
Abstract
The split product of the complement protein, C5, is C5a and is an extremely potent pro-inflammatory peptide that interacts with two C5a receptors, C5aR and C5L2, present on surfaces of phagocytes as well as other cell types. The former is a well-established receptor that initiates G-protein-coupled signaling via mitogen-activated protein kinase pathways. Its in vivo blockade greatly reduces inflammatory injury. Much less is known about C5L2, occupancy of which by C5a does not initiate increased intracellular Ca2+. There are numerous conflicting reports suggesting that C5L2 is a “default receptor” that attenuates C5a-dependent biological responses by competing with C5aR for binding of C5a. However, there are other reports suggesting that C5L2 plays an active, positive role in inflammatory responses. Better definition of C5L2 is needed if its in vivo blockade, along with C5aR, is to be considered in complement-dependent inflammatory diseases.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
The initial structural characterization in 1991 [1, 2] of the rhodopsin-like receptor for C5a, C5aR [1], and the receptor for N-formyl Met-Leu-Phe [2] provided key biochemical information that would permit development of antibodies and synthetic inhibitors to these receptors, for which C5aR binds C5a with high affinity and initiates G-protein-dependent cascade of cell responses (increased intracellular Ca2+, granule fusion with the cell membrane, enzyme release, on oxidative burst [H2O2 production], etc.). Similar signaling events occur with receptor–ligand interaction involving the formyl peptide receptor. C5aR is now known to be crucial in the initiation of acute inflammatory responses [3, 4]. Both C5a receptors, C5aR and C5L2, have been the subject of a recent review [5]. In the early 2000s, a second receptor for C5a, C5L2, was described, based on its ability to bind C5a and C5a des Arg with high affinity in the absence of an intracellular Ca2+ signal [6]. However, signaling as assessed by phosphoprotein appearance in myeloid-derived cells (neutrophils [PMNs], macrophages, monocytes, and dendritic cells) could not be measured [7]. Functional responses, such as chemotaxis, enzyme release, the respiratory burst, etc., were also undetectable after ligation of C5L2 with C5a, leading to the designation of C5L2 as a “default” or “scavenger” receptor [6]. In other words, C5L2 competed with C5aR for binding of C5a, and the balance in C5a occupancy between the two receptors would determine the outcome (pro-inflammatory or anti-inflammatory).
C3a des Arg as a controversial ligand for C5L2
Between 2002 and 2005, C5L2 was described as having the ability to bind with C3a des Arg with high affinity [8–10]. Under such conditions, C3a des Arg caused cell lines transfected with C5L2 as well as normal adipose cells to show increased synthesis of triglycerides and increased uptake of glucose [9]. Accordingly, C3a des Arg was named “acylation stimulating protein.” It should be pointed out that C3a des Arg was isolated from activated human serum and required >1 μM for the observed activity. The possibility of contaminating C5a or C5a des Arg needs to be considered. In addition, recombinant C3a des Arg or the truncated form of the peptide should be employed in order to determine if the functional activity on adipocytes can be confirmed. It was also suggested that C5L2 would bind C4a, and it was confirmed that C5L2 bound C5a and C5a des Arg with high affinity [6, 10, 11]. In 2006, investigators from Germany and the UK, using transfected cell lines (HEK 293 and RBL cells), described their inability to demonstrate the binding of either C3a or C3a des Arg C5L2 to these cells. It was argued that “…C5L2 is not a receptor C3a or C3a des Arg 77” [11]. One possible explanation may be that adipocytes have unique binding and functional responses to C3a and C3a des Arg that are not seen in other cell types. Resolution of this controversy is critically needed.
Biological responses of C5aR and C5L2
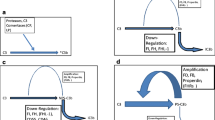
Evidence for the roles of C5aR and C5L2 is summarized in Table 1 and Fig. 1. Using a model of acute lung injury (ALI) in mice following distal airway deposition of IgG immune complexes, the inflammatory outcome was measured by morphological changes, albumin leak, and mediator presence (e.g., interleukin 6 [IL-6], tumor necrosis factor alpha [TNF-α]). Genetic absence of C5aR in C57Bl/6 mice was associated with greatly reduced evidence of ALI as well as reduced PMN accumulation [12] and reduced levels of pro-inflammatory mediators. C5L2−/− mice undergoing the same type of lung inflammatory injury (IgG immune complexes) showed amplified inflammation injury [7]. In these C5L2−/− mice, increased PMN content in bronchoalveolar lavage fluids was found, together with higher levels of IL-6 and TNF-α. When blocking antibody to C5L2 was employed in mice with cecal ligation and puncture (CLP), plasma levels of IL-6 were nearly threefold higher than in mice in which C5L2 was not blocked [13]. Collectively, these two experimental systems suggested that the lack of availability of C5L2 results in enhanced inflammatory responses via engagement of C5a exclusively with C5aR, in line with C5L2 acting as a “scavenger” or “default” receptor, functioning as a counterbalance to the pro-inflammatory effects of C5aR. However, in a recent report also using ovalbumin–anti-ovalbumin IgG immune complexes, reduced inflammatory injury in airways has been described in C5L2−/− mice [14].

Function of C5aR receptors
In vitro signaling in C5L2−/− macrophages or PMNs showed reduced activation (phosphorylation) of mitogen-activated protein kinases (MAPKs; p38, ERK1/2, JNK) after incubation with C3a or C5a [14]. Production of TNF-α or IL-6 in C5L2−/− cells stimulated with lipopolysaccharide (LPS) or LPS + C5a was also reduced. This is in contrast to an earlier report in which cells transfected with C5L2 showed little, if any, activation of MAPKs after incubation with C5a [7]. While there remains great confusion regarding the relationship between C5L2 and activation of signaling pathways, it is possible that differences in the types of cells being studied, problems with transgenic expression of C5L2 and linkage with signaling pathways, and the possibility that C5aR and C5L2 may interact in some unknown manner (such as heterodimerization) are issues that have not been adequately addressed but may be important in resolving the current confusion about the functionality of C5L2.
In the setting of experimental asthma in mice several years ago, it was described that C5−/− mice are protected against experimental asthma [15], suggesting that activation products of C5 are harmful in this model. Recent studies suggest that, in C5aR−/− mice, the upper airway inflammatory response and airway hyper-responsiveness (AHR) are significantly diminished, suggesting that C5aR induces “…marked enhancement of TH2 polarized responses” [16]. In C5L2−/− mice undergoing ovalbumin-induced asthma, development of AHR and peribronchiolar inflammation was reduced, suggesting that C5L2 contributes positively to the lung inflammatory response [14]. Finally, in the same study, PMN accumulation in dorsal subcutaneous air pouches of C5L2−/− mice was reduced (by more than twofold). Collectively, these data from the Toronto group suggest a linkage between C5L2, MAPK signaling, and phagocyte activation and mobilization. However, essentially, none of these reported phenotypes are seen in the mice studied in the Gerard lab (C. Gerard, J. Köhl, unpublished data).
Substantial work has been done to define the possible role of C5aR and C5L2 in the setting of experimental sepsis, using knockout mice or mice in which C5aR or C5L2 was blocked with an antibody (to the N-terminal region of each receptor) or with a synthetic inhibitor of both receptors. Using survival as the endpoint, evidence was obtained that both receptors contributed to adverse events resulting in mortality after CLP [17]. Absence or blockade of either receptor greatly reduced plasma levels of pro-inflammatory cytokines (IL-1β, IL-6, macrophage inflammatory protein [MIP]-1α, and MIP-2), but interestingly, there was no effect on TNF-α levels in the plasma of CLP mice, suggesting that cytokines and chemokines are individually regulated. These studies also showed that C5L2 absence either in vivo or in vitro resulted in little, if any, expression of HMGB1, either in plasma of CLP mice or in supernatant fluids from mouse macrophages or human blood PMNs or peripheral mononuclear cells stimulated with C5a, LPS, or the combination [17]. Collectively, these data suggest that, in the setting of sepsis, C5L2 contributes to adverse events related to the “cytokine storm,” lethality, and production of HMGB1, which is known to be linked to the adverse events in CLP-induced sepsis [18].
As indicated above, C5L2−/− mice have been reported to be hypersusceptible to lethal endotoxemia, although the explanation for this observation is not apparent [14]. This would seem to be reverse of the role of C5L2 in the setting of CLP-induced sepsis in which C5L2 contributes to pro-inflammatory, harmful events [17]. In the setting of septic shock in humans, C5L2 content on blood PMNs falls, likely because of internalization of C5aC5L2 complexes [19]. Similar changes occur in blood PMNs from CLP rats. Loss of C5L2 on blood PMNs after CLP was prevented if animals were given blocking antibody to C5a at the time of CLP. It has been suggested that C5L2 may have anti-inflammatory properties in the central nervous system [20]. The biological role of C5L2 has been the subject of a recent review [21]. Finally, a recent study suggests that C5a and C5a des Arg binding to cells results in a clathrin-dependent pathway of rapid internalization of these ligands that then become degraded, whereas internalization involving C5aR is slower, with suggestions that undegraded C5a and C5a des Arg may be released into the extravascular environment [22].
Conclusions
It is clear that there are two C5a receptors, C5aR and C5L2, each of which has high affinity binding for C5a and C5a des Arg. What is confusing is the extent to which C3a and C3a des Arg bind to C5L2. It is also known that C5aR interacts with C5a in a G-protein-dependent manner, which results in increased intracellular Ca2+ together with MAPK and Akt activation, followed by a series of functional responses such as chemotaxis, enzyme release, Mac-1 upregulation, degranulation, a respiratory burst, etc. The role of C5aR in the setting of sepsis also seems obvious: C5aR interacting with C5a contributes to adverse events in ALI after deposition of IgG immune complexes and in CLP-induced sepsis, including the intensified pro-inflammatory state and lethality. As for C5L2, there is much confusion and less certainty about its role. Clearly, it binds both C5a and C5a des Arg, but no increased intracellular Ca2+ occurs. Beyond this, its role is controversial. Interestingly, it has been recently shown that the vast majority of C5L2 exists intracellularly in endosomal compartments and is translocated to the cell surface after cell activation (Gerard, C., personal communication). It remains to be determined what signaling pathway is engaged by C5L2. Whether C3a des Arg is a ligand for C5L2 is strongly debated (see above). There is disagreement as to whether C5a reacting with C5L2 activates MAPK signaling. There is mixed evidence about the role of C5L2 in cytokine and chemokine production. This may be a matter of the mediator in question (e.g., IL-6 vs. IL-1β vs. TNF-α, etc.) It seems likely that C5a interaction with C5L2 triggers HMGB1 release, which is known to participate in the adverse consequences of experimental sepsis. As mentioned above, it is possible that some type of interaction between C5aR and C5L2 occurs, such as heterodimerization, or that C5a interaction with C5aR results in signaling events that alter cell responsiveness of C5L2 to C5a.
References
Gerard NP, Gerard C (1991) The chemotactic receptor for human C5a anaphylatoxin. Nature 349:614–617
Boulay F, Tardif M, Brouchon L, Vignais P (1990) Synthesis and use of a novel N-formyl peptide derivative to isolate a human N-formyl peptide receptor cDNA. Biochem Biophys Res Commun 168:1103–1109
Strainic MG, Liu J, Huang D, An F, Lalli PN, Muqim N, Shapiro VS, Dubyak GR, Heeger PS, Medof ME (2008) Locally produced complement fragments C5a and C3a provide both costimulatory and survival signals to naïve CD4+ T cells. Immunity 28:425–435
Hawlisch H, Wills-Karp M, Karp CL, Köhl J (2004) The anaphylatoxins bridge innate and adaptive immune responses in allergic asthma. Mol Immunol 41:123–131
Lee H, Whitfeld PL, Mackay CR (2008) Receptors for complement C5a. The importance of C5aR and the enigmatic role of C5L2. Immunol Cell Biol 86:153–160
Okinaga S, Slattery D, Humbles A, Zsengeller Z, Morteau O, Kinrade MB, Brodbeck RM, Krause JE, Choe HR, Gerard NP, Gerard C (2003) C5L2, a nonsignaling C5a binding protein. Biochemistry 42:9406–9415
Gerard N, Lu B, Liu P, Craig S, Fujiwara Y, Okinaga S, Gerard C (2005) An anti-inflammatory function for the complement anaphylatoxin C5a-binding protein, C5L2. JBC 280:39677–39680
Kalant D, Cain SA, Maslowska M, Sniderman AD, Cianflone K, Monk PN (2003) The chemoattractant receptor-like protein C5L2 binds the C3a des-arg77/acylation-stimulating protein. J Biol Chem 278:11123–11129
Kalant D, MacLaren, Cui W, Samanta R, Monk PN, Laporte SA, Cianflone K (2005) C5L2 is a functional receptor for acylation-stimulating protein. J Biol Chem 280:23936–23944
Cain SA, Monk PN (2002) The orphan receptor C5L2 has high affinity binding sites for complement fragments C5a and C5a des-arg74. J Biol Chem 277:7165–7169
Johswich K, Martin M, Thalmann J, Rheinheimer C, Monk PN, Klos A (2006) Ligand specificity of the anaphylatoxin C5L2 receptor and its regulation on myeloid and epithelial cell lines. J Biol Chem 281:39088–39095
Hopken UE, Lu B, Gerard NP, Gerard C (1997) Impaired inflammatory responses in the reverse Arthus reaction through genetic deletion of the C5a receptor. J Exp Med 186:749–756
Gao H, Neff TA, Guo TA, Speyer CL, Sarma JV, Tomlins S, Man Y, Riedemann NC, Hoesel LM, Younkin E, Zetoune FS, Ward PA (2005) Evidence for a functional role of the second C5a receptor C5L2. FASEB J 19:1003–1005
Chen NJ, Mirtsos C, Suh D, Lu YC, Lin WJ, McKerlie C, Lee T, Baribault H, Tian H, Yeh WC (2007) C5L2 is critical for the biological activities of the anaphylatoxins C5a and C3a. Nat Lett 446:203–207
Karp CL, Grupe A, Schadt E, Ewart SL, Keane-Moore M, Cuomo PJ, Köhl J, Wahl L, Kuperman D, Germer S, Aud D, Peltz G, Wills-Karp (2000) Identification of complement factor 5 as a susceptibility locus for experimental allergic asthma. Nat Immunol 1:221–226
Köhl J, Baelder R, Lewkowich IP, Pandey MK, Hawlisch H, Wang L, Best J, Herman NS, Sproles AA, Zwirner J, Whitsett JA, Gerard C, Sfyroera G, Lambris JD, Wills-Karp M (2006) A regulatory role for the C5a anaphylatoxin in type 2 immunity in asthma. J Clin Invest 116:628–632
Rittirsch D, Flierl MA, Nadeau BA, Day DE, Huber-Lang M, Mackay CR, Zetoune FS, Gerard NP, Cianflone K, Koehl J, Gerard C, Sarma JV, Ward PA (2008) Functional roles for C5a receptors in sepsis. Nat Med 14:551–557
Yang H, Wang H, Czura CJ, Tracey KJ (2005) The cytokine activity of HMGB1. J Leuk Biol 78:1–8
Huber-Lang M, Sarma JV, Rittirsch D, Schreiber H, Weiss M, Flierl M, Younkin E, Schneider M, Suger-Wiedeck H, Gebhard F, McClintock SD, Neff T, Zetoune F, Bruckner U, Guo RF, Monk PN, Ward PA (2005) Changes in the novel orphan, C5a receptor (C5L2), during experimental sepsis and sepsis in humans. J Immunol 174:1104–1110
Gavrilyuk V, Kalinin S, Hilbush BS, Middlecamp A, McGuire S, Pelligrino D, Weinberg G, Feinstien DL (2005) Identification of complement 5a-like receptor (C5L2) from astrocytes: characterization of anti-inflammatory properties. J Neurochem 92:1140–1149
Johswich K, Kos A (2007) C5L2 – an anti-inflammatory molecule or a receptor for acylation stimulating protein (C3a-desArg)? Review. Advances in experimental medicines and biology. In: Lambris JD (ed) Current topics in innate immunity. Springer, New York, pp 159–180
Scola A-M et al (2008) The human complement fragment receptor, C5L2, is a recycling decoy receptor. Mol Immunol. doi:10.1016/j.molimm.2008.11.001
Acknowledgment
We thank Professor Craig Gerard (Dept. Pediatrics, Children’s Hospital of Boston, MA) for helpful suggestions for the manuscript. This work is supported in part by National Institute of Health grants: HL-31963, GM-61656 and GM-029507.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Ward, P.A. Functions of C5a receptors. J Mol Med 87, 375–378 (2009). https://doi.org/10.1007/s00109-009-0442-7
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00109-009-0442-7