
Abstract
A novel autoantigen named GW182 was recently identified when the serum from a patient with a sensory ataxic polyneuropathy was used to immunoscreen a HeLa cDNA library. Unique features of the GW182 protein include 39 repeats of glycine (G) and tryptophan (W) residues, binding to a subset of messenger RNA and localization to unique structures within the cytoplasm that were designated GW bodies (GWBs). The goal of the present study was to identify the clinical features of patients with anti-GW182 antibodies and to characterize the B cell anti-GW182 response by defining the epitopes bound by human autoantibodies. The most common clinical diagnosis of patients with anti-GW182 antibodies was Sjögren’s syndrome followed by mixed motor/sensory neuropathy, and systemic lupus erythematosus. Of interest, 5 (28%), 9 (50%), and 3 (17%) of the 18 sera that react with GWBs had autoantibodies to the GW182 and the 52 kDa and 60 kDa SS-A/Ro autoantigens, respectively. Epitopes bound by the human autoantibodies were mapped to the GW-rich middle part of the protein, the non-GW rich region, and the C-terminus of GW182 protein. None of the GW182 epitopes had significant sequence similarities to other known proteins. GW182 represents a new category of ribonucleoprotein autoantigens.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The sera from patients with systemic autoimmune disease have been used to isolate and identify novel autoantigens that are components of macromolecular complexes that have a variety of cellular functions including transcription, translation, and ribosomal processing [1, 2, 3]. In addition, the identification of autoantigens and the characterization of their respective epitopes are used as diagnostic tools to assist in the clinical evaluation of autoimmune diseases [4, 5, 6]. For example, the presence of autoantibodies to double-stranded DNA and the Sm small nuclear ribonucleoproteins (RNPs) are highly specific serological markers for systemic lupus erythematosus (SLE) [7]. Sjögren’s syndrome (SjS) is characterized by the presence of autoantibodies to SS-A/Ro, and/or SS-B/la [8]. In addition, the identification of autoantigens and their association with autoimmune disease is a key approach to understanding the autoimmune disease state [9, 10].
Recently a novel autoantigen named GW182 was discovered when the serum from a patient with ataxic sensory polyneuropathy was used to immunoscreen a HeLa cDNA library [11]. Interesting features of the GW182 protein include 39 repeats of glycine (G) and tryptophan (W) residues and its localization in unique cytoplasmic structures that have been designated as GW bodies (GWBs). The GW182 protein, which has an RNA recognition motif and binds specific mRNAs, is thought to be part of a mRNA-protein macromolecular complex. It has been postulated that GWBs provide an additional level of posttranscriptional gene regulation and function in mRNA processing in a cell compartment referred to as the ribosome or posttranscriptional operon [12, 13]. More recent evidence implicates the GW182 protein and GWBs in mRNA degradation pathways [14]. The goal of the present study was to characterize the B-cell immune response in patients with antibodies to GWBs and the GW182 protein which resides within the GWBs and to assess the clinical features of these patients. This is the first report of the clinical features of patients with anti-GWB antibodies and a description of the GW182 epitopes bound by these sera.
Materials and methods
Patient serum and antibodies
All human sera used in this study were obtained from serum banks at the Advanced Diagnostics Laboratory (University of Calgary, Calgary, Canada), the W.M. Keck Autoimmune Disease Center (Scripps Research Institute, La Jolla, Calif., USA), and Juntendo University (Tokyo, Japan). The index human serum used in this study was selected based on its reactivity to an apparently unique cytoplasmic domain and its reactivity with the native and recombinant GW182 protein [11]. Clinical information was obtained by contacting the referring physician and retrospective chart review.
Indirect immunofluorescence
The presence of anti-GW182 antibodies in the human sera were initially tested by Indirect immunofluorescence (IIF) using HEp-2 cell substrates (Immuno Concepts, Sacramento, Calif., USA) and had a cytoplasmic staining pattern that was characteristic of anti-GWB antibodies [11]. Reactivity with GWBs was confirmed by IIF colocalization studies on HEp-2 cells where a monoclonal antibody (4B6) to the recombinant GW182 protein which stains GWBs was used as the marker antibody [15]. Secondary antibodies for colocalization studies were fluorescein isothiocyanate conjugated anti-mouse IgG (Jackson ImmunoResearch, West Grove, Pa.,USA) and fluorescein isothiocyanate or Cy3-conjugated anti-human IgG (Jackson ImmunoResearch). Nuclei in the cell substrates were stained with 4′,6-diamidino-2-phenylindole that was included in the glycerol mounting medium (VectaShield, Vector, Burlingame, Calif., USA).
In vitro transcription/translation and immunoprecipitation
Reactivity of the sera with recombinant GW182 protein was confirmed by immunoprecipitation (IP) of the recombinant protein. The full-length GW182 cDNA was used as a template to synthesize the protein in an in vitro transcription and translation (TnT) protocol that used a rabbit reticulocyte lysate kit (TnT, Promega Biotec, Madison, Wis., USA) in the presence of [35S]methionine at 30°C for 3–4 h as previously described [11, 16]. To confirm the presence of TnT products 2- to 5-µl samples were separated by sodium dodecyl sulfate (SDS) polyacrylamide gel electrophoresis and analyzed by autoradiography. The TnT products were then used in IP reactions by combining 100 µl of a 10% protein A Sepharose bead suspension (Sigma, catalog no. P-3391), 10 µl human serum, 500 µl NET2 buffer (50 mM Tris-HCl, pH 7.4, 150 mM NaCl, 5 mM EDTA, 0.5% Nonidet P-40, 0.5% deoxycholic acid, 0.1% SDS, 0.02% sodium azide), and 10 µl of radiolabeled protein product. After incubation for 1 h at 4–8°C the suspension was washed five times in NET2, the proteins eluted in 10 µl sample buffer, and analyzed by 10% gel SDS polyacrylamide gel electrophoresis as described [17].
Epitope mapping
Epitope mapping employed sequential peptides of 15 amino acids offset by five amino acids, representing the full-length GW182 protein, were synthesized on membranes using the SPOT technology as previously described [18, 19, 20]. The membranes containing the peptides were processed for immunoblotting by soaking the membrane in Tris-buffered saline (TBS; 10 mM Tris-HCl pH7.6, 150 mM NaCl) for 10 min and then blocking with 2% milk/TBS for 1 h at room temperature. The human sera were diluted 1/100 in 2% milk/TBS and applied to the membrane. After 2 h of incubation at room temperature the membrane was washed three times with TBS. A horseradish-peroxidase conjugated goat anti-human IgG (Jackson ImmunoResearch) was diluted according to the manufacturer’s protocol, and reactivity was visualized using enhanced chemiluminescence western blotting detection reagents (Amersham International). After reactive epitopes were identified a BLAST search of the GenBank using the reactive sequences as the query was conducted to identify homologous sequences in other proteins.
Purified recombinant GW182
The GW182 cDNA insert encoding a partial length of the GW182 protein was subcloned into pET28 (Novagen, Madison, Wis., USA). Escherichia coli JM109 (DE3) was transformed with this subclone, and the recombinant protein produced was purified using Ni2+ affinity chromatography as per the manufacturer’s instructions (Qiagen, Valencia, Calif., USA). This recombinant protein was subsequently used in the laser bead immunoassay described below.
Laser bead immunoassay
A set of addressable beads bearing laser reactive dyes (Luminex, Austin, Tex.,USA) were selected to couple the recombinant purified GW182 protein. Unless otherwise specified, all incubations and reactions were conducted at room temperature. Ten micrograms of 1-ethyl-3-(3-dimethylaminopropyl) carboiimide hydrochloride (Pierce, Rockford, Ill., USA) and N-hydroxysuccinimide (Pierce) was placed in separate microcentrifuge tubes (USA Scientific) and dissolved in 200 µl activation buffer (0.1 M sodium phosphate, pH 7.2). Of the laser bead suspension 100 µl was placed into a microcentrifuge tube and centrifuged at 10,000 rpm in a microcentrifuge for 1 min, and the fluid was decanted. Forty microliters of activation buffer was added to the pelleted beads, and they were gently resuspended by brief sonication and vortexing. Five microliters of 1-ethyl-3-(3-dimethylaminopropyl) carboiimide hydrochloride and N-hydroxysuccinimide was added in sequence to the resuspended microspheres, followed by brief sonication and vortexing. The suspended spheres were incubated in the dark for 20 min before the purified recombinant GW182 protein, dissolved in coupling buffer (0.14 M NaCl, 0.01 M NaPO4, pH approx. 7.2: PBS), was added to the mixture. After an additional incubation in the dark for 3 min, the suspension was centrifuged at 13,000 rpm for 3 min. The fluid was decanted and 125 µl coupling buffer added. The spheres were resuspended by sonication and vortexing as above before repelleting by centrifugation at 13,000 rpm for 3 min. The supernatant was decanted, 125 µl the protein solution (50 µg/ml) added, and the beads resuspended by sonication and vortexing. The protein and sphere suspension were incubated for 1 h at room temperature in the dark. The protein-coupled microspheres were pelleted by centrifugation at 10,000 rpm for 2 min and then resuspended in 125 µl washing buffer (PBS pH 7.2, 0.05% Tween 20). After two cycles of resuspension and pelleting in 125 µl blocking/storage buffer (0.5% BSA in PBS), the beads were stored as a suspension in 100 µl of blocking buffer at 2–8°C until required for use.
To analyze reactivity of the sera with the bound GW182, patient sera were diluted in Quanta Plex sample diluent (INOVA, San Diego, Calif., USA) to a final dilution of 1/1,000. To each well 40 µl of bead stock (1 part microspheres in blocking buffer to 40 parts Quanta Plex sample diluent) and 10 µl of diluted patient sera were added and incubated for 30 min on an orbital shaker. Then 50 µl phycoerythrin-conjugated goat anti-human IgG (Jackson ImmunoResearch) diluted 1/50 was added to each well and incubated on the orbital shaker for an additional 30 min. The reactivity of the antigen-coated beads was determined on a Luminex 100 dual-laser flow cytometer (Luminex). Control negative and standard positive sera were included in each assay. The tests were semiquantitative, and the results were expressed as median fluorescent intensity of the test sample.
Line immunoassay
The serum samples were tested for reactivity to other autoantigens using a “line” assay that includes recombinant and native SmB, SS-A/Ro52, SS-A/Ro60, SS-B/la, U1-RNP, Scl-70, ribo P antigens located on a solid phase strip (INNO-LIA, Innogenetics, Norcross, Ga., USA). The assays were performed according to the manufacturer’s instructions, and at the completion of the assay the strips were dried and were interpreted based on visual comparison of the intensity of the bands on the test strip to the cutoff control on another strip.
Results
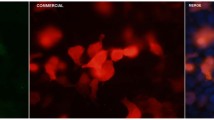
IIF using the index human serum on HEp-2 cells showed a pattern of distinctive cytoplasmic dots and what was previously described as GWBs (Fig. 1a). The number of GWBs present in HEp-2 cells varied from zero in mitotic cells to more than 30 in interphase cells. Previously it was shown that GWBs containing the GW182 autoantigen are distinguished from other cytoplasmic organelles, including the Golgi complex, lysosomes, endosomes, and proteasomes [11]. Over a 14-month period the clinical reference laboratory (Advanced Diagnostics Laboratory, University of Calgary) received approximately 5,000 sera for autoantibody analysis as requested by physicians who were investigating the presence of autoimmune disease, such as SLE and SjS, in their patients. From these 5,000 serum samples approx. 200 sera showed a cytoplasmic speckled staining pattern on HEp-2 cells. Of these 200 sera 18 (9%) had autoantibodies to the GWBs as determined by colocalization with the monoclonal antibody 4B6 that reacts with the GW182 protein and stains the GWBs (Fig. 1). The other sera had antibodies to early endosome antigen 1 [21], ribosomal RNP [22], mitochondria [23], cytoplasmic linker protein (CLIP-170) [24], and other as yet unknown endosome or lysosome antigens. None of the 18 sera that bound GWBs had antibodies to dsDNA, chromatin, U1-RNP, topoisomerase I (Scl-70), fibrillarin (U3 RNP), or centromeres/kinetochores [10]. The immunoglobulin isotype of all sera with antibodies to GWBs was IgG as shown by isotype-specific staining of HEp-2 cells, immunoblotting, and protein A Sepharose immunoprecipitation of recombinant GW182 protein. The anti-GWB titers as determined by IIF on HEp-2 cell substrates ranged from 1/320–1/5,120. A study of 2500 healthy female blood donors showed that none of these samples contained anti-GWB antibodies as determined by IIF using HEp-2 cells [25].

IIF colocalization studies using human and murine monoclonal anti-GWB antibodies. Cytoplasmic bodies in HEp-2 cells detected with the index patient serum diluted 1/100 (a, b) colocalized with the staining of a monoclonal antibody 4B6 to GW182 (c). The nuclei are stained blue with 4′,6-diamidino-2-phenylindole (d) and the merged images are shown in e. White bar (b) 5 µm
Although all 18 sera had antibodies to the GW body, the multiplexed laser bead assay indicated that 4 of the 18 sera (nos. 1, 3, 9, 10) recognized the recombinant GW182 protein which is one of several proteins found within GWBs (Table 1). When the reactivity of the 18 sera was also tested by IP using in vitro transcribed/translated protein, it was observed that 4 sera (nos. 1, 3, 8, 10) IP the GW182 protein (Fig. 2). Therefore when the data of the two assays that used recombinant protein are combined, 5 of the 18 sera recognized GW182.

Immunoprecipitation of the 35S-labeled GW182 TnT recombinant GW182 protein with patient sera. Four sera (lanes 1–4) that stained GWBs, immunoprecipitation IP the recombinant GW182 (TnT), but normal human serum (NHS) did not. MW 14C molecular weight markers
The clinical data obtained on the 18 patients who had the GWB staining pattern are summarized in Table 1. Of the 18 patients 17 (39%) with autoantibodies to the GWBs were women and ranged in age from 46 to 85 years (mean 58). The clinical diagnoses could be stratified into three groups: group A composed of 9 patients had predominantly mixed motor and/or sensory neuropathy, although other disease manifestations were also noted; 3 patients in group B had SjS in addition to some neurological features that overlapped with group A; in group C there were 6 patients who had SLE and/or SjS without documented evidence of neurological disease. When the various diagnoses or clinical conditions were tabulated individually, SjS was the most common, seen in 7 of 18 (39%), followed by patients with neurological disease (motor and sensory neuropathy and/or ataxia) in 6 (33%), followed by SLE in 4 (22%).
When it was observed that some of the patients had SLE and SjS, we were interested to determine whether autoantibodies to known autoantigens that are typical markers of SLE and SjS were present. Autoantibodies to SS-A/Ro and SS-B/la were correlated with the diagnosis of SjS in 6/7 patients diagnosed with SjS (Table 1). However, four patients in group A had anti-SSA/Ro52 antibodies but did not have a clinical diagnosis of SjS or SLE. Interestingly, 9 sera had antibodies to the 52-kDa SS-A/Ro antigen, but 7 did not have coexisting antibodies to the 60 kDa SS-A/Ro antigen. One patient (no. 4) had a malar rash, arthralgia, and antibodies to the SmB protein but did not fulfill criteria [26] for classification as definite SLE.
Only 4 of the 18 patient sera (22%; nos. 1, 3, 8, 10) with anti-GWB antibodies as defined by colocalization, IP the GW182 protein. Three of these four sera (nos. 1, 3, 10; Table 1) were used for epitope mapping due to limited quantity available for the fourth serum (no. 8). Multiple epitopes over the entire length of GW182 were recognized by the patient sera (Figs. 3, 4). Four overlapping reactive peptides were shared between patient no. 1 and patient no. 10: amino acids 666–695, 951–970, 1676–1690, 1691–1705. Several peptides were in common between patient no. 1 and patient no. 3: amino acids 431–450, 766–780, 921–945, 951–970, 1101–1115, 1161–1185, 1191–1205, 1391–1410, 1431–1445, 1616–1630. Interestingly, only one peptide (1511–1525) was bound by both patient no. 10 and patient no. 3. The reactive epitopes mapped to the GW-rich, the middle portion, the non-GW rich, and the C-terminal domains of the GW182 protein (Fig. 4). When the reactive peptides were subjected to a BLAST analysis, only the published GW182 protein and related EST clones, KIAA1460, KIAA1582, and KIAA1093 showed more than 60% amino acid sequence identity. The KIAA1460 EST is known to be partial-length GW182 [11].

Epitope mapping obtained using sequential 15mer peptides offset by five amino acids that represented the full-length GW182 protein were spotted on membranes and then probed with a normal human serum (NHS) and three sera with anti-GWB antibodies (patients 1, 3, 10)

Amino acid sequence and position of the GW182 protein synthetic peptides and their reactivity with three patient sera with anti-GWB antibodies. Gradient of white to black Increasing intensity of reaction of antibodies with peptide
Discussion
In this study we report the clinical features of 18 patients who have autoantibodies to a unique structure within the cytoplasm which we previously named GW bodies or GWBs. Five of the 18 sera that colocalized with the murine monoclonal antibody to GW182 (monoclonal antibody 4B6) which stains the GW bodies, reacted with the GW182 protein in two different immunoassays. This suggests that either the epitopes reactive by IIF are not present in the recombinant proteins used in these assays, or that GWBs contain target autoantigens that remain to be defined. Studies are underway to define additional autoantigens in GWBs.
The incidence of anti-GWB antibodies reported in this study was 0.36%. In the Advanced Diagnostics Laboratory at the University of Calgary, this approaches the incidence of autoantibodies to Sm and Golgi antigens (0.4% and 0.5%, respectively) and was higher than antibodies to proliferating cell nuclear antigen (0.1%), Jo-1 (histidyl t-RNA synthetase (0.1%) and Scl-70 (topoisomerase I, 0.3%; unpublished data).
It is not clear whether the autoantibodies directed against GWBs and GW182 are pathogenic. The clinical diagnosis of patients with GWB autoantibodies included SjS, motor or sensory neuropathies, SLE, and a variety of other clinical conditions. Observations of mRNA processing may be relevant because particles containing mRNA and the human protein staufen have been observed in neurons [27, 28]. Staufen binds double-stranded RNA and was visualized in RNA containing particles in rat hippocampal neurons after transient transfection experiments [28, 29]. It may be relevant that the GW182 autoantigen was also shown to bind mRNA through its RNA binding motif [11]. It is interesting to speculate that the storage of mRNA by GWBs may be an important process in maintenance of neurons and neurotransmission and that disruption of GW182 function by autoantibodies may affect neural integrity and subsequent motor/sensory neurological disease. This view is supported by preliminary data suggesting that GW182 is highly expressed in neural tissues (unpublished observations). Recent evidence suggests that GW182 and GW bodies are involved in mRNA decapping and subsequent mRNA degradation [14]. It is interesting to speculate that disruption of the GW182 protein and/or GW bodies by the presence of autoantibodies affect one aspect of the mRNA degradation pathway vital in the overall maintenance and function of the cell. Although we have not determined whether mRNA degradation in the GWBs is directly related GW182 function in nonstop [30] or nonsense-mediated mRNA decay [31, 32], the failure to degrade problematic mRNAs with no stop codons or premature termination codons may have pathological consequences on the function of the cell and subsequently be manifest as a disease state.
Our study shows that multiple epitopes of the GW182 protein are recognized by the human antibodies. The SPOT method of epitope mapping has been validated, and the majority of studies has shown that each patient displays an individual epitope pattern [6]. The diverse and heterogenic epitope recognition pattern among the patients observed in this study is not unlikely since the fine specificity of B-cell immune processes strongly depends on the MHC system. Epitope mapping followed by BLAST analysis confirmed that the autoantibody targets are unique to the GW182 protein because sequence similarity to other known eukaryotic or prokaryotic proteins or expressed sequence tags was not observed. This suggests that the GW182 protein drives the autoimmune response and reactivity to endogenous or exogenous proteins with similar sequence motifs and molecular mimicry is less likely. This also raises the possibility that, as with many other autoantibody systems, autoreactivity to GW182 demonstrates intramolecular epitope spreading [33, 34]. A study using more sera and different methods in an extended epitope mapping study should shed more light on the epitope distribution on GW182.
The association of anti-GWB antibodies with antibodies to the 52 kDa SS-A/Ro antigen, particularly in the patients with no evidence of SjS and SLE was an unexpected finding. Although the 52-kDa SS-A/Ro antigen has been localized to both the nucleus and cytoplasm, antibodies from a variety of sources directed to the 52-kDa SS-A/Ro autoantigen do not produce a GWB staining pattern [35, 36]. The function of the 52 kDa SS-A/Ro antigen is not clear [37], and the observation that it is associated with GWB antibodies may help clarify its function.
In summary, GWBs are a novel class of RNP autoantigens that are specifically recognized by human autoantibodies. Over the past three decades several autoantigens that are part of RNP macromolecular complexes have been described, and we propose that autoantibodies to GWBs and GW182 now join this growing list (Table 2). Some of these autoantigens, including Sm, U1-RNP, and Hu, have been shown to have a central role in mRNA splicing, mRNA processing, and mRNA translation [37, 38, 39]. In this study we observe that the diseases associated with autoantibodies to GWBs overlap with those associated with other RNPs but extend to patients who appear to have primary neurological disorders.
Abbreviations
- GWB :
-
Glycine tryptophan-rich cytoplasmic structure
- IIF :
-
Indirect immunofluorescence
- IP :
-
Immunoprecipitation
- NET2 :
-
NaCl, EDTA, Tris buffer
- SDS :
-
Sodium dodecyl sulfate
- SjS :
-
Sjögren’s syndrome
- SLE :
-
Systemic lupus erythematosus
- TBS :
-
Tris-buffered saline
- TnT :
-
Transcription and translation
References
Hassfeld W, Steiner G, Studnicka-Benke A, Skriner K, Graninger W, Fischer I, Smolen JS (1995) Autoimmune response to the spliceosome. An immunologic link between rheumatoid arthritis, mixed connective tissue disease, and systemic lupus erythematosus. Arthritis Rheum 38:777–785
Tan EM (1999) Autoantibodies in diagnosis and identifying autoantigens. Immunologist 7:85–92
Eenennaam H van, Vogelzangs JHP, Lugtenberg D, van den Hoogen FHJ, Van Venrooij WJ, Pruijn GJM (2002) Identity of the RNase MRP- and RNase P-associated Th/To autoantigen. Arthritis Rheum 46:3266–3272
Muhlen CA von, Chan EKL, Angles-Cano E, Mamula MJ, Garcia-de la Torre I, Fritzler MJ (1998) Advances in autoantibodies in SLE. Lupus 7:507–514
Fritzler MJ, Schoenroth LJ (2003) Advances in understanding and use of autoantibodies as markers of diseases. In: Sticherling M, Christophers E (eds) Treatment of autoimmune diseases. Springer, Vienna New York, pp 29–42
Mahler M, Blüthner M, Pollard KM (2003) Advances in B-cell epitope analysis of autoantigens in connective tissue diseases. Clin Immunol 107:65–79
Muhlen CA von, Tan EM (1995) Autoantibodies in the diagnosis of systemic rheumatic disease. Semin Arthritis Rheum 24:323–358
Chan EKL, Andrade LEC (1992) Antinuclear antibodies in Sjögren’s syndrome. Rheum Dis Clin North Am 18:551–570
Tan EM, Chan EKL, Sullivan KF, Rubin RL (1988) Short analytical review—antinuclear antibodies (ANAs): diagnostically specific immune markers and clues toward the understanding of systemic autoimmunity. Clin Immunol Immunopathol 47:121–141
Fritzler MJ (1997) Autoantibodies: diagnostic fingerprints and etiologic perplexities. Clin Invest Med 20:50–66
Eystathioy T, Chan EKL, Tenenbaum SA, Keene JD, Griffith KJ, Fritzler MJ (2002) A phosphorylated cytoplasmic autoantigen, GW182, associates with a unique population of human mRNAs within novel cytoplasmic speckles. Mol Biol Cell 13:1338–1351
Tenenbaum SA, Lager PJ, Carson CC, Keene JD (2002) Ribonomics: identifying mRNA subsets in mRNP complexes using antibodies to RNA-binding proteins and genomic arrays. Methods 26:191–198
Keene JD, Tenenbaum SA (2002) Eukaryotic mRNPs may represent post-transcriptional operons. Mol Cell 9:1161–1167
Eystathioy T, Jakymiw A, Chan EKL, Séraphin B, Cougot N, Fritzler MJ (2002) The GW182 protein co-localizes with mRNA degradation associated proteins hDcp1 and hLSm4 in cytoplasmic GW bodies. RNA 9:1171–1173
Eystathioy T, Chan EKL, Mahler M, Luft LM, Fritzler ML, Fritzler MJ (2003) A panel of monoclonal antibodies to cytoplasmic GW bodies and the mRNA binding protein GW182. Hybridoma Hybridomics 22:79–86
Griffith KJ, Chan EKL, Hamel JC, Miyachi K, Fritzler MJ (1997) Molecular characterization of a novel 97\kDa Golgi complex autoantigen recognized by autoimmune antibodies from patients with Sjögren’s syndrome. Arthritis Rheum 40:1693–1702
Laemmli UK (1970) Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227:680
Frank R (1992) SPOT-synthesis: an easy technique for the postionally addressable, parallel chemical synthesis on a membrane support. Tetrahedron 48:9217–9232
Mahler M, Mierau R, Blüthner M (2000) Fine-specificity of the anti-CENP-A B-cell autoimmune response. J Mol Med 78:460–467
Gausepohl H, Behn C (2002) Automated synthesis of solid-phase bound peptides. In: Koch J, Mahler M (eds) Peptide arrays on membranes-synthesis and applications. Springer, Berlin Heidelberg New York, pp 55–69
Selak S, Schoenroth L, Senécal J-L, Fritzler MJ (1999) Early endosome antigen 1: an autoantigen associated with neurological diseases. J Investig Med 47:311–318
Mahler M, Kessenbrock K, Raats J, Williams RC Jr, Fritzler MJ (2003) Characterization of the human autoimmune response to the major C-terminal epitope of the ribosomal P proteins. J Mol Med 81:194–204
Fritzler MJ, Manns MP (2002) Anti-mitochondrial antibodies. Clin Appl Immunol Rev 3:87–113
Griffith KJ, Ryan JP, Senécal J-L, Fritzler MJ (2002) The cytoplasmic linker protein CLIP-170 is a human autoantigen. Clin Exp Immunol 127:533–538
Fritzler MJ, Pauls JD, Kinsella TD, Bowen TJ (1985) Antinuclear, anticytoplasmic and anti-Sjögren’s syndrome antigen-A (SS-A/Ro) antibodies in female blood donors. Clin Immunol Immunopathol 36:120–128
Tan EM, Cohen AS, Fries JF, Masi AT, McShane DJ, Rothfield NF, Schaller JG, Talal N, Winchester RJ (1982) The 1982 revised criteria for the classification of systemic lupus erythematosus. Arthritis Rheum 25:1271–1277
Kuhl D, Skehel P (1998) Dendritic localization of mRNAs. Curr Opin Cell Biol 8:600–806
Kiebler MA, Hemraj I, Verkade P, Kohrmann M, Fortes P, Marion RM, Ortin J, Dotti CG (1999) The mammalian staufen protein localizes to the somatodendritic domain of cultured hippocampal neurons: implications for its involvement in mRNA transport. J Neurosci 19:288–297
Kohrmann M, Luo M, Kaether C, DesGroseillers L, Dotti CG, Kiebler MA (1999) Microtubule-dependent recruitment of staufen-green fluorescent protein into large RNA-containing granules and subsequent dendritic transport in living hippocampal neurons. Mol Biol Cell 10:2945–2953
Vasudevan S, Peltz SW, Wilusz CJ (2002) Non-stop decay-a new mRNA surveillance pathway. Bioessays 24:785–788
Danckwardt S, Neu-Yilik G, Thermann R, Frede U, Hentze MW, Kulozik AE (2002) Abnormally spliced beta-globin mRNAs: a single point mutation generates transcripts sensitive and insensitive to nonsense-mediated mRNA decay. Blood 99:1811–1816
Wilusz CJ, Wormington M, Peltz SW (2001) The cap-to-tail guide to mRNA turnover. Nat Rev Mol Cell Biol 2:237–246
Arbuckle MA, Reichlin M, Harley JB, James JA (1999) The development of lupus humoral autoimmunity for anti-Sm autoantibodies is consistent with predictable sequential B-cell epitope spreading. Scand J Immunol 50:447–455
Monneaux F, Muller S (2002) Epitope spreading in systemic lupus erythematosus-Identification of triggering peptide sequences. Arthritis Rheum 46:1430–1438
Schmitz M, Bachmann M, Laubinger J, Thijssen JP, Pruijn GJ (1997) Characterization of murine monoclonal antibodies against the Ro52 autoantigen. Clin Exp Immunol 110:53–62
Kelekar A, Saitta MR, Keene JD (1994) Molecular composition of Ro small ribonucleoprotein complexes in human cells: intracellular localization of the 60- and 52-kD proteins. J Clin Invest 93:1637–1644
Rhodes DA, Ihrke G, Reinicke AT, Malcherek G, Towey M, Isenberg DA, Trowsdale J (2002) The 52 000\MW Ro/SS-A autoantigen in Sjögren’s syndrome/systemic lupus erythematosus (Ro52) is an interferon-gamma inducible tripartite motif protein associated with membrane proximal structures. Immunology 106:246–256
Lerner MR, Steitz JA (1979) Antibodies to small nuclear RNAs complexed with proteins are produced by patients with systemic lupus erythematosus. Proc Natl Acad Sci USA 76:5495–5499
Tan EM (1989) Antinuclear antibodies: diagnostic markers for autoimmune diseases and probes for cell biology. Adv Immunol 44:93–151
Acknowledgements
We acknowledge the technical assistance of Joan Miller, Cheryl Hanson, Jill Wenger (University of Calgary) and Dr. Zheng Yang (Scripps Research Institute). This work was supported in part by the Canadian Institutes for Health Research Grant MOP-57674 and the National Institutes of Health Grants AR42455, AI47859 and AI39645. M.J.F holds the Arthritis Society Chair at the University of Calgary.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Eystathioy, T., Chan, E.K.L., Takeuchi, K. et al. Clinical and serological associations of autoantibodies to GW bodies and a novel cytoplasmic autoantigen GW182. J Mol Med 81, 811–818 (2003). https://doi.org/10.1007/s00109-003-0495-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00109-003-0495-y