenetic DNA and histone modifications to treat kidney disease. Nephrol Dial Transplant 33:1875–1886
Chun P (2017) Therapeutic effects of histone deacetylase inhibitors on kidney disease. Arch Pharm Res 41:162–183
Brilli LL, Swanhart LM, de Caestecker MP, Hukriede NA (2012) HDAC inhibitors in kidney development and disease. Pediatr Nephrol 28:1909–1921
Levine MH, Wang Z, Bhatti TR, Wang Y, Aufhauser DD, McNeal S, Liu Y, Cheraghlou S, Han R, Wang L et al (2015) Class-specific histone/protein deacetylase inhibition protects against renal ischemia reperfusion injury and fibrosis formation. Am J Transplant 15:965–973
Choi HS, Song JH, Kim IJ, Joo SY, Eom GH, Kim I, Cha H, Cho JM, Ma SK, Kim SW et al (2018) Histone deacetylase inhibitor, CG200745 attenuates renal fibrosis in obstructive kidney disease. Sci Rep 8:11546
Xiong C, Guan Y, Zhou X, Liu L, Zhuang MA, Zhang W, Zhang Y, Masucci MV, Bayliss G, Zhao TC et al (2019) Selective inhibition of class IIa histone deacetylases alleviates renal fibrosis. FASEB J 33:8249–8262
Pang M, Kothapally J, Mao H, Tolbert E, Ponnusamy M, Chin YE, Zhuang S (2009) Inhibition of histone deacetylase activity attenuates renal fibroblast activation and interstitial fibrosis in obstructive nephropathy. Am J Physiol-Renal Physiol 297:F996–F1005
Yang M, Chen G, Zhang X, Guo Y, Yu Y, Tian L, Chang S, Chen ZK (2019) Inhibition of class I HDACs attenuates renal interstitial fibrosis in a murine model. Pharmacol Res 142:192–204
Kang SW, Lee SM, Kim JY, Kim SY, Kim YH, Kim TH, Kang MS, Jang WH, Seo SK (2017) Therapeutic activity of the histone deacetylase inhibitor SB939 on renal fibrosis. Int Immunopharmacol 42:25–31
Na Liu SH, Ma L, Ponnusamy M, Tang J, Tolbert E, Bayliss G, Zhao TC, Yan H, Zhuang S (2013) Blocking the class I histone deacetylase ameliorates renal fibrosis and inhibits renal fibroblast activation via modulating TGF-beta and EGFR signaling. PLoS One 8:e54001
Zhang Y, Zou J, Tolbert E, Zhao TC, Bayliss G, Zhuang S (2020) Identification of histone deacetylase 8 as a novel therapeutic target for renal fibrosis. FASEB J 34:7295–7310
Lai L, Cheng P, Yan M, Gu Y, Xue J (2019) Aldosterone induces renal fibrosis by promoting HDAC1 expression, deacetylating H3K9 and inhibiting klotho transcription. Mol Med Rep 19:1803–1808
Chen F, Gao Q, Wei A, Chen X, Shi Y, Wang H, Cao W (2020) Histone deacetylase 3 aberration inhibits Klotho transcription and promotes renal fibrosis. Cell Death Differ. https://doi.org/10.1038/s41418-020-00631-9
Chung AC, Lan HY (2015) MicroRNAs in renal fibrosis. Front Physiol 6:50
Lu TX, Rothenberg ME (2018) MicroRNA. J Allergy Clin Immunol 141:1202–1207
Dey BK, Mueller AC, Dutta A (2014) Long non-coding RNAs as emerging regulators of differentiation, development, and disease. Transcription 5:e944014. https://doi.org/10.4161/21541272.2014.944014
Chen LL (2016) The biogenesis and emerging roles of circular RNAs. Nat Rev Mol Cell Biol 17:205–211
Abolghasemi M, Yousefi T, Maniati M, Qujeq D (2019) The interplay of Klotho with signaling pathway and microRNAs in cancers. J Cell Biochem 120:14306–14317
Morii K, Yamasaki S, Doi S, Irifuku T, Sasaki K, Doi T, Nakashima A, Arihiro K, Masaki T (2019) microRNA-200c regulates KLOTHO expression in human kidney cells under oxidative stress. PLoS One 14:e0218468
Mehi SJ, Maltare A, Abraham CR, King GD (2014) MicroRNA-339 and microRNA-556 regulate Klotho expression in vitro. Age (Dordr) 36:141–149
Liu Y, Lai P, Deng J, Hao Q, Li X, Yang M, Wang H, Dong B (2019) Micro-RNA335-5p targeted inhibition of sKlotho and promoted oxidative stress-mediated aging of endothelial cells. Biomark Med 13:457–466
Rodrigues CE, Capcha JMC, de Bragança AC, Sanches TR, Gouveia PQ, de Oliveira PAF, Malheiros DMAC, Volpini RA, Santinho MAR, Santana BAA et al. (2017) Human umbilical cord-derived mesenchymal stromal cells protect against premature renal senescence resulting from oxidative stress in rats with acute kidney injury. Stem Cell Res Ther 8. https://doi.org/10.1186/s13287-017-0475-8
Liang H, Yang K, Xin M, Liu X, Zhao L, Liu B, Wang J (2017) MiR-130a protects against lipopolysaccharide-induced glomerular cell injury by upregulation of Klotho. Pharmazie 72:468–474
Grange C, Papadimitriou E, Dimuccio V, Pastorino C, Molina J, O'Kelly R, Niedernhofer LJ, Robbins PD, Camussi G, Bussolati B (2020) Urinary extracellular vesicles carrying klotho improve the recovery of renal function in an acute tubular injury model. Mol Ther 28:490–502
Shilo V, Mor-Yosef Levi I, Abel R, Mihailović A, Wasserman G, Naveh-Many T, Ben-Dov IZ (2017) Let-7 and microRNA-148 regulate parathyroid hormone levels in secondary hyperparathyroidism. J Am Soc Nephrol 28:2353–2363
Liu Y, Bi X, Xiong J, Han W, Xiao T, Xu X, Yang K, Liu C, Jiang W, He T et al (2019) MicroRNA-34a promotes renal fibrosis by downregulation of Klotho in tubular epithelial cells. Mol Ther 27:1051–1065
Wu C, Lv C, Chen F, Ma X, Shao Y, Wang Q (2015) The function of miR-199a-5p/Klotho regulating TLR4/NF-kappaB p65/NGAL pathways in rat mesangial cells cultured with high glucose and the mechanism. Mol Cell Endocrinol 417:84–93
Jia Y, Zheng Z, Xue M, Zhang S, Hu F, Li Y, Yang Y, Zou M, Li S, Wang L et al (2019) Extracellular vesicles from albumin-induced tubular epithelial cells promote the M1 macrophage phenotype by targeting Klotho. Mol Ther 27:1452–1466
Kang WL, Xu GS (2016) Atrasentan increased the expression of klotho by mediating miR-199b-5p and prevented renal tubular injury in diabetic nephropathy. Sci Rep 6:19979
Ye H, Su B, Ni H, Li L, Chen X, You X, Zhang H (2018) microRNA-199a may be involved in the pathogenesis of lupus nephritis via modulating the activation of NF-kappaB by targeting Klotho. Mol Immunol 103:235–242
Shilo V, Ben-Dov IZ, Nechama M, Silver J, Naveh-Many T (2015) Parathyroid-specific deletion of dicer-dependent microRNAs abrogates the response of the parathyroid to acute and chronic hypocalcemia and uremia. FASEB J 29:3964–3976
Lv W, Fan F, Wang Y, Gonzalez-Fernandez E, Wang C, Yang L, Booz GW, Roman RJ (2018) Therapeutic potential of microRNAs for the treatment of renal fibrosis and CKD. Physiol Genomics 50:20–34
Butz H, Racz K, Hunyady L, Patocs A (2012) Crosstalk between TGF-beta signaling and the microRNA machinery. Trends Pharmacol Sci 33:382–393
Almaani S, Meara A, Rovin BH (2017) Update on lupus nephritis. Clin J Am Soc Nephrol 12:825–835
Yung S, Yap DY, Chan TM (2017) Recent advances in the understanding of renal inflammation and fibrosis in lupus nephritis. F1000Res 6:874
Cen H, Zhou M, Leng RX, Wang W, Feng CC, Li BZ, Zhu Y, Yang XK, Yang M, Zhai Y et al (2013) Genetic interaction between genes involved in NF-kappaB signaling pathway in systemic lupus erythematosus. Mol Immunol 56:643–648
Jiang T, Tian F, Zheng H, Whitman SA, Lin Y, Zhang Z, Zhang N, Zhang DD (2014) Nrf2 suppresses lupus nephritis through inhibition of oxidative injury and the NF-kappaB-mediated inflammatory response. Kidney Int 85:333–343
Rupaimoole R, Slack FJ (2017) MicroRNA therapeutics: towards a new era for the management of cancer and other diseases. Nat Rev Drug Discov 16:203–222
Chen J, Ning Y, Zhang H, Song N, Gu Y, Shi Y, Cai J, Ding X, Zhang X (2019) METTL14-dependent m6A regulates vascular calcification induced by indoxyl sulfate. Life Sci 239:117034
Liu L, Zou J, Guan Y, Zhang Y, Zhang W, Zhou X, Xiong C, Tolbert E, Zhao TC, Bayliss G, Zhuang S (2019) Blocking the histone lysine 79 methyltransferase DOT1L alleviates renal fibrosis through inhibition of renal fibroblast activation and epithelial-mesenchymal transition. FASEB J 33:11941–11958
Han X, Sun Z (2020) Epigenetic regulation of KL (Klotho) via H3K27me3 (histone 3 lysine [K] 27 trimethylation) in renal tubule cells. Hypertension 75:1233–1241
Li Y, Ren D, Xu G (2019) Long noncoding RNA MALAT1 mediates high glucose-induced glomerular endothelial cell injury by epigenetically inhibiting klotho via methyltransferase G9a. IUBMB Life 71:873–881
Shah RR (2019) Safety and tolerability of histone deacetylase (HDAC) inhibitors in oncology. Drug Saf 42:235–245
Filì C, Candoni A, Zannier ME, Olivieri J, Imbergamo S, Caizzi M, Nadali G, Di Bona E, Ermacora A, Gottardi M et al (2019) Efficacy and toxicity of decitabine in patients with acute myeloid leukemia (AML): a multicenter real-world experience. Leuk Res 76:33–38
Stancheva TCaI (2008) Methyl-CpG binding proteins: specialized transcriptional repressors or structural components of chromatin? Cell Mol Life Sci 65:1509–1522
Acknowledgments
We thank former and current laboratory members Lin Liu, Qin Zhang, Wenjun Lin, Shasha Yin, Dawei Cai, Fangfang Bi, Yanning Li, Fang Chen, Ai Wei, Qi Gao, Xiaobo Zhu, Xingren Chen, and Lijun Zhang for their contributions to the publications substantiating this article.
Funding
This work is supported by research grants from National Nature Science Foundation of China General Program 81970577 and 81670762 to W.C.
Author information
Authors and Affiliations
Contributions
J.X. searched the literatures drafted the manuscript. W.C. reviewed, edited, and wrote the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Xia, J., Cao, W. Epigenetic modifications of Klotho expression in kidney diseases. J Mol Med 99, 581–592 (2021). https://doi.org/10.1007/s00109-021-02044-8
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00109-021-02044-8