
Abstract
Geminin, a key regulator of DNA replication licensing in the cell cycle, plays an essential role in determining the fate of cells via suppression of cell proliferation and cellular differentiation. Neuropeptide Y (NPY) intensifies the proliferation of vascular smooth muscle cells (VSMCs) directly by binding with Y1 receptors. In vitro experiments have shown that stimulation of NPY on VSMCs via regulation of geminin is a double-edged sword. Given that the proliferation and the phenotypic transformation of VSMCs increase the risk for progression of atherosclerosis, we focus on the role of geminin interference in determining the fate of VSMCs. Furthermore, we discuss the therapeutic potential of peripheral neurotransmitter interference, thus pointing toward future research directions in the treatment of atherosclerosis.
Zusammenfassung
Geminin, eine wesentlicher Regulator der Lizensierung der DNA-Replikation im Zellzyklus, spielt eine wichtige Rolle in der Bestimmung des Zellschicksals via Suppression der Zellproliferation und Zelldifferenzierung. Neuropeptid Y (NPY) verstärkt die Proliferation vaskulärer glattmuskulärer Zellen („vascular smooth muscle cells“, VSMC) unmittelbar durch Bindung an Y1-Rezeptoren. In-vitro-Versuche haben gezeigt, dass die Stimulation von NPY auf VSMC via Regulation von Geminin ein zweischneidiges Schwert darstellt. Angesichts dessen, dass die Proliferation und die phänotypische Transformation von VSMC das Risiko der Progression von Atherosklerose erhöht, richteten die Autoren ihr Augenmerk auf die Rolle der Beeinflussung von Geminin bei der Bestimmung des Schicksals von VSMC. Darüber hinaus erörtern die Autoren das therapeutische Potenzial der Beeinflussung peripherer Neurotransmitter, um eine Richtung zukünftiger Forschung für die Therapie der Atherosklerose aufzuzeigen.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Atherosclerosis, a major cause of cardiovascular disease [1, 2], involves multiple processes including endothelial cell dysfunction, inflammation, vascular smooth muscle cell (VSMC) proliferation, and matrix alteration.
VSMC proliferation is closely connected to other cellular processes such as inflammation and matrix alterations [3]. More specifically, the switch of VSMCs from contractile type (differentiation) to synthetic type (dedifferentiation) is modulated by growth factors and cytokines secreted during the process of atherosclerosis in vivo. Recent studies showed that the proliferative potential of VSMCs was increased through silencing of geminin [4], which was first identified by Kirschner and colleagues as an inhibitor of DNA replication licensing and a neuralizing factor related to neural cell fate [5, 6]. Geminin, a nuclear protein, is involved in differentiation and cell fate decisions mainly through binding with Cdt1 and Brahma/Brg1 to control the suppression of cellular proliferation and to maintain the undifferentiated state of cells, respectively [7,8,9,10].
Sympathetic nervous activity is also implicated in VSMC proliferation. Neuropeptide Y (NPY), a ubiquitous sympathetic neurotransmitter originally recognized as an appetite stimulant and a vasoconstrictor, recently emerged as a growth factor for a variety of cells including VSMCs. An intriguing observation is that the proliferation-promoting effect of NPY only functions in contractile VSMCs [11]. Moreover, there are multiple receptors, Y1–Y5, mediating the pleiotropic activities of NPY, of which the Y1 receptor is the predominant NPY vascular receptor responsible for accelerating the proliferation of VSMCs. Moreover, norepinephrine coexisting with NPY can also affect VSMC growth by promoting the expression of geminin [12,13,14,15].
With improved understanding of the molecular mechanism of atherosclerosis, the suppression of VSMC growth by targeting cell cycle regulation may be a feasible strategy. In addition, new insights into the role that geminin plays in VSMCs may generate new ideas on treating arteriosclerotic cardiovascular disease in clinical practice.
In this review, we focus on the molecular mechanism of the neurotransmitters of sympathetic postganglionic neurons, especially NPY, that interfere with the proliferation of different VSMCs via regulation of geminin; we also discuss the therapeutic potential of targeting NPY in atherosclerosis from a different perspective.
Molecular function of geminin
In metazoan cells, DNA replication is a complicated process that involves a variety of protein factors. The process must occur once per cell cycle in order to maintain genome integrity. It has been reported that geminin acts as a key regulator of DNA replication licensing through direct binding to Cdt1 and by inhibiting the incorporation of the mini-chromosome maintenance (MCM) complex into the pre-replication complex (pre-RC), thus preventing re-replication in the S phase [8, 16, 17]. The underlying mechanism of cell cycle regulation by geminin is complicated, but there is evidence that a high expression of geminin plays an essential role in suppressing cell proliferation [8]. In addition, geminin can also govern cellular differentiation, not only by regulation of DNA replication, but also through binding of transcription factors such as the Homeobox family [18] and by preventing stem cell differentiation by maintaining the transcription of specific target genes via inhibition of chromatin remodeling activity [7]. Geminin expression determines the fate of a wide variety of cells, for instance, downregulation of geminin protein expression sustains the proliferation potential of hematopoietic stem cells (HSC) and hematopoietic progenitor cells (HPC; [19,20,21]). Besides, the level of geminin expression in cancer cells is significantly higher than in normal cells in order to prevent the accumulation of stalled replication forks caused by DNA re-replication and avoid apoptosis [22, 23]. Notably, previous studies suggested that there were similar correlations in VSMCs.
Geminin and vascular smooth muscle cells
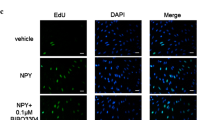
Zhang and colleagues [4] found that geminin silencing markedly increased the incorporation of 3H-thymidine and EdU in A10, a rat thoracic aortic smooth muscle cell line; this demonstrated that knockdown of geminin can promote DNA synthesis and proliferation of VSMCs. Furthermore, flow cytometry analysis showed that compared with control groups, silencing geminin in VSMCs led to a decrease of G0/G1 cells and an increase of S‑phase cells. Considering that CDK1 is a binding partner of geminin and CDK1 degradation can inhibit replication licensing [24, 25], the researchers investigated whether geminin gene silencing could prevent the degradation of CDK1 and enhance the proliferation potential of VSMCs based on protein and mRNA levels. Collectively, the results of the study indicated that downregulation of geminin facilitated G0/G1–S cell-cycle progression and upregulation of CDK1, which ultimately stimulated the proliferation of VSMCs. In another study, however, the depletion or overexpression of geminin did not significantly affect the proliferation of VSMCs compared with controls [26]. The reason for such diverse results may be due to the different ways of silencing geminin or differences in cell samples.
VSMCs can acquire different functions through phenotypic transformation from a contractile type (differentiation) to a synthetic type (dedifferentiation). VSMC shuttling between the different phenotypes is based on the specific morphology, particularly the varying levels of the marker proteins, and on changes of proliferation potential and migration rates that are higher in synthetic VSMCs than in contractile VSMCs [27, 28]. Interestingly, it has also been reported that geminin, always located in the nucleus, can be excluded from the nucleus to the cytoplasm when contractile VSMCs transformed into synthetic VSMCs, and the same subcellular regulation of geminin was reported in Michigan Cancer Foundation-7 (MCF7) cells as well [29]. Taken together, these data reveal that cell growth and specific cell types are interrelated with the expression levels and subcellular location of geminin, respectively, offering new insight into the shaping of cell destiny by geminin.
Neuronal axis of geminin involved in atherosclerosis
Neurotransmitters of sympathetic postganglionic neurons
As previously noted, increased sympathetic nervous system activity is increasingly regarded as a crucial mechanism underlying cardiovascular complications in humans [30]. In the periphery, elevated levels of circulating norepinephrine, the main neurotransmitter of sympathetic postganglionic neurons, prompt an increase in heart rate, cardiac contractility, vascular tone, and renin–angiotensin system activity [31, 32], making norepinephrine a major contributor to cardiovascular disease. It has been proven that norepinephrine can promote the expression levels of geminin in VSMCs and affect their growth [26]. Interestingly, NPY coexisting with norepinephrine at sympathetic nerve endings in peripheral regions is an endogenous vasoconstrictor and is released with norepinephrine in response to direct stimulation of cardiac sympathetic neurons [12, 13]. On the one hand, in the central nervous system, an increase in NPY in the paraventricular nucleus (PVN) or lateral ventricle can reduce the release of norepinephrine and decrease the activity of peripheral sympathetic nerves, heart rate, and blood pressure. On the other hand, NPY directly binds with the Y1 receptor located on the surface of VSMCs to strengthen the vasoconstrictor effect of norepinephrine. Consequently, sympathetic nervous activity correlates strongly with the pathophysiological process of atherosclerosis via neurotransmitters directly or indirectly affecting vascular endothelial dysfunction and the proliferation of VSMCs.
Geminin and NPY-Y1 receptor system
Besides potentiating norepinephrine activity, NPY significantly enhances the angiotensin II-induced vasoconstrictor response and intensifies the proliferation of VSMCs directly by binding with Y1 receptors to promote atherosclerosis [14, 33]. Moreover, it has been demonstrated that the switch of VSMCs from contractile type (differentiation) to synthetic type (dedifferentiation) plays a critical role in atherosclerosis, and these cell type changes are regulated by cytokines released in atherosclerosis lesions or serum concentrations in vitro [11, 34]. Jiang and colleagues [11] stimulated A10 cells with NPY in different concentrations of serum in order to explore the role of NPY in the proliferation of different VSMC phenotypes in the pathogenesis of atherosclerosis. The results indicated that NPY only promoted the proliferation of VSMCs incubated with 0.5% rather than 10% serum, while VSMCs remained in a differentiated and dedifferentiated state, respectively. Moreover, the authors observed a steep increase of geminin in synthetic-type VSMCs with the presence of NPY, which was attenuated by antagonism of Y1 receptors. Furthermore, geminin prevented DNA replication during the S phase of the cell cycle, and upregulation of geminin induced cell cycle arrest in the S phase. Flow cytometry analysis indicated that more cells remained in the S phase after administration of NPY in synthetic-type VSMCs, which may be mediated by increased expression of geminin.
In the cardiovascular system, harmful vasoconstrictive and proliferative effects of NPY are primarily mediated by Y1 receptors [35]. In in vitro experiments, the proliferative effects of NPY on human pulmonary arterial smooth muscle cells were inhibited in a concentration-dependent way by selective Y1 receptor antagonists [36], while the percentage of cells in the S phase significantly increased via activation of the NPY-Y1 receptor agonist on VSMCs. Thus, this set of results clearly identifies the close connection between VSMC growth induced by NPY and the underlying mechanisms of geminin involvement in atherosclerosis.
Multiple factors released by different cell types such as endothelial cell, platelets, and inflammatory cells induce an aberrant phenotypic switch of VSMCs during atherosclerosis. Additionally, NPY release is long-lastingly activated during the development of atherosclerosis; thus, understanding the relationship between geminin and the NPY-Y1 receptor system may provide exciting insights into treatments for atherosclerosis (Fig. 1).

Effects of the NPY-Y1 receptor system on VSMC proliferation in atherosclerosis via regulation of geminin. In atherosclerosis lesions, multiple regulatory factors are released by different cell types, such as endothelial cell, platelets, and inflammatory cells, which induce an aberrant phenotypic switch of VSMCs from contractile type to synthetic type. Geminin expression in synthetic VSMCs is upregulated by binding of NPY and Y1 receptors that prevents DNA replication during the S phase and leads to cell cycle arrest in the S phase. Thus, stimulation of NPY on VSMCs could be a double-edged sword in the pathogenesis of atherosclerosis. NPY neuropeptide Y, VSMCs vascular smooth muscle cells
Discussion
Atherosclerosis and its clinical complications such as stroke are common diseases that pose a health threat worldwide. Since Ross proposed that atherosclerosis was a chronic inflammatory disease [37], it has been gradually widely accepted that inflammation as the response to injury represents the pathogenesis of atherosclerosis. On the one hand, the sympathetic nervous system plays a central role in atherosclerosis through release of neurotransmitters to induce inflammation. On the other hand, neurotransmitters such as norepinephrine can interfere with geminin and ultimately affect VSMC proliferation that is linked to atherosclerotic inflammation. Most importantly, the proliferative effects of another neurotransmitter, NPY, on VSMCs can be inhibited by the selective Y1 receptor antagonist [36].
Conclusion
New insights into the neuronal axis of geminin and Y1 receptor antagonist to regulate levels of geminin in VSMCs may provide important knowledge for the treatment of atherosclerosis. This therapy would be aimed at alleviating the side effects of current treatments and lead to a more rational strategy in preventing diseases related to the abnormal transformation of VSMCs.
Abbreviations
- HPC:
-
Hematopoietic progenitor cell
- HSCs:
-
Hhematopoietic stem cells
- MCM:
-
Mini-chromosome maintenance complex
- NPY:
-
Neuropeptide Y
- pre-RC:
-
Pre-replication complex
- VSMCs:
-
Vascular smooth muscle cells
References
Legein B et al (2013) Inflammation and immune system interactions in atherosclerosis. Cell Mol Life Sci 70(20):3847–3869
Yahagi K et al (2016) Pathophysiology of native coronary, vein graft, and in-stent atherosclerosis. Nat Rev Cardiol 13(2):79–98
Dzau VJ, Braun-Dullaeus RC, Sedding DG (2002) Vascular proliferation and atherosclerosis: new perspectives and therapeutic strategies. Nat Med 8(11):1249–1256
Zhang Y et al (2014) Geminin interference facilitates vascular smooth muscle cell proliferation by upregulation of CDK-1. Cardiovasc Drugs Ther 28(5):407–414
McGarry TJ, Kirschner MW (1998) Geminin, an inhibitor of DNA replication, is degraded during mitosis. Cell 93(6):1043–1053
Kroll KL et al (1998) Geminin, a neuralizing molecule that demarcates the future neural plate at the onset of gastrulation. Development 125(16):3247–3258
Seo S et al (2005) Geminin regulates neuronal differentiation by antagonizing Brg1 activity. Genes Dev 19(14):1723–1734
Yasunaga S et al (2016) Role of Geminin in cell fate determination of hematopoietic stem cells (HSCs). Int J Hematol 104(3):324–329. https://doi.org/10.1007/s12185-016-2060-9
Wohlschlegel JA et al (2000) Inhibition of eukaryotic DNA replication by geminin binding to Cdt1. Science 290(5500):2309–2312
Saxena S, Dutta A (2005) Geminin-Cdt1 balance is critical for genetic stability. Mutat Res 569(1–2):111–121
Jiang ZQ et al (2017) Different effects of neuropeptide Y on proliferation of vascular smooth muscle cells via regulation of Geminin. Mol Cell Biochem 433(1-2):205–211. https://doi.org/10.1007/s11010-017-3028-7
Warner MR et al (1991) Sympathetic stimulation-evoked overflow of norepinephrine and neuropeptide Y from the heart. Circ Res 69(2):455–465
Herring N (2015) Autonomic control of the heart: going beyond the classical neurotransmitters. Exp Physiol 100(4):354–358
Zhu P et al (2016) The role of neuropeptide Y in the pathophysiology of atherosclerotic cardiovascular disease. Int J Cardiol 220:235–241
Zukowska Z (2005) Atherosclerosis and angiogenesis: what do nerves have to do with it? Pharmacol Rep 57(Suppl):229–234
Karamitros D et al (2014) Life without geminin. Cell Cycle 9(16):3201–3205
Suchyta M, Miotto B, McGarry TJ (2015) An inactive geminin mutant that binds cdt1. Genes (Basel) 6(2):252–266
Zhou B et al (2012) Structural basis for homeodomain recognition by the cell-cycle regulator Geminin. Proc Natl Acad Sci USA 109(23):8931–8936
Ohtsubo M et al (2008) Polycomb-group complex 1 acts as an E3 ubiquitin ligase for Geminin to sustain hematopoietic stem cell activity. Proc Natl Acad Sci USA 105(30):10396–10401
Ohno Y et al (2010) Hoxb4 transduction down-regulates Geminin protein, providing hematopoietic stem and progenitor cells with proliferation potential. Proc Natl Acad Sci USA 107(50):21529–21534
Ohno Y et al (2013) Hoxa9 transduction induces hematopoietic stem and progenitor cell activity through direct down-regulation of geminin protein. PLoS ONE 8(1):e53161
Haruki T et al (2011) Geminin expression in small lung adenocarcinomas: implication of prognostic significance. Lung Cancer 71(3):356–362
DePamphilis ML (2011) Spotlight on geminin. Breast Cancer Res 13(3):109
Liu E et al (2004) Cyclin-dependent kinases phosphorylate human Cdt1 and induce its degradation. J Biol Chem 279(17):17283–17288
Nishitani H et al (2001) The human licensing factor for DNA replication Cdt1 accumulates in G1 and is destabilized after initiation of S‑phase. J Biol Chem 276(48):44905–44911
Guo J, Sun N (2013) Cell cycle regulator geminin is dispensable for the proliferation of vascular smooth muscle cells. Sci China Life Sci 56(8):731–738
Rensen SS, Doevendans PA, van Eys GJ (2007) Regulation and characteristics of vascular smooth muscle cell phenotypic diversity. Neth Heart J 15(3):100–108
Hao H, Gabbiani G, Bochaton-Piallat ML (2003) Arterial smooth muscle cell heterogeneity: implications for atherosclerosis and restenosis development. Arterioscler Thromb Vasc Biol 23(9):1510–1520
Dimaki M et al (2013) Cell cycle-dependent subcellular translocation of the human DNA licensing inhibitor geminin. J Biol Chem 288(33):23953–23963
Masuo K et al (2010) The role of sympathetic nervous activity in renal injury and end-stage renal disease. Hypertens Res 33(6):521–528
Schroeder C, Jordan J (2012) Norepinephrine transporter function and human cardiovascular disease. Am J Physiol Heart Circ Physiol 303(11):H1273–H1282
Katz DP et al (2016) Benzylpiperazine: “a messy drug”. Drug Alcohol Depend 164:1–7
Zukowska-Grojec Z et al (1998) Mechanisms of vascular growth-promoting effects of neuropeptide Y: role of its inducible receptors. Regul Pept 75–76:231–238
Saleh Al-Shehabi T, Iratni R, Eid AH (2016) Anti-atherosclerotic plants which modulate the phenotype of vascular smooth muscle cells. Phytomedicine 23(11):1068–1081
Abe K, Tilan JU, Zukowska Z (2007) NPY and NPY receptors in vascular remodeling. Curr Top Med Chem 7(17):1704–1709
Crnkovic S et al (2014) NPY/Y(1) receptor-mediated vasoconstrictory and proliferative effects in pulmonary hypertension. Br J Pharmacol 171(16):3895–3907
Ross R (1993) The pathogenesis of atherosclerosis: a perspective for the 1990s. Nature 362(6423):801–809
Acknowledgements
This work was supported by Natural Science Foundation of China (grant numbers 81670402, 815703960).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
S.-y. Liang, Y.-l. Zhou, M.-q. Shu, and S. Lin declare that they have no competing interests.
This article does not contain any studies with human participants or animals performed by any of the authors.
Additional information
Author contributions
Shu Lin and Maoqin Shu are responsible for revising the manuscript and approving the final version. All authors read and approved the final manuscript.
Rights and permissions
About this article

Cite this article
Liang, Sy., Zhou, Yl., Shu, Mq. et al. Regulation of geminin by neuropeptide Y in vascular smooth muscle cell proliferation. Herz 44, 712–716 (2019). https://doi.org/10.1007/s00059-018-4721-3
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00059-018-4721-3