
Abstract
Metabolic homeostasis is essential for cellular survival and proper tissue function. Multi-systemic metabolic regulation is therefore vital for good health. A number of tissues have the task of maintaining appropriate metabolism, and skeletal muscle is the most abundant of them. Muscle possesses a remarkable plasticity and is able to rapidly adapt to changes in energetic demands by fine-tuning the balance between catabolic and anabolic processes. Autophagy is a catabolic process responsible for the degradation of protein aggregates and damaged organelles, through the autophagosome–lysosome system. Proper regulation of autophagy flux is fundamental for organism homeostasis under physiological conditions and even more in response to metabolic stress, such as during physical activity and nutritional deficits. Both deficient and excessive autophagy are harmful for health and have devastating consequences in a myriad of pathologies. The regulation of autophagy flux in various tissues, and in particular in skeletal muscle, is of great importance for health and tissue homeostasis and represents a feasible mechanism by which physical exercise exerts its beneficial effects on muscle and whole body metabolism. This review is focused on the key molecular mechanisms regulating macromolecule and organelle turnover in muscle during alterations in nutrient availability and energetic demands, as well as their involvement in disease pathogenesis.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Cellular homeostasis is essential for tissue function and cell survival. Several biological processes contribute to this purpose and autophagy is likely the most dynamic and prominent of these mechanisms. Autophagy is a lysosome-mediated bulk degradation process, which is constantly ongoing at low levels in all cells and tissues [1, 2]. This process is of particular importance in long-lived post-mitotic cells, such as skeletal muscle fibers and neurons, as this is the sole known mechanism for these cells to rid themselves of dysfunctional and harmful organelles. Three main forms of autophagy have been described thus far, namely macroautophagy, microautophagy, and chaperone-mediated autophagy. The major distinction between these forms is the substrate selection and delivery mechanisms involved. Microautophagy and chaperone-mediated autophagy degrade small portions of cytosol and selected proteins by directly delivering them to lysosomes in a chaperone-independent or chaperone-dependent manner. Macroautophagy (autophagy) is a more complex process involving the formation of double-membrane vesicles termed autophagosomes, which engulf large portion of cytoplasm, as well as entire organelles, and transport them to the lysosome for degradation [3].
At first glance, autophagy was considered a coarse, nonselective, degradative system, but closer investigation revealed a different truth. Autophagy represents an extremely refined collector of altered organelles, abnormal protein aggregates, and pathogens, similar to a selective recycling center rather than a general landfill [4]. Indeed, new terms have been adopted to describe the selective nature of autophagy, corresponding to the specific cargo being degraded, such as mitophagy, reticulophagy, ribophagy, xenophagy, and pexophagy [4]. The selectivity of the autophagy process is conferred by a growing number of specific cargo receptors, including p62, Nbr1, Bnip3, Nix (Bnip3L), and optineurin [5]. These adaptor proteins are equipped with both a cargo-binding domain, with the capability to recognize and attach directly to molecular tags on organelles, and at the same time a LC3-interacting region domain, able to recruit and bind essential autophagosome membrane proteins. Adaptor proteins are able to recognize their targets by specific flag molecules or posttranslational modifications, such as ubiquitination, presented on the surface of the cargo [6]. In addition, substrate recognition may also depend on the physiological properties of the cargo to be degraded. For instance, mitochondrial depolarization induces the stabilization of Pink1 and the recruitment of the E3 ubiquitin ligase Parkin to the surface of the organelle, acting as a functional “eat me” signal, ultimately resulting in the engulfment and subsequent demise of the damaged mitochondria [7]. In mammals, autophagy is active in almost all tissues and provides an effective mechanism for protein quality control, elimination of defective organelles, regulation of metabolism, and pathogen removal. Thus, any perturbation to autophagy flux may lead to derailments in tissue homeostasis. This is mostly critical during aging, cancer, neurodegenerative and metabolic diseases, as well as infections [1, 3, 8].
De novo formation of autophagosomes is regulated by at least three molecular complexes: the LC3 conjugation system and the regulatory complexes governed by ULK1 and Beclin 1 [8]. The conjugation complex is composed of different proteins encoded by autophagy-related genes (Atg), which are highly conserved between species and act in a hierarchical manner [8, 9]. The Atg12–Atg5–Atg16L1 complex, along with Atg7, plays an essential role in the conjugation of LC3 to phosphatidylethanolamine, which is required for the elongation and closure of the isolation membrane [8] (Fig. 1). This system is under the regulation of at least two major cellular energy sensing complexes, able to ascertain the metabolic status of the cell. Under basal conditions, the ULK1 complex is inactivated by phosphorylation through the mTOR complex 1 (mTORC1), whereas during autophagy induction mTORC1 is inhibited thus enhancing the formation of a complex between ULK1, Atg13 and FIP200. mTORC1 is a nutrient sensor downstream of the insulin/AKT pathway, however mTORC1 can also be negatively regulated independently of AKT by energy stress sensors such as AMPK [10] and, in a mechanical-activity-dependent manner through tuberous sclerosis complex (TSC)1/2 [11, 12]. Moreover, AMPK can also directly phosphorylate ULK1 and Beclin 1, thus promoting autophagy induction [13]. During autophagy, the ULK1 complex is localized to the isolation membrane, where it facilitates the formation of autophagosomes through interaction with the Beclin 1 complex (Fig. 1). The two complexes are strictly linked through protein–protein interactions. Beclin 1 is directly phosphorylated by ULK1 [14], which also interacts with Ambra1 [15]. At the same time, Ambra1 is inhibited by mTORC1 phosphorylation [15]. Moreover, AKT can also regulate autophagy through the inhibition of transcription factors of the forkhead box (FoxO) class. Several genes, such as LC3, GABARAP, Bnip3, and glutamine synthetase, are under FoxO3 control. Indeed, in skeletal muscle the effect of FoxO3 on autophagy appears to be, at least in part, mediated by its downstream target Bnip3. Bnip3 alone is sufficient to induce autophagy, while its knockdown attenuates FoxO3-mediated autophagy induction [16]. Furthermore, FoxO3 also mediates the upregulation of glutamine synthetase expression, which can indirectly induce autophagy by inhibiting mTOR [17]. Recently, the role of the Beclin 1 complex in the formation and elongation of the isolation membrane has been challenged. Vacuolar protein sorting 15 (Vps15) muscle-specific knockout mice display normal LC3 lipidation and vesicle formation, despite decreased Vps34 and Beclin 1 levels. Interestingly, these mice present an impaired autophagosome–lysosome fusion similar to that observed in Vps34-depleted neurons, suggesting a role for Vps proteins in this process [18]. Therefore, the Beclin 1 complex may play a more important role in autophagosome docking rather than in autophagosome formation. A more detailed analysis of the in vivo role of Beclin 1 is required to further address this important concept.

Autophagy modulation during metabolic alterations. Upon nutrient deprivation, an increase in cellular AMP/ATP ratio and a rise in ROS production activate sestrins and sirtuins (SIRT1), culminating in the activation of AMPK and inhibition of AKT. AMPK in turn inhibits mTORC1, releasing the block on the autophagy initiation factors composed of the ULK1 and Beclin 1 complexes and thus leading to autophagosome formation. Inhibition of AKT/mTOR pathway also allows for the nuclear translocation of TFEB and FoxO, enhancing the transcription of several autophagy and lysosome-related genes (Atgs). The molecular mechanisms activated by a high fat diet are still poorly understood, but the net result is a block in autophagy due to the phosphorylation and activation of mTOR as well as impaired autophagosome–lysosome fusion, which initiates a vicious feed-forward loop between mTORC1 and p62
Autophagy in muscle
Skeletal muscle is an indispensable metabolic center and possesses a remarkable capacity to rapidly and effectively adapt to variations in contractile activity, a process known as muscle plasticity. Moreover, muscle organelles can be easily damaged following strenuous physical activity, improper nutrition, and aging. A finely tuned system for protein degradation and organelle removal is therefore required for the proper function and contractility of skeletal muscle [19–21]. Indeed, autophagic flux is increased in several physiological and pathological conditions, such as fasting, atrophy, and exercise [16, 22, 23]. Conversely, an impairment in autophagy results in the accumulation of unfolded and aggregate-prone proteins and dysfunctional organelles, which are typical features of several myopathies [24, 25]. Generation of Atg5 and Atg7 muscle-specific knockout mice confirmed the physiological importance of this system in muscle mass maintenance [26, 27]. The muscle-specific Atg7 knockout mice are characterized by the presence of abnormal mitochondria, oxidative stress, accumulation of polyubiquitinated proteins, and consequent sarcomere disorganization [27]. Moreover, the central role of the autophagy-lysosome system in muscle homeostasis is highlighted by lysosomal storage diseases, a group of debilitating muscle disorders characterized by alterations in lysosomal proteins and autophagosome buildup. Pompe disease is a myopathy caused by a defect in lysosomal acid α-glucosidase [28, 29], Danon disease is caused by the lack of lysosome-associated membrane protein 2 (LAMP-2) [30, 31], whereas X-linked myopathy with excessive autophagy is triggered by mutations in an essential assembly chaperone of the V-ATPase [32]. All of these myopathies portray an accumulation of autophagic vacuoles inside myofibers due to defects in their clearance. Several other animal models, which are characterized by aberrant autophagy flux, confirmed the essential role of autophagy in skeletal muscle (Table 1).
These findings paved the way to explore the role of autophagy in inherited muscle diseases such as congenital muscular dystrophies. The first evidence of impaired autophagy was provided by studies in mice and patients with mutations in collagen VI [25, 42]. A similar block in autophagy progression was later described in dystrophin-deficient (mdx) and lamin A/C null mice [33, 35, 36]. Conversely, laminin mutated (dy/dy) animals display excessive levels of autophagy, which is equally detrimental [34]. Importantly, restoring normal autophagy flux through pharmacological and dietary tools rescued the muscle pathology of collagen VI knockout (Col6a1 −/−) animals, as well as of mdx and dy/dy mice [25, 34, 35] (Table 1).
In addition to congenital muscular dystrophies, perturbations in autophagy flux have also been described in other myopathies. For instance, evidence of impaired autophagy were reported in myofibrillar myopathy, which is characterized by myofibril disassembly and the accumulation of protein aggregates inside myofibers [37, 38]. Additionally, hereditary myopathy with early respiratory failure presents a buildup of p62 positive aggregates due to a disturbance in the Nbr1/p62/MuRF2 pathway [39, 40], whereas X-linked myotubular/centronuclear myopathy is instead characterized by an overabundance of autophagosomes due to a defect in autophagosome–lysosome fusion [41].
The first inherited disease caused by a mutation in an autophagy-related gene was recently discovered. Vici syndrome is a multisystemic disorder, associated with a mutation in the autophagy gene EPG5 and is also characterized by the presence of a myopathy. The EPG5 gene is evolutionary conserved and plays a critical role in the regulation of autophagy in Caenorhabditis elegans and mice [43, 44]. Thus, autophagy is a vital process the aberrant regulation of which results in various myopathies and muscular dystrophies.
Autophagy, nutrients, and energy balance
The energetic status of a cell is one of the most potent stimuli known to regulate autophagy. This regulation is highly dependent on the specific cell type and on the type and duration of the stimulus. Starvation is the most well-studied condition for the induction of autophagy, and the importance of the autophagy pathway is most evident during the short period of nutrient deprivation immediately after birth. Indeed, mice deficient for autophagy essential genes, such as Atg3, Atg5, or Atg7, die shortly after birth [1, 2, 8]. Both increased reactive oxygen species (ROS) production and nutrient stress during starvation were reported to activate autophagy through the intricate interplay between FoxO, AMPK, AKT, and mTOR [16, 45–47].
Activation of autophagy ensures optimal energy utilization efficiency, which is beneficial during periods of scarce nutrients (Fig. 2). It is therefore no wonder that this process is conserved on the evolutionary scale. To this effect, lifelong caloric restriction has been documented to prolong life span and improve health, as well as reduce the frequency of chronic ailments [48], although this issue is controversial [49]. The benefits deriving from nutrient deprivation are thought to be mediated by mild energetic stress, which inevitably activates autophagy as a cellular stress responder. An age-related decline in autophagic degradation has been shown to occur concomitantly with age-related increases in oxidative damage and apoptosis, both of which negatively correlate with autophagy. Interestingly, a chronic autophagic stimulus, such as caloric restriction, was found to ameliorate the pathological state of muscle during aging [50]. Moreover, caloric restriction has been documented to reverse the muscle hypertrophy phenotype characteristic of mice lacking myostatin, thus helping restore muscle function and force-generating capability in this model [51].

Autophagy and tissue homeostasis during energetic imbalance. During metabolic stress, autophagy is initiated in the major metabolic centers of the body: liver, muscle, and adipose tissue. This likely occurs through AMPK activation and inhibition of the insulin/AKT/mTOR axis. Activation of autophagy in adipose tissue results in the release of free fatty acids through lipophagy. Free fatty acids in turn can fuel muscle, heart, and liver function. In muscle, autophagy results in protein catabolism and the generation of amino acids, which are delivered to the liver. AMPK activation in muscle also promotes the translocation of FoxOs and PGC-1α to the nucleus, where they activate a transcriptional program to increase autophagy and mitochondrial biogenesis, while dysfunctional mitochondria can stimulate the secretion of FGF21 to induce lipophagy in adipose tissue. In liver, the activation of TFEB and FoxO results in the upregulation of lipid metabolism, gluconeogenesis, and glycogen breakdown. AA amino acids, FFA free fatty acids, GNG gluconeogenesis
Autophagy in substrate utilization
In addition to its catabolic role, autophagy actively mobilizes several cellular energy stores to provide nutritional support in times of need. The interplay between autophagy, lipid metabolism, and carbohydrate metabolism illustrates the existence of a dynamic crosstalk between autophagy and cellular energy balance (Fig. 2). During fasting, a selective form of autophagy (lipophagy) drives cellular lipids, stored as triglycerides in lipid droplets, to lysosomes, where they are hydrolysed into fatty acids and subsequently broken down to replenish ATP [52]. A block in autophagy results in reduced rates of β-oxidation. This is evident in liver specific Atg7 −/− mice, which develop an accumulation of triglycerides and cholesterol in lipid droplets, with no apparent alteration in lipogenesis [53]. The interplay between autophagy and lipid metabolism is endowed with different levels of complexity, where lipids themselves have an important role in autophagy regulation [54, 55]. Moreover, autophagy has been found to mediate adipose tissue development by modulating adipocyte differentiation. White adipose tissue-specific deletion of Atg7 in mice results in browning of adipocytes, characterized by decreased adipose mass, increased mitochondrial content and multilocular lipid droplets. These mice are remarkably lean and present improved glucose tolerance [56, 57]. Various molecules are involved in the autophagic control of lipid metabolism, including the autophagy substrate p62/SQSTM1. Indeed, one of the most important players, mTORC1, is controlled by a feed forward loop, where p62 activates mTORC1 resulting in augmented p62 protein levels [58].
Animals exposed to a high fat diet (HFD) first experience an increase in autophagic activity, but prolonged over-feeding decreases autophagy flux. This decrease in autophagy is speculated to occur due to impaired autophagosome–lysosome fusion, likely stemming from a pathological remodeling of membrane lipids [59]. In another study, HFD was shown to cause a slight suppression of autophagy in muscle, while mice deficient in stress-activated autophagy (Bcl2AAA knock-in mutants) were more sensitive to HFD-induced obesity. This is somewhat of a paradox, since suppression of autophagy was observed with HFD and yet its deficiency appears to sensitize these mice to diet-mediated weight gain. However, this may be due to alterations in other tissues (e.g., liver), which were not examined in the above study [60]. Moreover, studies focusing on autophagy deficiency specifically in muscle show conflicting results. In fact, lack of Atg7 results in protection from HFD-induced insulin resistance due to mitochondrial dysfunction and intercellular crosstalk through the mitokine FGF21 [61]. Accordingly, feeding a HFD to muscle-specific HDAC1/2 mutant mice, which also present an autophagy-deficient phenotype, actually restores autophagy flux and prevents myopathy in adult mice [62]. Therefore, further studies are required to address the conflicting consequences of HFD on autophagy and its role in glucose and lipid metabolism during overfeeding.
Other than its role in lipid mobilization and glucose homeostasis, autophagy is also involved in glycogen breakdown, a process playing a critical role in tissues, such as liver and muscle. Glycogen hydrolysis is regulated primarily by phosphorylases and debranching enzymes, but lysosomal acid glycosidases also contribute to glucose homeostasis. Indeed, cells can activate glycogen-specific autophagy (glycophagy) in order to maintain glucose homeostasis [63]. Dynamic autophagosome formation was described in the β-cells of obese and insulin-resistant db/db mice [64], illustrating that impaired insulin signaling drives autophagy in the pancreas [65]. Similarly to lipids, the interplay between autophagy and carbohydrate metabolism is bidirectional as a variety of glycidic groups have regulatory roles in autophagic signaling. The activation of autophagy during glucose deprivation has been proposed to be, in part, a consequence of the associated oxidative stress. In line with this, a recent study demonstrated a role for the TSC2 complex in regulating autophagy in response to ROS [45]. Conversely, increased intracellular glucose results in reduced phosphorylation of the AKT/FoxO/mTOR pathways, leading to increased protein degradation via autophagy [66].
Altered autophagic degradation of glycogen stores underlie the basis of different muscular disorders previously described as lysosomal storage diseases. Glycogen granules accumulate in muscles of patients affected by Pompe disease or Danon disease. Several studies have revealed that the primary defects manifest in abnormal delivery of the material sequestered in autophagosomes, including glycogen, to lysosomes for degradation [67]. Interestingly, muscle-specific Vps15 knockout mice develop a severe myopathy with hallmarks of autophagic vacular myopathies, including accumulation of autophagosomes and glycogen. This “myophagy” (autophagy-related myopathy) occurs due to a defect in the fusion of autophagosomes with lysosomes. Accordingly, over-expression of the Vps34–Vps15 complex in myoblasts of Danon disease patients results in a partial amelioration of glycogen overload [31]. Recently an alternative route for the clearance of accumulated glycogen from cells has emerged to occur through exocytosis. Indeed, overexpression of TFEB, a key transcription factor for lysosome biogenesis, was shown to result in lysosomal fusion with the plasma membrane, and thus in effective lysosomal exocytosis and significant glycogen clearance [68]. Whether releasing toxic lysosomal enzymes in the extracellular space, which might trigger inflammation and damage of matrix proteins, is beneficial or detrimental should be addressed in future studies.
Autophagy and physical exercise
The merits of regular physical activity on lipid and glucose homeostasis and in muscle mass maintenance, have been known for decades. In light of the rising obesity epidemic, a sedentary lifestyle is now considered a risk factor for various chronic ailments [69]. Exercise training has been documented to enhance metabolism, improve oxidative capacity as well as overall health and well-being. Six to eight weeks of endurance training are sufficient to elicit measurable elevations in aerobic capacity and to render undisputed cardioprotection as well as insulin sensitizing benefits [70, 71]. Exercise training has also been demonstrated to delay the loss of muscle mass that accompanies various pathologies including age-dependent muscle loss, also known as sarcopenia. Five months of exercise training was shown to completely reverse the premature aging phenotype of the mtDNA mutator mice, which harbor a mitochondrial DNA polymerase gamma with defective proofreading-exonuclease activity [72]. These mice experience premature aging and multisystemic degeneration stemming from mitochondrial dysfunction. Forced endurance exercise was sufficient to induce mitochondrial rejuvenation and prevent mtDNA depletion and mutations, thus blunting wholesale cell death in multiple organs and resulting in protection from multisystemic derailment. Most impressively, exercise-induced benefits were sufficient to increase the lifespan of these progeroid mice.
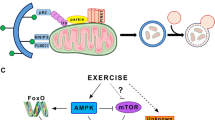
Many attempts have been made to elucidate the molecular mechanisms underlying the pleiotrophic effects of physical activity, as the ability to harness these benefits holds great therapeutic potential for the treatment and prevention of various morbidities as well as for delaying both senescence and mortality. Exercise confers many of its metabolic benefits by augmenting mitochondrial density as well as vitality. This is thought to be achieved mainly by mitochondrial biogenesis and remodeling, governed largely by the transcriptional co-activator PGC-1α [73]. It has been established that acute muscle contraction results in mild oxidative and metabolic stress (Fig. 3). Indeed, increases in metabolic demands during muscle contraction result in increased ROS production coupled with elevated AMP/ATP ratio along with a rise in NAD+, which activate various downstream signaling kinases as well as stress responders. Energetic sensors, such as AMPK, SIRT1, and p38-MAPK, are well-established factors activated by muscle contraction. These proteins work in unison to restore energetic homeostasis both acutely and chronically through the activation of downstream transcription factors like FoxOs and transcriptional activators such as PGC-1α. AMPK and SIRT1 lead to enhanced activity of FoxOs and PGC-1α by phosphorylation and deacetylation, respectively [74–76]. Moreover, p38-MAPK has been documented to regulate PGC-1α and this regulation has been deemed necessary for exercise-induced adaptations in skeletal muscle [77, 78]. Other than its role in regulating mitochondrial vigor, PGC-1α over-expression has been demonstrated to be sufficient to prevent muscle loss during various muscle wasting conditions, including aging [79, 80]. Similarly, inhibition of FoxOs blocks muscle loss in different catabolic conditions [81]. In addition, muscle contraction results in calcium surges that may lead to endoplasmic reticulum stress and thus induction of the unfolded protein response, a process partially mediated by PGC-1α [82]. Rapid increases in intracellular calcium also lead to the activation of calcium/calmodulin-dependent protein kinases and calcineurin, which may participate in exercise-induced muscular remodeling, possibly through facilitating HDAC export from the nucleus [83]. With this in mind, muscle contraction-induced ROS production can lead to pathological intermediates as well as cause damage to membrane lipids and proteins. If accumulated, these modified intermediates may hinder mitochondrial function and perturb proper cellular homeostasis, resulting in impaired muscle health. An efficient system, for the removal of damaged and carbonylated proteins, is therefore critical in maintaining overall muscle health and function [27] and may be crucial for achieving exercise-induced benefits. Indeed, it has been recently demonstrated that autophagy is activated following an acute bout of exercise [23, 60] and may play a vital role in exercise-induced muscular remodeling, as well as physical activity-mediated protection against HFD-induced glucose intolerance [60, 84].

Exercise-induced signaling in skeletal muscle. During exercise, acute muscle contraction results in an increase in intracellular calcium as well as mild oxidative and metabolic stress. This initiates a cellular stress cascade, where elevated calcium levels activate calcium/calmodulin-dependent protein kinases (CaMK) and calcineurin, resulting in HDAC export from the nucleus thus allowing for contraction-activated muscular remodeling. ROS activate various stress responders, such as sestrins, JNK, p38, p53, the unfolded protein response (UPR), and AMPK, which may also be activated by the elevated energetic demands elicited by an increase in AMP/ATP ratio. Sirtuins (SIRT1) are also activated due to elevations of NAD+. Activation of these pathways in response to muscle contraction culminates in autophagy induction. More chronically, these proteins activate muscular remodeling through a downstream transcriptional program largely mediated by PGC-1α. TF transcription factors
The activation of autophagy during muscle contraction is important for maintaining cellular energy homeostasis acutely, as well as for efficient organelle and protein turnover following exercise. In fact, autophagy markers are up-regulated following ultra-endurance exercise [85] as well as during acute bouts of treadmill running, and more chronically following exercise training [23, 60, 86]. Autophagy may also enhance cellular turnover following exercise in order to prime the cell for more efficient energy utilization and production with repeated bouts. He et al. reported that mice with Bcl2 knock-in mutations (Bcl2AAA), displaying intact basal autophagy but defective stress-induced autophagy, fail to achieve exercise-mediated protection against HFD-induced glucose intolerance, supporting beneficial metabolic effects of stimulus-induced autophagy [60]. These autophagy-mediated, exercise-induced metabolic benefits presumably occur through the activation of AMPK followed by the up-regulation of the glucose transporter Glut4 on the surface of muscle fibers. However, the upstream signals transducing this cascade remain a much sought after mystery. Moreover, Bcl2AAA mice also exhibit reduced adiponectin levels following exercise, when compared with wild-type animals. In agreement with this, a recent study suggested that adiponectin deficiency may aggravate HFD-induced obesity, metabolic derangement, cardiac hypertrophy, and contractile dysfunction, possibly through decreased myocardial autophagy [87]. In contrast to the aforementioned findings, skeletal muscle-specific Atg7 −/− mice actually show an improved metabolic profile, including glucose homeostasis, owing to mitochondrial inefficiency and the increased release of the mitokine FGF21 [61]. Although not yet explored, it is conceivable that deficient autophagy may alter the release of muscle-derived cytokines (myokines) or affect p62-dependent signaling. Myokines have been documented to play an important role in mediating whole-body adaptations to exercise through the regulation of skeletal muscle metabolism and signaling to distant metabolic organs. In addition to its canonical role as an autophagy substrate, p62 has also been found to participate in whole body metabolic regulation and indeed p62-deficient animals are obese [58].
The implication of autophagy in exercise is quite expected, as acute muscle contraction mimics a form of energetic stress and a temporary increase in ROS similar to that observed in other forms of nutrient deprivation, such as caloric restriction and starvation. All of these impinge on a common axis of metabolically sensitive enzymes, such AMPK and SIRT1, as well as on the activation of redox sensitive pathways involving p38, JNK, and cellular stress response elements, such as p53 and FoxOs, all of which were associated with the activation of autophagy in one way or another [88]. An additional family of factors that may be activated during exercise and were already found to play a role in autophagy induction are sestrins, although this remains to be established [89]. In addition, the downstream transcriptional executer PGC-1α has also been recently documented to play a role in lysosomal biogenesis [90] and in the regulation of autophagy-related protein expression [84]. Moreover, AKT is dephosphorylated following an acute bout of exercise [23, 60], suggesting that FoxOs and potentially mTOR could be another possible route to explore. The signaling events leading to the activation of autophagy during exercise and the role of autophagy in exercise-induced metabolic benefits warrant further examination, as the molecular mechanisms responsible for these events remain largely elusive.
The progression of autophagy during aging is controversial, and although some studies indicate an increase of various autophagic markers [22], autophagy flux is most likely compromised during senescence, leading to the accumulation of dysfunctional organelles and harmful protein aggregates. Aged rats display an up-regulation of some autophagy genes (Beclin 1), but exhibit no changes in other autophagy regulators (Atg7 and Atg9) and a decrease in the lysosomal marker LAMP-2. In this context, life-long caloric restriction alone, or combined with voluntary exercise, resulted in a mild reduction of LC3 expression and lipidation coupled with an increased LAMP-2 expression, suggesting a potential increase in autophagy flux. The age-related increase in oxidative damage and apoptosis were also attenuated by caloric restriction and exercise, perhaps through autophagy induction [50].
While physical activity stimulates autophagy in normal muscles and may hold therapeutic benefit in some disease models, exercise therapy should be considered with caution. Neither long-term nor shorter spurts of intense physical activity stimulated autophagy in Col6a1 −/− mice. Indeed, in this model of muscular dystrophy, where autophagy flux is compromised, exercise resulted in severe signs of myofiber death, mitochondrial abnormalities, degeneration and exacerbation of the dystrophic phenotype [23]. This further demonstrates the importance of autophagy for exercise-induced benefits. Thus, although some studies suggest a role for autophagy in physical activity-induced adaptations [60], very few have actually examined the mechanisms behind autophagy induction after an acute bout of exercise, and even fewer have evaluated the mechanisms involved in this activation.
Concluding remarks and future directions
Autophagy is essential for the metabolic homeostasis of the whole body under physiological conditions, and it can be further augmented during nutritional imbalance. This process has also been deemed essential for the proper maintenance and function of skeletal muscle, a central metabolic organ. More recently, autophagy has also been implicated in exercise-induced metabolic adaptations. The autophagic cascade therefore presents an enticing pharmaceutical target for the treatment of metabolic abnormalities as well as myopathies, and its roles in this perspective are starting to be uncovered. With this in mind, much investigation is still required in order to discern the function and molecular regulation of autophagy during conditions of elevated energetic balance, such as during HFD and overfeeding. Moreover, despite recent data revealing a role for autophagy following exercise, the mechanisms upstream and downstream of autophagy, during short- and long-term contractile activity in the different metabolic organs, should be further characterized. Finally, as autophagy is responsible for endosome trafficking as well as for some forms of exo- and endo-cytosis, it is conceivable that autophagy may have a role in myokine release and receipt. These processes represent an attractive endeavor that requires further exploration. Thus, the dissection of autophagy in muscle physiology and metabolic regulation promises to be an exhilarating journey full of discovery and therapeutic potential in the near future.
References
Levine B, Kroemer G (2008) Autophagy in the pathogenesis of disease. Cell 132:27–42
Klionsky DJ, Emr SD (2000) Autophagy as a regulated pathway of cellular degradation. Science 290:1717–1721
Mizushima N, Levine B, Cuervo AM, Klionsky DJ (2008) Autophagy fights disease through cellular self-digestion. Nature 451:1069–1075
Park C, Cuervo AM (2013) Selective autophagy: talking with the UPS. Cell Biochem Biophys 67:3–13
Shaid S, Brandts CH, Serve H, Dikic I (2013) Ubiquitination and selective autophagy. Cell Death Differ 20:21–30
McEwan DG, Dikic I (2011) The three musketeers of autophagy: phosphorylation, ubiquitylation and acetylation. Trends Cell Biol 21:195–201
Youle RJ, Narendra DP (2011) Mechanisms of mitophagy. Nat Rev Mol Cell Biol 12:9–14
Mizushima N, Komatsu M (2011) Autophagy: renovation of cells and tissues. Cell 147:728–741
Suzuki K, Ohsumi Y (2007) Molecular machinery of autophagosome formation in yeast, Saccharomyces cerevisiae. FEBS Lett 581:2156–2161
Inoki K, Kim J, Guan KL (2011) AMPK and mTOR in cellular energy homeostasis and drug targets. Ann Rev Phamacol Toxicol 52:381–400
Miyazaki M, McCarthy JJ, Fedele MJ, Esser KA (2011) Early activation of mTORC1 signalling in response to mechanical overload is independent of phosphoinositide 3-kinase/Akt signalling. J Physiol 589:1831–1846
Jacobs BL, You JS, Frey JW, Goodman CA, Gundermann DM, Hornberger TA (2013) Eccentric contractions increase the phosphorylation of tuberous sclerosis complex-2 (TSC2) and alter the targeting of TSC2 and the mechanistic target of rapamycin to the lysosome. J Physiol 591:4611–4620
Kim J, Kim YC, Fang C, Russell RC, Kim JH, Fan W, Liu R, Zhong Q, Guan KL (2013) Differential regulation of distinct Vps34 complexes by AMPK in nutrient stress and autophagy. Cell 152:290–303
Russell RC, Tian Y, Yuan H, Park HW, Chang YY, Kim J, Kim H, Neufeld TP, Dillin A, Guan KL (2013) ULK1 induces autophagy by phosphorylating Beclin-1 and activating VPS34 lipid kinase. Nature Cell Biol 15:741–750
Nazio F, Strappazzon F, Antonioli M, Bielli P, Cianfanelli V, Bordi M, Gretzmeier C, Dengjel J, Piacentini M, Fimia GM et al (2013) mTOR inhibits autophagy by controlling ULK1 ubiquitylation, self-association and function through AMBRA1 and TRAF6. Nature Cell Biol 15:406–416
Mammucari C, Milan G, Romanello V, Masiero E, Rudolf R, Del Piccolo P, Burden SJ, Di Lisi R, Sandri C, Zhao J et al (2007) FoxO3 controls autophagy in skeletal muscle in vivo. Cell Metab 6:458–471
van der Vos KE, Eliasson P, Proikas-Cezanne T, Vervoort SJ, van Boxtel R, Putker M, van Zutphen IJ, Mauthe M, Zellmer S, Pals C et al (2012) Modulation of glutamine metabolism by the PI(3)K-PKB-FOXO network regulates autophagy. Nature Cell Biol 14:829–837
Zhou X, Wang L, Hasegawa H, Amin P, Han BX, Kaneko S, He Y, Wang F (2010) Deletion of PIK3C3/Vps34 in sensory neurons causes rapid neurodegeneration by disrupting the endosomal but not the autophagic pathway. Proc Natl Acad Sci U S A 107:9424–9429
Sandri M (2010) Autophagy in skeletal muscle. FEBS Lett 584:1411–1416
Sandri M (2013) Protein breakdown in muscle wasting: role of autophagy-lysosome and ubiquitin–proteasome. Int J Biochem Cell Biol 45:2121–2129
Bonaldo P, Sandri M (2013) Cellular and molecular mechanisms of muscle atrophy. Dis Mod Mech 6:25–39
O’Leary MFN, Vainshtein A, Carter HN, Zhang Y, Hood DA (2012) Denervation-induced mitochondrial dysfunction and autophagy in skeletal muscle of apoptosis-deficient animals. Am J Physiol Cell Phsyiol 304:422–430
Grumati P, Coletto L, Schiavinato A, Castagnaro S, Bertaggia E, Sandri M, Bonaldo P (2011) Physical exercise stimulates autophagy in normal skeletal muscles but is detrimental for collagen VI deficient muscles. Autophagy 7:1415–1423
Nogalska A, D’Agostino C, Terracciano C, Engel WK, Askanas V (2010) Impaired autophagy in sporadic inclusion-body myositis and in endoplasmic reticulum stress-provoked cultured human muscle fibers. Am J Pathol 177:1377–1387
Grumati P, Coletto L, Sabatelli P, Cescon M, Angelin A, Bertaggia E, Blaauw B, Urciuolo A, Tiepolo T, Merlini L et al (2010) Autophagy is defective in collagen VI muscular dystrophies, and its reactivation rescues myofiber degeneration. Nat Med 16:1313–1320
Raben N, Hill V, Shea L, Takikita S, Baum R, Mizushima N, Ralston E, Plotzet P (2008) Suppression of autophagy in skeletal muscle uncovers the accumulation of ubiquitinated proteins and their potential role in muscle damage in Pompe disease. Hum Mol Genet 17:3897–3908
Masiero E, Agatea L, Mammucari C, Blaauw B, Loro E, Komatsu M, Metzger D, Reggiani C, Schiaffino S, Sandri M (2009) Autophagy is required to maintain muscle mass. Cell Metab 10:507–515
Fukuda T, Ahearn M, Roberts A, Mattaliano RJ, Zaal K, Ralston E, Plotz PH, Raben N (2006) Autophagy and mistargeting of therapeutic enzyme in skeletal muscle in Pompe disease. Mol Ther 14:831–839
Nascimbeni AC, Fanin M, Masiero E, Angelini C, Sandri M (2012) The role of autophagy in the pathogenesis of glycogen storage disease type II (GSDII). Cell Death Differ 19:1698–1708
Sugie K, Noguchi S, Kozuka Y, Arikawa-Hirasawa E, Tanaka M, Yan C, Saftig P, von Figura K, Hirano M, Ueno S et al (2005) Autophagic vacuoles with sarcolemmal features delineate Danon disease and related myopathies. J Neuropathol Exp Neurol 64:513–522
Nemazanyy I, Blaauw B, Paolini C, Caillaud C, Protasi F, Mueller A, Proikas-Cezanne T, Russell RC, Guan KL, Nishino I et al (2013) Defects of Vps15 in skeletal muscles lead to autophagic vacuolar myopathy and lysosomal disease. EMBO Mol Med 5:870–890
Ramachandran N, Munteanu I, Wang P, Aubourg P, Rilstone JJ, Israelian N, Naranian T, Paroutis P, Guo R, Ren ZR et al (2009) VMA21 deficiency causes an autophagic myopathy by compromising V-ATPase activity and lysosomal acidification. Cell 137:235–246
Ramos FJ, Chen SC, Garelick MG, Dai DF, Liao CY, Schreiber KH, MacKay VL, An EH, Strong R, Ladiges WC et al (2012) Rapamycin reverses elevated mTORC1 signaling in lamin A/C-deficient mice, rescues cardiac and skeletal muscle function, and extends survival. Sci Transl Med 4:144ra103
Carmignac V, Svensson M, Körner Z, Elowsson L, Matsumura C, Gawlik KI, Allamand V, Durbeej M (2011) Autophagy is increased in laminin alpha2 chain-deficient muscle and its inhibition improves muscle morphology in a mouse model of MDC1A. Hum Mol Genet 20:4891–4902
De Palma C, Morisi F, Cheli S, Pambianco S, Cappello V, Vezzoli M, Rovere-Querini P, Moggio M, Ripolone M, Francolini M et al (2012) Autophagy as a new therapeutic target in Duchenne muscular dystrophy. Cell Death Dis 3:e418
Choi JC, Muchir A, Wu W, Iwata S, Homma S, Morrow JP, Worman HJ (2012) Temsirolimus activates autophagy and ameliorates cardiomyopathy caused by lamin A/C gene mutation. Sci Transl Med 4:144ra102
Kley RA, Serdaroglu-Oflazer P, Leber Y, Odgerel Z, van der Ven PFM, Olivé M, Ferrer I, Onipe A, Mihaylov M, Bilbao JM et al (2012) Pathophysiology of protein aggregation and extended phenotyping in filaminopathy. Brain 135:2642–2660
Kley RA, van der Ven PFM, Olivé M, Höhfeld J, Goldfarb LG, Fürst DO, Vorgerd M (2013) Impairment of protein degradation in myofibrillar myopathy caused by FLNC/filamin C mutations. Autophagy 9:422–423
Lange S, Xiang F, Yakovenko A, Vihola A, Hackman P, Rostkova E, Kristensen J, Brandmeier B, Franzen G, Hedberg B et al (2005) The kinase domain of titin controls muscle gene expression and protein turnover. Science 308:1599–1603
Gotthardt M, Hammer RE, Hübner N, Monti J, Witt CC, McNabb M, Richardson JA, Granzier H, Labeit S, Herz J (2003) Conditional expression of mutant M-line titins results in cardiomyopathy with altered sarcomere structure. J Biol Chem 278:6059–6065
Al-Qusairi L, Prokic I, Amoasii L, Kretz C, Messaddeq N, Mandel JL, Laporte J (2013) Lack of myotubularin (MTM1) leads to muscle hypotrophy through unbalanced regulation of the autophagy and ubiquitin–proteasome pathways. FASEB J 27:3384–3394
Irwin WA, Bergamin N, Sabatelli P, Reggiani C, Megighian A, Merlini L, Braghetta P, Columbaro M, Volpin D, Bressan GM et al (2003) Mitochondrial dysfunction and apoptosis in myopathic mice with collagen VI deficiency. Nat Genet 35:367–371
Cullup T, Kho AL, Dionisi-Vici C, Brandmeier B, Smith F, Urry Z, Simpson MA, Yau S, Bertini E, McClelland V et al (2012) Recessive mutations in EPG5 cause Vici syndrome, a multisystem disorder with defective autophagy. Nat Genet 45:83–87
Zhao H, Zhao YG, Wang X, Xu L, Miao L, Feng D, Chen Q, Kovacs AL, Fan D, Zhang H (2013) Mice deficient in Epg5 exhibit selective neuronal vulnerability to degeneration. J Cell Biol 200:731–741
Zhang J, Kim J, Alexander A, Cai S, Tripathi DN, Dere R, Tee AR, Tait-Mulder J, Di Nardo A, Han JM et al (2013) A tuberous sclerosis complex signalling node at the peroxisome regulates mTORC1 and autophagy in response to ROS. Nat Cell Biol 15:1186–1196
Li L, Chen Y, Gibson SB (2013) Starvation-induced autophagy is regulated by mitochondrial reactive oxygen species leading to AMPK activation. Cell Signal 25:50–65
Zhao J, Brault JJ, Schild A, Cao P, Sandri M, Schiaffino S, Lecker SH, Goldberg AL (2007) FoxO3 coordinately activates protein degradation by the autophagic/lysosomal and proteasomal pathways in atrophying muscle cells. Cell Metab 6:472–483
Colman RJ, Anderson RM, Johnson SC, Kastman EK, Kosmatka KJ, Beasley TM, Allison DB, Cruzen C, Simmons HA, Kemnitz JW et al (2009) Caloric restriction delays disease onset and mortality in rhesus monkeys. Science 325:201–204
Mattison JA, Roth GS, Beasley TM, Tilmont EM, Handy AM, Herbert RL, Longo DL, Allison DB, Young JE, Bryant M et al (2012) Impact of caloric restriction on health and survival in rhesus monkeys from the NIA study. Nature 489:318–321
Wohlgemuth SE, Seo AY, Marzetti E, Lees HA, Leeuwenburgh C (2010) Skeletal muscle autophagy and apoptosis during aging: effects of calorie restriction and life-long exercise. Exp Gerontol 45:138–148
Matsakas A, Romanello V, Sartori R, Masiero E, Macharia R, Otto A, Elashry M, Sandri M, Patel K (2013) Food restriction reverses the hyper-muscular phenotype and force generation capacity deficit of the myostatin null mouse. Int J Sports Med 34:223–231
Singh R, Cuervo AM (2011) Autophagy in the cellular energetic balance. Cell Metab 13:495–504
Singh R, Kaushik S, Wang Y, Xiang Y, Novak I, Komatsu M, Tanaka K, Cuervo AM, Czaja MJ (2009) Autophagy regulates lipid metabolism. Nature 458:1131–1135
Dall’Armi C, Devereaux KA, Di Paolo G (2013) The role of lipids in the control of autophagy. Curr Biol 23:33–45
Singh R, Cuervo AM (2013) Lipophagy: connecting autophagy and lipid metabolism. Int J Cell Biol (in press)
Singh R, Xiang Y, Wang Y, Baikati K, Cuervo AM, Luu YK, Tang Y, Pessin JE, Schwartz GJ, Czaja MJ (2009) Autophagy regulates adipose mass and differentiation in mice. J Clin Invest 119:3329–3339
Zhang Y, Goldman S, Baerga R, Zhao Y, Komatsu M, Jin S (2009) Adipose-specific deletion of autophagy-related gene 7 (atg7) in mice reveals a role in adipogenesis. Proc Natl Acad Sci U S A 106:19860–19865
Moscat J, Diaz-Meco MT (2011) Feedback on fat: p62-mTORC1-autophagy connections. Cell 147:724–727
Koga H, Kaushik S, Cuervo AM (2010) Altered lipid content inhibits autophagic vesicular fusion. FASEB J 24:3052–3065
He C, Bassik MC, Moresi V, Sun K, Wei Y, Zou Z, Loh J, Fisher J, Sun Q, Korsmeyer S et al (2012) Exercise-induced BCL2-regulated autophagy is required for muscle glucose homeostasis. Nature 481:511–515
Kim KH, Jeong YT, Oh H, Kim SH, Cho JM, Kim YN, Kim SS, Kim DH, Hur KY, Kim HK et al (2013) Autophagy deficiency leads to protection from obesity and insulin resistance by inducing Fgf21 as a mitokine. Nat Med 19:83–92
Moresi V, Carrer M, Grueter CE, Rifki OF, Shelton JM, Richardson JA, Bassel-Duby R, Olson EN (2012) Histone deacetylases 1 and 2 regulate autophagy flux and skeletal muscle homeostasis in mice. Proc Natl Acad Sci U S A 109:1649–1654
Kotoulas OB, Kalamidas SA, Kondomerkos DJ (2006) Glycogen autophagy in glucose homeostasis. Pathol Res Pract 202:631–638
Fujitani Y, Ebato C, Uchida T, Kawamori R, Watada H (2009) β-cell autophagy: a novel mechanism regulating β-cell function and mass: lessons from β-cell-specific Atg7-deficient mice. Islets 1:151–153
Ebato C, Uchida T, Arakawa M, Komatsu M, Ueno T, Komiya K, Azuma K, Hirose T, Tanaka K, Kominami E et al (2008) Autophagy is important in islet homeostasis and compensatory increase of beta cell mass in response to high-fat diet. Cell Metab 8:325–332
Ravikumar B, Stewart A, Kita H, Kato K, Duden R, Rubinsztein DC (2003) Raised intracellular glucose concentrations reduce aggregation and cell death caused by mutant huntingtin exon 1 by decreasing mTOR phosphorylation and inducing autophagy. Hum Mol Genet 12:985–994
Nishino I, Fu J, Tanji K, Yamada T, Shimojo S, Koori T, Mora M, Riggs JE, Oh SJ, Koga Y et al (2000) Primary LAMP-2 deficiency causes X-linked vacuolar cardiomyopathy and myopathy (Danon disease). Nature 406:906–910
Spampanato C, Feeney E, Li L, Cardone M, Lim JA, Annunziata F, Zare H, Polishchuk R, Puertollano R, Parenti G et al (2013) Transcription factor EB (TFEB) is a new therapeutic target for Pompe disease. EMBO Mol Med 5:691–706
Myers J, Atwood JE, Froelicher V (2003) Active lifestyle and diabetes. Circulation 107:2392–2394
Adhihetty PJ, Irrcher I, Joseph AM, Ljubicic V, Hood DA (2003) Plasticity of skeletal muscle mitochondria in response to contractile activity. Exp Physiol 88:99–107
Brown DA, Moore RL (2007) Perspectives in innate and acquired cardioprotection: cardioprotection acquired through exercise. J Appl Physiol 103:1894–1899
Safdar A, Bourgeois JM, Ogborn DI, Little JP, Hettinga BP, Akhtar M, Thompson JE, Melov S, Mocellin NJ, Kujoth GC et al (2011) Endurance exercise rescues progeroid aging and induces systemic mitochondrial rejuvenation in mtDNA mutator mice. Proc Natl Acad Sci U S A 108:4135–4140
Baar K, Wende AR, Jones TE, Marison M, Nolte LA, Chen M, Kelly DP, Holloszy JO (2002) Adaptations of skeletal muscle to exercise: rapid increase in the transcriptional coactivator PGC-1. FASEB J 16:1879–1886
Rodgers JT, Lerin C, Haas W, Gygi SP, Spiegelman BM, Puigserver P (2005) Nutrient control of glucose homeostasis through a complex of PGC-1alpha and SIRT1. Nature 434:113–118
Jäger S, Handschin C, St-Pierre J, Spiegelman BM (2007) AMP-activated protein kinase (AMPK) action in skeletal muscle via direct phosphorylation of PGC-1alpha. Proc Natl Acad Sci U S A 104:12017–12022
Brunet A, Sweeney LB, Sturgill JF, Chua KF, Greer PL, Lin Y, Tran H, Ross SE, Mostoslavsky R, Cohen HY et al (2004) Stress-dependent regulation of FOXO transcription factors by the SIRT1 deacetylase. Science 303:2011–2015
Akimoto T, Pohnert SC, Li P, Zhang M, Gumbs C, Rosenberg PB, Williams RS, Yan Z (2005) Exercise stimulates Pgc-1alpha transcription in skeletal muscle through activation of the p38 MAPK pathway. J Biol Chem 280:19587–19593
Pogozelski AR, Geng T, Li P, Yin X, Lira VA, Zhang M, Chi JT, Yan Z (2009) p38gamma mitogen-activated protein kinase is a key regulator in skeletal muscle metabolic adaptation in mice. PloS One 4:e7934
Sandri M, Lin J, Handschin C, Yang W, Arany ZP, Lecker SH, Goldberg AL, Spiegelman BM (2006) PGC-1alpha protects skeletal muscle from atrophy by suppressing FoxO3 action and atrophy-specific gene transcription. Proc Natl Acad Sci U S A 103:16260–16265
Wenz T, Rossi SG, Rotundo RL, Spiegelman BM, Moraes CT (2009) Increased muscle PGC-1alpha expression protects from sarcopenia and metabolic disease during aging. Proc Natl Acad Sci U S A 106:20405–20410
Schiaffino S, Dyar KA, Ciciliot S, Blaauw B, Sandri M (2013) Mechanisms regulating skeletal muscle growth and atrophy. FEBS J 280:4294–4314
Wu J, Ruas JL, Estall JL, Rasbach KA, Choi JH, Ye L, Bostrom P, Tyra HM, Crawford RW, Campbell KP et al (2011) The unfolded protein response mediates adaptation to exercise in skeletal muscle through a PGC-1α/ATF6α complex. Cell Metab 13:160–169
McGee SL, Fairlie E, Garnham AP, Hargreaves M (2009) Exercise-induced histone modifications in human skeletal muscle. J Physiol 587:5951–5958
Lira VA, Okutsu M, Zhang M, Greene NP, Laker RC, Breen DS, Hoehn KL, Yan Z (2013) Autophagy is required for exercise training-induced skeletal muscle adaptation and improvement of physical performance. FASEB J 27:4184–4193
Jamart C, Francaux M, Millet GY, Deldicque L, Frère D, Féasson L (2012) Modulation of autophagy and ubiquitin–proteasome pathways during ultra-endurance running. J Appl Physiol 112:1529–1537
Salminem A, Vihko V (1984) Autophagic response to strenuous exercise in mouse skeletal muscle fibers. Virchows Arch Cell Pathol Incl Mol Pathol 45:97–106
Guo R, Zhang Y, Turdi S, Ren J (2013) Adiponectin knockout accentuates high fat diet-induced obesity and cardiac dysfunction: role of autophagy. Biochim Biophys Acta 1832:1136–1148
O’Neill HM, Maarbjerg SJ, Crane JD, Jeppesen J, Jørgensen SB, Schertzer JD, Shyroka O, Kiens B, van Denderen BJ, Tarnopolsky MA et al (2011) AMP-activated protein kinase (AMPK) beta1beta2 muscle null mice reveal an essential role for AMPK in maintaining mitochondrial content and glucose uptake during exercise. Proc Natl Acad Sci U S A 108:16092–16097
Sanchis-Gomar F (2013) Sestrins: novel antioxidant and AMPK-modulating functions regulated by exercise? J Cell Physiol 228:1647–1650
Takikita S, Schreiner C, Baum R, Xie T, Ralston E, Plotz PH, Raben N (2010) Fiber type conversion by PGC-1α activates lysosomal and autophagosomal biogenesis in both unaffected and Pompe skeletal muscle. PloS One 5:e15239
Acknowledgments
The authors apologize to their colleagues whose studies were not cited owing to space limitations. This work is supported by grants from Telethon-Italy (GGP10225 and GGP11082 to P.B.; TCP04009 to M.S), the European Union (MYOAGE, contract: 223576 of FP7 to M.S.; BIO-NMD FP7-HEALTH-241665 to P.B.), ERC (MYOPHAGY, contract: 282310 to M.S.), the Italian Ministry of Education, University and Research (to P.B. and M.S.), Foundation Leducq (to M.S.), and CARIPARO (to M.S. and P.B.). A.V. is supported by scholarships from Natural Sciences and Engineering Research Council of Canada-CGS and NSERC Michael Smith Foreign Study Supplement. The authors declare no conflict of interest.
Author information
Authors and Affiliations
Corresponding authors
Additional information
Anna Vainshtein and Paolo Grumati equally contributed to this work.
Rights and permissions
About this article
Cite this article
Vainshtein, A., Grumati, P., Sandri, M. et al. Skeletal muscle, autophagy, and physical activity: the ménage à trois of metabolic regulation in health and disease. J Mol Med 92, 127–137 (2014). https://doi.org/10.1007/s00109-013-1096-z
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00109-013-1096-z